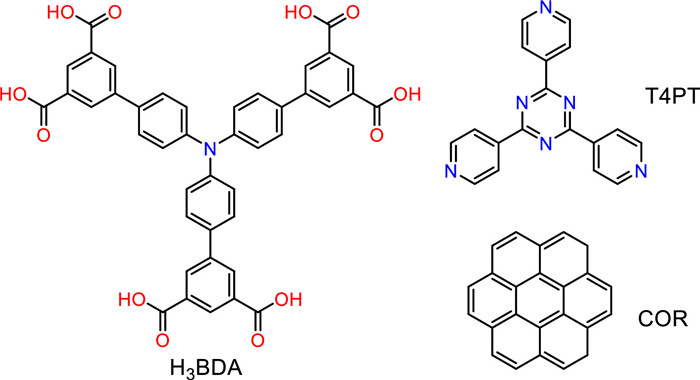
Figure 1.
Chemical structures of H3BDA, T4PT and COR.
Site-selection and recognition of aromatic carboxylic acid in response to coronene and pyridine derivative
Yuanjiao Liu , Xiaoyang Zhao , Songyao Zhang , Yi Wang , Yutuo Zheng , Xinrui Miao , Wenli Deng

Supramolecular self-assembly through a bottom-up strategy on solid surfaces is growing expeditiously due to potential applications in nanotechnology and molecular devices [1-7]. Diverse nanoporous networks with high crystallinity have been fabricated and are used as ideal templates to enclose guest molecules of various dimensions and contours [8-13]. In general, the host networks are connected by noncovalent interactions including van der Waals (vdW) interactions, hydrogen bonds, or coordinate bonds [12-16]. The guest molecules such as fullerene or coronene (COR) often interact with the host network and surface via attractive dispersion interactions and are trapped in the suitable host cavities according to the size-matching effect [17-21]. The solution–solid interface is a favorable condition for investigating the dynamic behavior of guest inclusion complex, where the guest molecule binding takes place at or close to equilibrium conditions [22-24]. In order to precisely control the deposition of chemical functionality at the nanoscale, various methods have been explored, including molecular structure design and environmental factors [25-27]. In the study area of host-guest chemistry, the flexible network is of great significance due to its potential advantage, which can provide high guest selectivity and obtain target multi-component patterns [28,29].
Pyridine derivatives are common ligands and are frequently used to modulate the self-assembly of aromatic carboxylic acids, showing a wide range of potential applications in constructing surface functional materials [30-34]. The N atoms are equipped at the end of the pyridine molecule, which can replace the O–H···O hydrogen bonds between the acid molecules and form stronger intermolecular O–H···N hydrogen bonds with the aromatic carboxylic acid molecule [35-37]. As a result, the original acid-based self-assembly system is destroyed, and a new co-assembled nanostructure is generated. In most cases, pyridine molecules act as a bridge between aromatic carboxylic acid molecules to expand their self-assembly structures on the substrate [30,38]. Through co-assembly with different guest molecules and pyridine molecules, abundant supramolecular nanostructures can be obtained. For example, Zeng’s group [39] has reported that minor adjustments in the molecular structure of pyridine derivatives may directly determine the intermolecular bonding sites and further affect the co-adsorption of aromatic carboxylic acids with various pyridine derivatives. However, there are few studies about the reconstruction of the host-guest system by breaking the carboxylic acid–pyridine heterosynthon.
Herein, the self-assembled structures are fabricated by the 4,4″,4″″-nitrilotris(([1,1′-biphenyl]−3,5-dicarboxylic acid)) (H3BDA) molecule at the HA/HOPG interface, which is successfully regulated by coronene (COR) and 2,4,6-tri(4-pyridyl)-1,3,5-triazine (T4PT). The self-assembled and co-assembled nanostructures are recorded by STM. Fig. 1 displays the chemical structures of the compounds employed in this work. H3BDA molecule has a triphenylamine unit in the center and three pairs of meta-carboxyl substituents. With the help of DFT calculations, the optimized molecule conformation is displayed in Fig. S1 (Supporting information). Apparently, the H3BDA molecule does not undergo planarization when adsorbed on the graphite surface, because the triphenylamine group presents a three-dimensional propeller structure. However, the carboxyl groups at the end of the H3BDA molecule are in the same plane, which means that saturated hydrogen bonds can be formed. Besides, T4PT contains multiple nitrogen atoms, which can also provide multiple hydrogen bond binding sites. Density functional theory (DFT) calculations and molecular dynamics simulations (MD) revealed the reconstruction of two-dimensional nanoporous networks induced by co-adsorbed guest molecules.
After depositing a droplet of H3BDA solution with a high concentration (1.0 × 10−4 mol/L) on HOPG, it is worth noting that two types of structures can be observed (Fig. S2a in Supporting information). One is a close arrangement named linear structure (Fig. 2), and the other is a loose-packed network named flower-like structure (Fig. 3). When the concentration is diluted to 1.0 × 10−5 mol/L, the adsorption region of the linear structure became smaller, while the flower-like structure become larger (Fig. S2b in Supporting information). These experimental results show that the assembly structure is transformed from a close arrangement to a loose-packed pattern with the decrease of the solution concentration, which is consistent with the typical concentration effect [40].
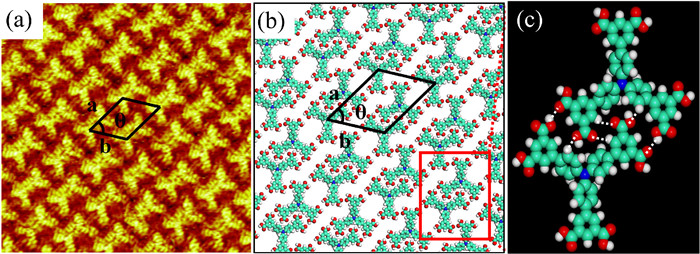
The high-resolution STM image of the linear structure is displayed in Fig. 2a, which manifests more arrangement details. Two H3BDA molecules form a dimer with an antiparallel mode, in which each H3BDA molecule rotates 180° against the other one along the axis. The dimers of two adjacent rows are arranged alternately to form the liner structure. A corresponding molecular model is presented in Fig. 2b. The dimers in the same row are connected by weak hydrogen bonds formed between carboxyl groups. The optimized dimer conformation is presented in Fig. 2c. It can be clearly seen that there are multiple bonding sites between the two molecules in the dimer, forming multiple C=O···H hydrogen bonds marked by white dotted. The cell parameters of the unit cell (marked by black lines) are a = 3.1 ± 0.1 nm and b = 2.7 ± 0.1 nm with an angle of ~51°. Each unit cell contains two H3BDA molecules, and the calculated nanostructure density is 0.307 molecule/nm2.
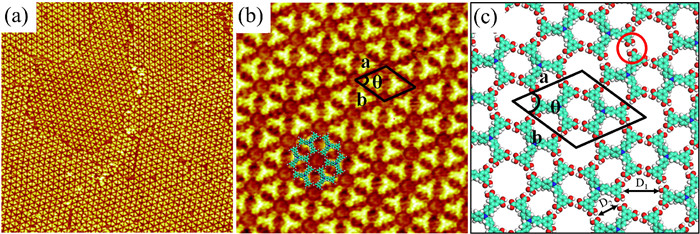
The large-scale and high-resolution STM images of the flower-like structure are detected at the interface of heptanoic acid/HOPG, as shown in Figs. 3a and b. As indicated in Fig. 3a, H3BDA molecules form multiple domains with different sizes and orientations. In each domain, all H3BDA molecules self-assemble to form a flower-like nanostructure. More details are revealed in the high-resolution STM image (Fig. 3b) and the optimized molecular model is presented in Fig. 3c. Obviously, the flower-like structure contains two types of nanopores. The center hexamer cavity of the flower-like structure is six-fold symmetrical and surrounded by six pairs of meta-carboxyl groups of six H3BDA molecules via hydrogen bonds. The measured inner diameter of hexamer cavity is about 1.4 ± 0.1 nm (D1). The small-type pore is connected by two pairs of carboxyl groups in an edge-edge mode with a thin width of 0.9 ± 0.1 nm (D2). A unit cell of the molecular model is marked in Fig. 3c, a = b = 3.1 ± 0.1 nm and θ = 60 ± 1°. The roughly calculated molecular packing density is 0.24 molecule/nm2. In above case, the carboxyl groups of the adjacent H3BDA molecules can form double hydrogen bonds, which evidently contributes to maximize the intermolecular forces and improves the stability of the self-assembled structure.
To evaluate the accommodation ability of the nanoporous of H3BDA structure, COR is chosen as the guest molecules in consideration of its suitable size and strong molecule–substrate interaction. Upon applying a drop of COR solution to the liquid/solid interface, a large-scale STM image showing the H3BDA/COR co-assembled structure is obtained (Fig. 4a and Fig. S3 in Supporting information). The introduction of the guest molecule COR does not affect the six-membered ring structures of H3BDA (Fig. 4a), but results in the disappearance of linear nanostructure. The high-resolution image (Fig. 4b) indicates that the cavity of each six-membered ring is filled with a guest COR molecule, which appears as a bright spot in STM images due to the highly delocalized electrons. A structural model for the adlayer is sketched in Fig. 4c and in good agreement with the STM results. On the basis of symmetry and intermolecular distance, a unit cell is superimposed on the high-resolution image in Fig. 4b with a = b = 3.1 ± 0.1 nm and θ = 60 ± 1°, which is similar to that of H3BDA adlayer. The entrapment of the guest molecules did not change the original flower-like template.
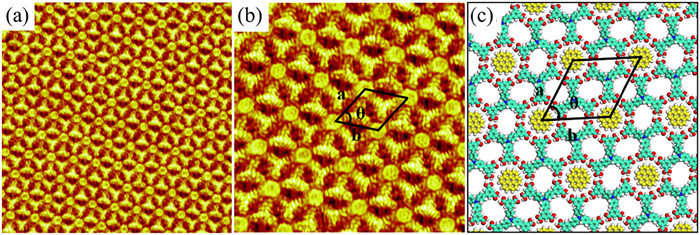
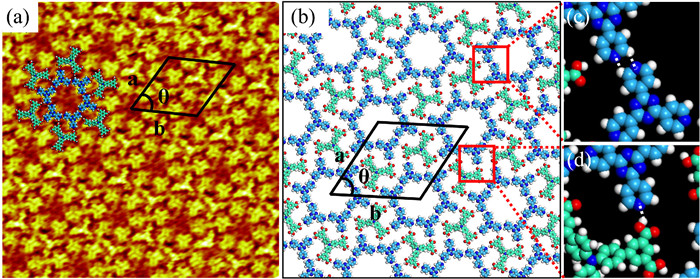
Pyridine derivatives are commonly used as ligands for regulating hydrogen bond-driven self-assembled structures. A Y-shaped bipyridine molecule (T4PT) is selected to mediate the network of H3BDA. When a droplet of heptanoic acid containing T4PT is deposited on the single-component adlayer of H3BDA, the original linear structure and flower-like structure are broken and completely replaced by an interesting two-component co-assembled structure (Fig. 5a and Fig. S4 in Supporting information). The solution concentration ratio of H3BDA to T4PT is 1:5, which is proved to be the most suitable ratio for obtaining large-area co-assembled structure. It is easy to distinguish the arrangement of the different molecules from the high-resolution STM image (Fig. 5a). The larger Y-shaped feature represents an H3BDA molecule, and the smaller Y-shaped feature corresponds to a T4PT molecule. Based on the STM images and optimized structures by DFT method, a corresponding molecular model of H3BDA/T4PT pattern is presented in Fig. 5b. As shown in the molecular model, the inner ring of the hexagon cavity is encircled by six T4PT molecules. The distance between the N atom of pyridine group and the adjacent central benzene ring of T4PT is measured to be approximately 0.275 nm, which is sufficient to form a C–H···N hydrogen bond (Fig. 5c). The outer ring of the hexagon cavity is also surrounded by six H3BDA molecules. It can be clearly seen that H3BDA molecules are not able to form intermolecular hydrogen bond due to the long distance. T4PT molecules act as the bridge to connect H3BDA molecules through O–H···N hydrogen bonds as shown in Fig. 5d. Careful analysis of this porous network reveals that six HO– in carboxyl group of each H3BDA molecule are linked with six N atoms of the surrounding six T4PT molecules. The cell parameters of the porous structure are a = 4.7 ± 0.1 nm, b = 4.7 ± 0.1 nm, θ = 60 ± 1°.
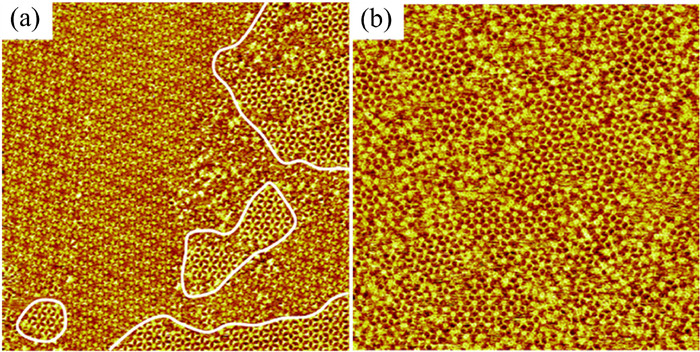
After adding a droplet of COR with a low solution concentration on the preorganized H3BDA/T4PT porous network, the structural transformation of binary network is shown Fig. 6a. Interestingly, the H3BDA/T4PT porous network and the H3BDA/COR co-adsorption structure coexist on the graphite surface. It is noteworthy that the COR molecules do not immobilize in the central porous of H3BDA/T4PT but give priority to the formation of the host-guest system with H3BDA (marked with white lines in Fig. 6a). After adding a high concentration of COR solution, all the H3BDA/T4PT binary networks disappear (Fig. 6b). Although excessive molecules cause some disorder, the H3BDA/COR host-guest structure can be clearly seen and stably exists on the HOPG surface. The addition of COR molecules results in the H3BDA/T4PT binary network to fracture, and then reconstruct a host-guest structure of H3BDA/COR system. This result can be attributed to the size and geometry selectivity of guest molecule, which determines the formation of the final assembled structure.
To further illustrate the noncovalent interactions and the response to guest molecules into these systems from theoretical perspectives, molecular dynamics simulation is performed based on the STM results (Fig. S5 in Supporting information). In the interface self-assembly system, the main forces include the adsorbate–adsorbate interaction and the adsorbate–substrate interaction, and their sum is equal to the total energy value in Table 1. Furthermore, the total energy per unit area is an essential value to compare thermodynamic stability of different structures. The smaller the value of total energy per unit area, the stronger the stability of the assembled system [41].
According to Table 1, the energy per unit area of the flower-like structure is −0.464 kcal mol−1 Å−2, which is much lower than that of the linear structure (−0.408 kcal mol−1 Å−2). Although the packing density of the linear structure is larger than that of the flower-like structure, more hydrogen bonds are formed in the latter structure, which means that the flower-like structure is more thermodynamically stable. When COR molecules are introduced into the above system, the energy of intermolecular interaction increases slightly, while the molecule−substrate interaction energy increases significantly. The energy per unit area (−0.544 kcal mol−1 Å−2) of the H3BDA/COR system is much lower than that of the rhombus structure, which is beneficial for the formation of the co-assembled structure. The calculation results agree well with our STM observations. After depositing the T4PT solution on the H3BDA adlayer, the formation of molecular self-assembled behavior undergoes dramatic changes containing the collapse of origin two kinds of structures and the construction of the new porous system. The H3BDA/T4PT binary system (−0.495 kcal mol−1 Å−2) is more stable compared with the H3BDA single component system. In the porous structure of H3BDA/T4PT adlayer, three N atoms at the vertex of T4PT molecules form O–H···N hydrogen bonds with the –OH in carboxyl groups of H3BDA molecules. In addition, adjacent T4PT molecules facilitate the formation of C–H···N hydrogen bonds due to the appropriate distance. The N atom of pyridine derivatives is more electronegative. The O–H···N hydrogen bond between N atom and the carboxy group is stronger than the O–H···O hydrogen bond between the carboxy groups, resulting in the formation of the H3BDA/T4PT two-component structure. After the introduction of COR molecules into the H3BDA/T4PT system, the porous structure of H3BDA/T4PT is regulated into the H3BDA/COR host-guest structure. The calculated results manifest that the total energy per unit area of H3BDA/COR nanostructure is the lowest energy system, indicating that the H3BDA/COR system is the most thermodynamically stable.
In summary, we investigated the self-assembly of H3BDA and its co-assembly behaviors with COR and T4PT at the HA/HOPG interface by STM. DFT calculations and MD simulations were carried out to illustrate the formation mechanism of the assembled structures. For single-component system of H3BDA molecule, two types of 2D nanostructures (linear structure and the flower-like network) could be observed and showed an obvious concentration effect. With the decrease of concentration, it was more conducive to the formation of flower-like network. In the above structures, the hydrogen bond and the vdW forces played the significant roles. After the introduction of the guest COR molecule, the original linear structure of H3BDA was destroyed and the flower-like network could act as a template to immobilize COR and form a stable host-guest system. However, the H3BDA adlayer was regulated into a new binary structure by the addition of T4PT molecule owing to the stronger intermolecular hydrogen bonds between carboxyl and pyridine groups. As a response to the addition of COR guest molecules, the H3BDA/T4PT porous structure breakdown and subsequently generated the H3BDA/COR host–guest system. MD simulation results showed that the H3BDA/COR co-assembled structure was the energetically favorable structure with the most thermodynamic stability, which explained the reason of structural transformation of the H3BDA/T4PT adlayer after the addition of COR. The present study guides the construction of novel 2D multi-component structures with highly selectively on the HOPG surface, which is conducive to the construction of functional nanostructures and applied to the molecular recognition technology.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from the National Natural Science Foundation of China (No. 22172055), the Natural Science Foundation of Guangdong Province (Nos. 2023B1515040026, 2022A1515011892), the Basic and Applied Basic Research Program of Guangzhou City (Nos. 202002030083, 202102080443) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Kikkawa, M. Nagasaki, E. Koyama, et al., Nanoscale Adv. 2 (2020) 4895–4901. doi: 10.1039/d0na00616e
M.Q. Dong, X.R. Miao, R. Brisse, et al., NPG Asia Mater. 12 (2020) 1–9. doi: 10.1038/s41427-019-0187-x
F. Silly, Y. Kervella, B. Jousselme, RSC Adv. 5 (2015) 101740–101744. doi: 10.1039/C5RA22117J
H.L. Dai, Y.F. Geng, Q.D. Zeng, et al., Chin. Chem. Lett. 28 (2017) 729–737. doi: 10.1016/j.cclet.2016.09.018
H. Zou, L. Liu, S. Zhang, et al., J. Phys. Chem. Lett. 14 (2023) 489–498. doi: 10.1021/acs.jpclett.2c03691
Y. Wang, X. Tan, P. Pang, et al., Chem. Commun. 56 (2020) 13991–13994. doi: 10.1039/d0cc05937d
C. Deng, Z. Liu, C. Ma, et al., Langmuir 36 (2020) 5510–5516. doi: 10.1021/acs.langmuir.0c00425
S.B. Khan, S.L. Lee, Molecules 26 (2021) 3995. doi: 10.3390/molecules26133995
Y. Wang, X.R. Miao, W.L. Deng, et al., Nanomaterials 12 (2022) 775. doi: 10.3390/nano12050775
J. Wang, L.M. Wang, C. Lu, et al., RSC Adv. 9 (2019) 11659–11663. doi: 10.1039/c9ra01493d
S. Yoshimoto, E. Tsutsumi, R. Narita, et al., J. Am. Chem. Soc. 129 (2007) 4366–4376. doi: 10.1021/ja0684848
S.J.H. Griessl, M. Lackinger, F. Jamitzky, et al., Langmuir 20 (2004) 9403–9407. doi: 10.1021/la049441c
M. Lackinger, W.M. Heckl, Langmuir 25 (2009) 11307–11321. doi: 10.1021/la900785f
D.L. Wisman, H. Kim, C. Kim, et al., Chemistry 27 (2021) 13887–13893. doi: 10.1002/chem.202101611
E. Khare, N. Holten-Andersen, M.J. Buehler, Nat. Rev. Mater. 6 (2021) 421–436. doi: 10.1038/s41578-020-00270-z
K. Tahara, S. Lei, J. Adisoejoso, et al., Chem. Commun. 46 (2010) 8507–8525. doi: 10.1039/c0cc02780d
J. Teyssandier, S.D. Feyter, K.S. Mali, Chem. Commun. 52 (2016) 11465–11487. doi: 10.1039/C6CC05256H
S.Q. Zhang, Z.Y. Liu, W.F. Fu, et al., ACS Nano 11 (2017) 11701–11713. doi: 10.1021/acsnano.7b06961
G. Eder, S. Kloft, N. Martsinovich, et al., Langmuir 27 (2011) 13563–13571. doi: 10.1021/la203054k
J. Liu, X. Zhang, H.J. Yan, et al., Langmuir 26 (2010) 8195–8200. doi: 10.1021/la904568q
X. Li, J. Li, C. Ma, et al., Chin. Chem. Lett. 32 (2021) 1077–1080. doi: 10.1016/j.cclet.2020.07.049
U. Mazur, K.W. Hipps, Chem. Commun. 51 (2015) 4737–4749. doi: 10.1039/C4CC09840D
G. Velpula, C. Martin, B. Daelemans, et al., Chem. Sci. 12 (2021) 13167–13176. doi: 10.1039/d1sc02950a
X. Gong, H. Zhang, N. Jiang, et al., Microchem. J. 145 (2019) 435–443. doi: 10.1016/j.microc.2018.11.011
X. Zeng, W. Ding, H. Zou, et al., J. Phys. Chem. C 126 (2022) 5777–5783. doi: 10.1021/acs.jpcc.2c00570
L. Cheng, Y. Li, C.Y. Zhang, et al., ACS Appl. Mater. Interfaces 8 (2016) 32004–32010. doi: 10.1021/acsami.6b10883
R. Chen, H.l. Song, H. Zhu, et al., J. Phys. Chem. C 126 (2022) 9567–9571. doi: 10.1021/acs.jpcc.2c02313
J. Xue, K. Deng, B. Liu, et al., RSC Adv. 5 (2015) 39291–39294. doi: 10.1039/C5RA01517K
J. Adisoejoso, K. Tahara, S. Okuhata, et al., Angew. Chem. Int. Ed. 48 (2009) 7353–7357. doi: 10.1002/anie.200900436
L.S. Pinheiro, M.L.A. Temperini, Surf. Sci. 601 (2007) 1836–1843. doi: 10.1016/j.susc.2007.02.009
Q. Xue, Y. Zhang, R. Li, et al., Chin. Chem. Lett. 30 (2019) 2355–2358. doi: 10.1016/j.cclet.2019.09.027
C. Shen, M. Haryono, A. Grohmann, et al., Langmuir 24 (2008) 12883–12891. doi: 10.1021/la8019974
J. Li, W. Luo, S. Zhang, et al., Nano Res. 15 (2022) 1691–1697. doi: 10.1007/s12274-021-3871-4
P. Lei, L. Ma, S. Zhang, et al., Chin. Chem. Lett. 34 (2023) 108005. doi: 10.1016/j.cclet.2022.108005
S. Zhang, C. Chen, J. Li, et al., Nanoscale 14 (2022) 2419–2426. doi: 10.1039/d1nr07840b
T. Meng, P. Lei, Y. Zhang, et al., Chin. J. Chem. 40 (2022) 2727–2733. doi: 10.1002/cjoc.202200336
Y. Qian, Y. Gu, J. Feng, et al., J. Phys. Chem. C 122 (2018) 24158–24163. doi: 10.1021/acs.jpcc.8b07723
Y. Hong, L. Wang, S.F. Wang, et al., CrystEngComm 23 (2021) 3849–3855. doi: 10.1039/d1ce00239b
P. Lei, W. Luo, B. Tu, et al., Chem. Commun. 58 (2022) 9914–9917. doi: 10.1039/d2cc03859e
S. Lei, K. Tahara, F.C. De Schryver, et al., Angew. Chem. Int. Ed. 47 (2008) 2964–2968. doi: 10.1002/anie.200705322
Q. Yan, T. Meng, W. Luo, et al., Langmuir 38 (2022) 8651–8656. doi: 10.1021/acs.langmuir.2c01076

Figure 2 (a) High-resolution STM image for the self-assembled adlayer of H3BDA at the HA/HOPG interface showing the linear structure (16 × 16 nm2; Vbias = 734 mV and Iset = 261 pA). (b) Corresponding molecular model of the linear structure. (c) The optimized dimer conformation and the intermolecular hydrogen bonds in the dimer marked by white dotted.

Figure 3 (a) Large-scale and (b) high-resolution STM images for the self-assembled adlayer of H3BDA at the HA/HOPG interface showing the flower-like structure (80 × 80 nm2, 20 × 20 nm2; Vbias = 734 mV, and Iset = 261 pA). (c) Corresponding molecular model of the flower-like structure. The main intermolecular interactions are a pair of O–H···O=C hydrogen bonds marked with a red circle.

Figure 4 (a) Small-scale and (b) high-resolution STM images for the co-assembled adlayer of H3BDA/COR at the HA/HOPG interface (50 × 50 nm2, 20 × 20 nm2; Vbias = 734 mV, and Iset = 261 pA). (c) Corresponding molecular model of the H3BDA/COR co-assembled structure.

Figure 5 (a) High-resolution STM image showing the porous structure of the H3BDA/T4PT adlayer at the HA/HOPG interface (20 × 20 nm2; Vbias = 654 mV and Iset = 261 pA). (b) Corresponding molecular model of the porous structure. (c) The C–H···N hydrogen bonds between T4PT molecules. (d) The O–H···N hydrogen bond between H3BDA and T4PT.

Figure 6 Large-scale STM images of the co-assembled nanostructure formed by H3BDA/T4PT/COR molecules on the HOPG surface (120 × 120 nm2, Vbias = 654 mV, and Iset = 261 pA). (a) At a lower COR concentration (1.0 × 10−5 mol/L). (b) At a higher COR concentration (1.0 × 10−4 mol/L).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
 下载:
下载: