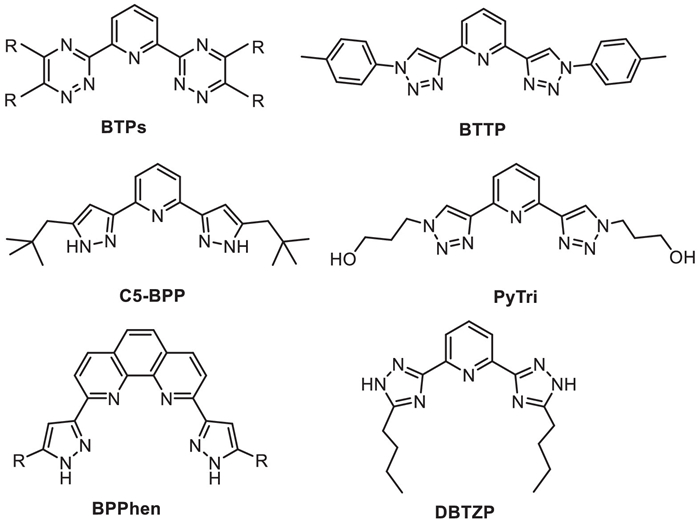
Scheme 1.
Structures of N-donor ligands.
Theoretical design of polyazole based ligands for the separation of Am(Ⅲ)/Eu(Ⅲ)
Lingling Su , Qunyan Wu , Congzhi Wang , Jianhui Lan , Weiqun Shi

Nuclear power as a clean energy source is now a strategically important option for many countries, which calls for the advanced nuclear fuel cycle models to achieve efficient utilization of nuclear fuel and reduce environmental hazards [1,2]. Although the majority of plutonium and uranium in spent nuclear fuels can be extracted through the well-established plutonium uranium reduction extraction process, its radioactive residue still contains long-term radiotoxic minor actinides (MAs) [3]. The partitioning and transmutation (P&T) strategy is widely accepted as an effective method to convert MAs into shorter-lived or stable nuclides by neutron irradiation [4]. The co-existing lanthanides, having large neutron absorption cross sections, can significantly reduce the efficiency of the P&T and therefore need to be separated from the MAs [5]. The trivalent actinide (An(Ⅲ)) and lanthanide (Ln(Ⅲ)) ions, however, have similar physicochemical properties, making their separation extremely challenging [6,7].
In general, solvent extraction is a popular f-element extraction method with scalability and robustness up to now [8–11]. An(Ⅲ) and Ln(Ⅲ) ions are "hard" Lewis acids based on the Hard-Soft Acid-Base theory (HSAB) [12], the former is slightly softer than the latter. Many soft S- or N-donor ligands have been studied for their selectivity for An(Ⅲ)/Ln(Ⅲ) ions through solvent extraction [13–15]. According to the CHON principle [16], N-donor ligands have received more attention, e.g., 2,6-bis(5,6-dialkyl-1,2,4-triazin-3-yl)pyridines derivatives (BTPs, Scheme 1) [17], and 2,9-bis(1,2,4-triazin-3-yl)-1,10-phenanthroline derivatives (BTPhens) [18]. Bis-triazinyl ligands have achieved excellent extraction ability, but there are some practical drawbacks, such as poor chemical and radiological stability, slow extraction kinetics, and difficulty in synthesis, which still limit their wide application [13]. In recent years, polyazole based ligands, especially those with 1,2,4-triazol [17,19], pyrazol [20–24], and 1,2,3-triazol [25–31] side chains, have received a lot of attention owing to their relatively easy synthesis [13] and high extraction kinetics [26]. Kolarik et al. reported that 2,6-bis(5–butyl–1,2,4-triazol-3-yl)pyridine (DBTZP, Scheme 1) in the presence of 2-bromohexanoic acid could extract Am(Ⅲ) with Am(Ⅲ)/Eu(Ⅲ) separation factors (SFAm/Eu) up to 150 [17]. Bremer et al. synthesized 2,6-bis(5-(2,2-dimethylpropyl)-1H-pyrazol-3-yl)pyridine (C5-BPP, Scheme 1), which could achieve SFAm/Eu about 100 under 1 mol/L HNO3 with the existence of 2-bromohexanoic acid [20]. Ding's group has done extensive works on the synthesis of pyrazol based ligands [21–23], in which 2,9-bis(5-alkyl-1H-pyrazol-3-yl)-1,10-phenanthroline (BPPhen, Scheme 1) [23] exhibits SFAm/Eu value of about 100. Kiefer et al. reported that the lipophilic 2,6-bis(1-(ptolyl)-1H-1,2,3-triazol-4-yl)pyridine (BTTP, Scheme 1) exhibits a high complexation selectivity with Cm(Ⅲ) than Eu(Ⅲ) in CH3CN [25]. Subsequently, Macerata's group synthesized hydrophilic pyridine-2,6-bis(1H-1,2,3-triazol-4-yl) (PyTri, Scheme 1) with high extraction kinetics and chemical/radiolytic stability, which has the SFEu/Am values of about 60–150 [26]. In addition, Ye's group proposed a process for An(Ⅲ)/Ln(Ⅲ) extraction separation, with N, N, N', N'-tetraoctyl diglycolamide (TODGA) as the extractant, tri-n–butyl phosphate (TBP) as the phase modifier and PyTri as hydrophilic stripping agent, the 'hot test' could achieve 99.92% removal of Am(Ⅲ) with SFEu/Am of 3.8 × 103 [31]. The first 1,10-phenanthroline-derived hydrophilic ligand 2,9-bis-(1H-1,2,3-triazol-4-yl)1,10-phenanthroline was reported to exhibit SFEu/Am ≈ 36–47 and SFCm/Am ≈ 2.5 [27]. Ossola's group synthesized five novel lipophilic derivatives of PyTri with different alkyl groups [28], among which 2,6-bis(1-(2-ethylhexyl)-1H-1,2,3-triazol-4-yl)pyridine (L2, Scheme 2) with ethylhexyl alkyl chain showed the best solubility and separation performance. Despite the great progress in the research of polyazole based ligands in this area, the understanding of how polyazole side chains affect separation performance is still very limited.
With the rapid development of actinide computational chemistry, theoretical calculations can provide reliable results to elucidate the solvent extraction of An(Ⅲ)/Ln(Ⅲ) ions [32–35], which further expand our understanding of the electronic structure, bonding nature and spectra of the complex actinide systems [36–39]. We have theoretically explored a series of studies on the Am(Ⅲ)/Eu(Ⅲ) separation by N-donor and N, O-mixed donor ligands [40–48] by considering the factors of hard-soft donor combined strategy [40], the variations of the central heterocyclic moieties [41], preorganization [42,43], and substitution effects [44,45]. In addition, we have also investigated the Am(Ⅲ)/Eu(Ⅲ) separation by hydrophilic ligands [46,47]. Recent work [48] reported the effect of different side chains containing imidazol, tetrazol, and 1,2,4-triazol, which showed that the phenanthroline derivative with 1,2,4-triazole side chains has the best Am(Ⅲ)/Eu(Ⅲ) selectivity. Experimentally, pyrazole, 1,2,3-triazol and 1,2,4-triazole have better Am(Ⅲ)/Eu(Ⅲ) separation than imidazol and tetrazol [49]. To further explore the effect of pyrazole, 1,2,3-triazole and 1,2,4-triazole side chains on the Am(Ⅲ)/Eu(Ⅲ) separation, we attempts to design polyazole-based ligands based on above works. Therefore, the Am(Ⅲ)/Eu(Ⅲ) selectivity of three similar pyridine-derived polyazole ligands, 2,6-bis(5-ethylhexyl-1,2,4-triazol-3-yl)pyridine (L1), L2, and 2,6-bis(5-(2,2-ethylhexyl)-1H-pyrazol-3-yl)pyridine (L3) (Scheme 2) were investigated with scalar relativistic density functional theory (DFT). L1 and L3 have similar structures to C5-BPP and DBTZP (Scheme 1), while their alkyl chains were replaced by ethylhexyl, the same as L2. We theoretically investigated the electronic structures of L1, L2, and L3, and evaluated their Am(Ⅲ)/Eu(Ⅲ) selectivity. Based on the results of the complexation and selectivity ability for Am(Ⅲ)/Eu(Ⅲ) ions of the three ligands, we designed a new ligand, 2,9-bis(5-(2-ethylhexyl)-1H-1,2,4-triazol-3-yl)-1,10-phenanthroline (L4) with 1,2,4-triazole side chains and a preorganized phenanthroline backbone. We first elucidated the selectivity and binding nature of three similar pyridine-derived polyazole ligands and then designed new preorganized phenanthroline derivatives. This work helps to understand how the polyazole side chains affect the Am(Ⅲ)/Eu(Ⅲ) selectivity of ligands and provides design strategies for highly efficient polyazole based ligands in the future.
The optimizations of all the structures were performed in the gas phase with B3LYP functional [50], applying Gaussian 16 software [51]. The scalar-relativistic effective core potentials (RECPs), which replace 28 and 60 core electrons for Eu [52] and Am [53] and the corresponding ECP28MWB-SEG [54] and ECP60MWB-SEG [55,56] valence basis sets were used, respectively. The 6–31G(d) basis set was used for the other atoms. Previous studies suggest that the structure is not sensitive to solvation [57,58], so the optimizations of the Am(Ⅲ)/Eu(Ⅲ) complexes were carried out at the B3LYP/RECP/6–31G(d) level of theory in the gas phase. The septet state is ground state for Am(Ⅲ)/Eu(Ⅲ) complexes and the spin contamination is negligible based on the results reported in previous works [40,45,59,60]. Spin-orbit coupling effects are not included considering the precision and computational cost [40,61]. To improve the accuracy of the energies, we performed single-point calculations for each species in solution using the B3LYP function with the larger 6-311G(d, p) basis set. The conductor-like polarizable continuum model (CPCM) [62,63] was applied for calculating the thermodynamic properties with Klamt's radii in cyclohexanone (ε = 15.619), nitrobenzene (ε = 34.809) and water (ε = 78.3553) solutions. The Gibbs free energy of each species in the solution is the sum of the solvation energy and thermal correction to the Gibbs free energy obtained at the B3LYP/RECP/6-31G(d) level of theory in the gas phase [64].
The molecular electrostatic potential (ESP) maps of ligands were carried out using Multiwfn 3.8 software [65]. The molecular orbitals (MOs) were obtained with the GaussView program [51]. Mulliken charge and Mayer bond order (MBO) [66] were performed by natural bond orbital (NBO) [67,68]. The topological analysis of electron density was performed through the quantum theory of atoms in molecules (QTAIM) [65,69,70]. Energy decomposition analysis (EDA) [71] and the extended transition state method with the natural orbital for the chemical valence (NOCV) theory were performed at the BP86/TZ2P/ZORA level of theory with Amsterdam Modeling Suite 2022 (AMS 2022) [72–74] without frozen core.
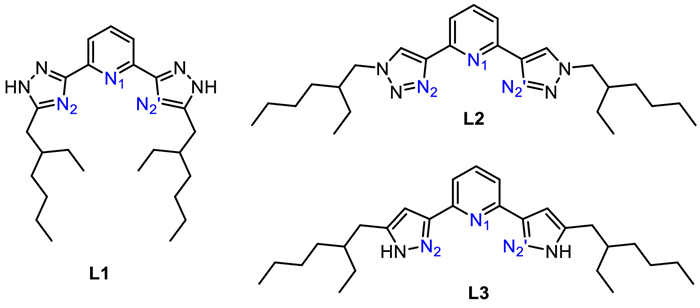
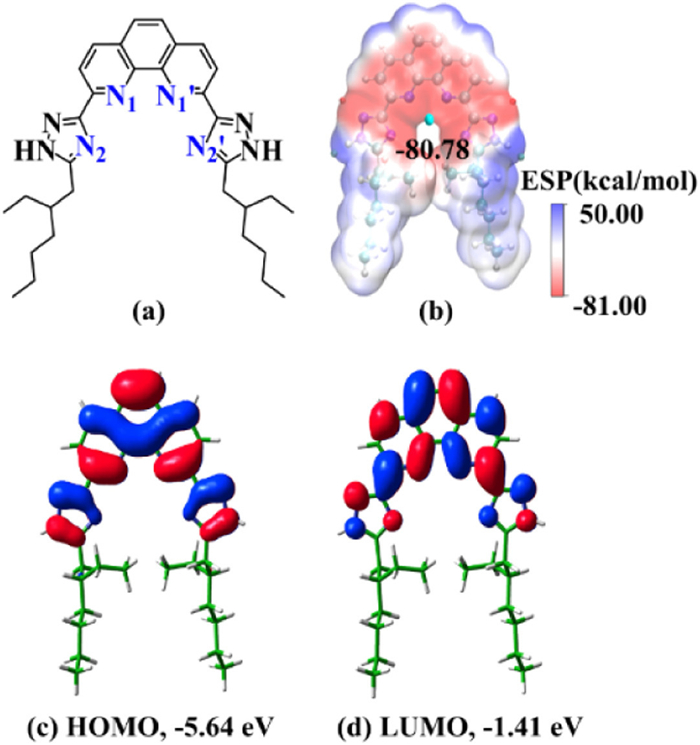
The ESP map can visualize the variably charged region of the ligand. The red and blue areas indicate the negative and positive values of ESP, respectively, as shown in Fig. 1. The negative minima of the ESP surface for L1, L2 and L3 are located in the cavities created by the N atoms of pyridine skeleton and polyazole side chains, indicating that the metal ion can be bound in the cavity. The more negative ESP for L1 and L3 implies that these two ligands are more able to bind with metal ions than L2.
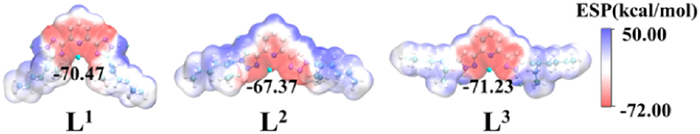
Fig. 2 shows the highest occupied MO (HOMO), the lowest unoccupied MO (LUMO), and HOMO-LUMO gap for ligands. The HOMOs have delocalized π bonding character with electron density distributed over pyridine and polyazole rings, reflecting that pyridine and polyazole ring are good electron donors. LUMOs for the three ligands have the anti-bonding nature. L1 has the lowest HOMO-LUMO gap (5.05 eV), suggesting that L1 is the most active ligand. According to the HSAB principle [12], the hardness (η) of the ligand is an important indicator for An(Ⅲ)/Ln(Ⅲ) separation. η is calculated by the equation η = (IP-EA)/2, where IP and EA represent the vertical ionization potential and electron affinity of the neutral ligand, respectively [75]. Table S1 (Supporting information) shows that the three ligands have almost the same η with the value of about 4.0, which indicating that the three ligands have potential selectivity for Am(Ⅲ)/Eu(Ⅲ) ions.
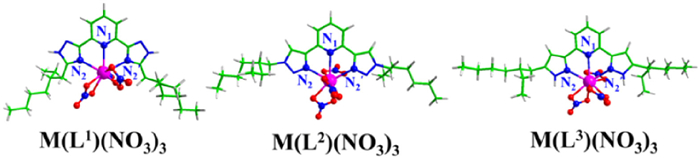
The neutral ML(NO3)3 (M = Am/Eu) complexes were considered, as shown in Fig. 3. The Am(Ⅲ)/Eu(Ⅲ) ions were coordinated to ligand and nitrate ions in a tridentate and bidentate modes, respectively. The M-N and M-O bond lengths of the complexes are shown in Table 1. The bond length of M-N bonds follows the order of M-N2 < M-N1 for each complex, suggesting that the N2 atoms of polyazoles binds metal ion more strongly than the N1 on pyridines. And the M-N2 bond lengths of ML(NO3)3 follow the order of M(L1)(NO3)3 > M(L2)(NO3)3 > M(L3)(NO3)3, which suggests that the coordinating ability of N2 atom and metal ions is in the order of 1,2,4-triazole < 1,2,3-triazole < pyrazole. Moreover, all Eu-N bonds are a little shorter than the corresponding Am-N ones, owing to the smaller ionic radius of Eu(Ⅲ) compared to Am(Ⅲ) [76]. Besides, the M-O bonds are significantly shorter than the M-N bonds, suggesting that the nitrate ions are more tightly coordinated to metal ions.
The extraction process was carried out in acidic environment and therefore the protonation ability of ligands was evaluated here. Polyazole and pyridine are easily protonated due to the lone pair of electrons on their N atom. The protonation capacity of N atom were calculated based on the reactions L + H+ → [LH]+ and L + [H3O]+ → [LH]+ + H2O. Table S2 (Supporting information) shows that the N1 atom on pyridine is more likely to be protonated compared to the N2 atom on polyazole.
The interactions between ligand and metal ion can be evaluated by MOs. The α-spin valence MOs of ML(NO3)3 complexes are given in Fig. 4, Figs. S1 and S2 (Supporting information). Similar bonding interactions between the ligands and Am(Ⅲ)/Eu(Ⅲ) ions were found, the lower MO energy levels of AmL(NO3)3 complexes compared to the corresponding EuL(NO3)3 ones indicates that the former has a stronger interaction. Moreover, the contribution of Am and Eu atoms for the MOs are dominated by their d and f atomic orbitals according to the composition listed in Table S3 (Supporting information).
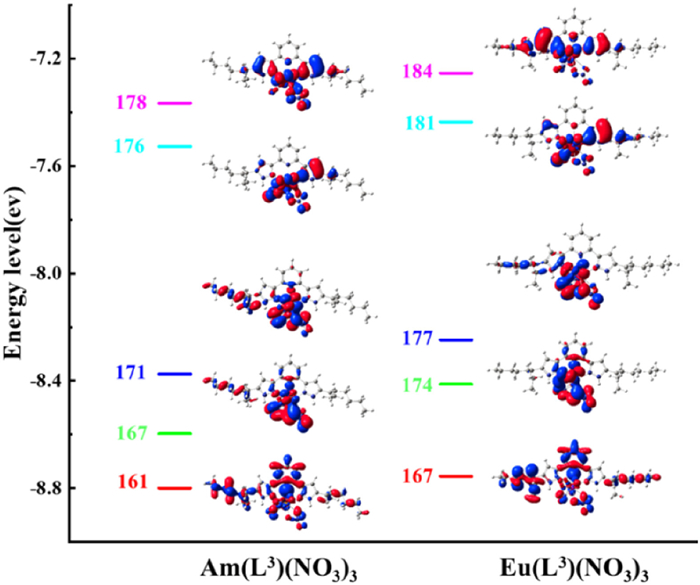
Table 2 shows the calculated Mulliken charges (Q) and charge transfer (ΔQ) in ML(NO3)3. It is obvious that electrons transferred from ligand and nitrate ion to Am and Eu, leaving the charge on them less than +3. In addition, ΔQ(Am) is greater than ΔQ(Eu) in each ML(NO3)3 complex, suggesting a tendency for the ligand to select Am(Ⅲ) ions. In addition, the charge transfer difference ΔΔQ(L) follows the trend of L1 > L2 > L3, which is related to the Am(Ⅲ)/Eu(Ⅲ) selectivity of the three ligands.
To investigate the bonding properties of ligand and metal, multiple theoretical approaches were applied. The Mayer bond order (MBO) could indicate the covalency of M-L interactions. Table 3 shows the M-N and M-O MBOs of complexes. The M-N2 MBOs are greater compared to the corresponding M-N1 ones, suggesting that the N2 atoms have significant interactions with metal ions. Moreover, all Am-N MBOs are greater than the corresponding Eu-N MBOs, reflecting a greater covalency interaction between ligand with Am(Ⅲ) ions. In addition, M-N MBOs are smaller than M-O MBOs because O atoms have stronger coordination ability with metal ions than that of N atoms, which is in accordance with the bond length results.
In QTAIM approach, the electron density (ρ), Laplacian density (∇2ρ), kinetic energy density (G) and energy density (H) at the bond critical point (BCP) are calculated to evaluate the bonding properties [77,78]. Generally, a value of ρ < 0.10 a.u. with ∇2ρ > 0 a.u. at a BCP refers to an ionic bond, while ρ > 0.20 a.u. with ∇2ρ < 0 a.u. reflects a typical covalent bond [77]. And usually, a ratio G/ρ > 1 and G/ρ < 1 indicates ionic interaction and covalent interaction, respectively. Table 4 shows that the ρ and ∇2ρ indicate that the M-N1, M-N2 and M-O bonds are ionic bond with weak covalency interaction, which is also supported by the G/ρ values. The covalency of the Am-N bond is larger than that of the Eu-N bond, and the M-N2 bond has more covalency compared to the M-N1 bond, which may explain the selectivity of Am(Ⅲ) ions compared to Eu(Ⅲ) ions and the more important role of N2 atoms than N1 atoms for the Am(Ⅲ)/Eu(Ⅲ) selectivity. What's more, ρ values at the M-N2 BCPs are in order of M(L1)(NO3)3 < M(L2)(NO3)3 < M(L3)(NO3)3, which reflects the polyazole side chains affect the covalent interaction of M-N2 bonds.
EDA was performed to study the bonding nature between the ligand and metal fragments. Each ML(NO3)3 complex was split into the [L] and [M(NO3)3] fragments. Table 5 lists the interaction energy (ΔEint) between the two fragments in the complex and gives three main terms [79], Pauli repulsive interaction (ΔEPauli), electrostatic interaction (ΔEelec), and orbital interaction (ΔEorb). The values of ΔEorb range from −46.00 kcal/mol to −65.46 kcal/mol and ΔEelec range from −85.32 kcal/mol to −116.27 kcal/mol, the latter being more negative than the former, reflecting a greater contribution of electrostatic interaction to the interaction between [L] and [M(NO3)3] fragments of each complex. The ΔEorb of each AmL(NO3)3 complex is more negative than the corresponding Eu-ones, which also suggests the greater covalent interactions between the [Am(NO3)3] and [L] fragments.
 DownLoad:
CSV
DownLoad:
CSV

|
NOCV can provide intuitive and detailed information about the interactions between ligand and metal ion. In this method, ΔEorb is decomposed into pairwise contributions of interacting orbitals. Figs. S3 and S4 (Supporting information) show the NOCVs contributing significantly to the ΔEorb of complexes. The red to blue regions indicate the direction of electron flow. The electrons transfer from the N1 and N2 atoms of ligand to the metal ions. In addition, most Am-complex NOCVs have more negative ΔEorb values than the corresponding Eu-complex NOCVs, which reflects that there are more electron transfer from ligand to [Am(NO3)3] fragment than that to [Eu(NO3)3] fragment. Fig. S5 (Supporting information) shows the donor-acceptor interactions of the main pairwise interacting orbitals in AmL1(NO3)3 complex. To compare the possible difference in orbital interaction between ligands L1, L2, and L3 and ML(NO3)3 fragments, the major donor-acceptor interactions of ΔEorb1 in complexes are given in Fig. 5. Noticeably, the acceptor orbitals for the [M(NO3)3] fragments are dominated by Am 5f, 6d orbitals and Eu 4f, 5d orbitals. Moreover, Am 5f contributes significantly more to the [Am(NO3)3] fragments than the corresponding Eu 4f to the [Eu(NO3)3] fragments, which may explain the ligands' Am-preference over Eu. The HOMO and HOMO-4 of [L] for AmL1(NO3)3 act as electron donor orbitals, and only HOMO for EuL1(NO3)3 provide electron to [Eu(NO3)3] fragment, reflecting that ligand L1 has a greater difference in electron donating ability for Am and Eu ions. The HOMO of [L] acted as donor in ML2(NO3)3 complexes, and both HOMO and HOMO-6 of [L] acted as donors in ML3(NO3)3 complexes, suggesting that ligand L3 has a stronger electron-donating ability.

The actual extraction process is very complex, TPH (tetrapropylhydrogen) [17], tert–butylbenzene (TBB) [21,23] and meta-nitrobenzotrifluoride [22] were usually used as organic solvents for the polyazole based ligands to extract Am(Ⅲ)/Eu(Ⅲ) ions. Here, we used nitrobenzene as an organic solvent and also tested cyclohexanone solvent. According to previous study [40], [M(NO3)(H2O)8]aq2+ + Lorg + 2NO3aq− = [ML(NO3)3]org + 8H2Oaq is the most probable reaction that occurs during the Am(Ⅲ)/Eu(Ⅲ) ions extraction process with ligands. Table 6 shows the Gibbs free energy (ΔG) and their differences (ΔΔG = ∆GAm - ∆GEu) for extraction reactions in cyclohexanone/water and nitrobenzene/water solutions at the B3LYP/RECP/6-311G(d, p) level of theory. All negative ΔG values for the reactions suggests that the three ligands can spontaneously complex with Am(Ⅲ) and Eu(Ⅲ) ions. The ΔG values of Am-extraction reactions are more negative than the corresponding Eu-ones, reflecting the extraction preference of the three ligands for Am(Ⅲ) ion. In addition, the ΔG values follows the order of L3 > L2 > L1 in both cyclohexanone/water and nitrobenzene/water solutions, reflecting that L3 has the strongest extraction ability for Am(Ⅲ) and Eu(Ⅲ) ions. However, the ΔΔG values show the opposite trend L1 > L2 > L3. To conclude, L1 has the highest selectivity for Am(Ⅲ)/Eu(Ⅲ) ions, although it has the lowest complexation ability with them. The coordination number of the Eu(Ⅲ) and Am(Ⅲ) complexes in the solution is generally 6–12, and the preferable coordination number is 8 and 9 based on the result of previous work [80]. So we also investigated the possible reactions with initial reactants [M(H2O)9]3+ and [M(NO3)(H2O)7]2+. The corresponding ΔG and ΔΔG values are provided in Table S4 (Supporting information), it shows that the trend of ΔG and ΔΔG values for the three ligands in Table 6 and Table S4 is similar, which is also consistent with our previous work [40]. Considering the protonation ability of ligands, N1 atom on pyridine is more likely to be protonated than N2 atoms on polyazole in highly acidic media. Thus, the reactions of the protonated ligands complexing with metal ions were also investigated in Table 6. We found that the ΔΔG values are the same with those of the unprotonated ones. However, the complexation abilities of the protonated ligands with Am(Ⅲ)/Eu(Ⅲ) ions are significantly lower compared to the unprotonated ligands, which suggests that the complexation ability of the polyazole ligands towards metal ions is sensitive to the acidic environment.
DownLoad:
CSV

|
According to the above results, it reveals that the polyazole side chains do affect the electronic structure and Am(Ⅲ)/Eu(Ⅲ) extraction performance of the three pyridine-derived polyazole ligands. Among them, L1 with 1,2,4-triazole side chains has the largest separation factor for Am(Ⅲ)/Eu(Ⅲ) ions according to the ΔΔG values for the reactions. Therefore, we conclude that the 1,2,4-triazole side chains obviously enhance the Am(Ⅲ)/Eu(Ⅲ) selectivity of ligand. As we know, the preorganized phenanthroline moiety could significantly increase the complexation ability of ligand and its selectivity of Am(Ⅲ)/Eu(Ⅲ) than the bipyridine backbone [18,40,43]. For example, phenanthroline-based ligand CyMe4-BTPhen could extract Cm(Ⅲ) and Am(Ⅲ) from lanthanide ions with significantly faster kinetics, efficiency and higher selectivity than CyMe4-BTBP [18]. Thus, we predict that if the side chain 1,2,4-triazol is grafted onto the phenanthroline moiety, a new ligand, 2,9-bis(5-(2-ethylhexyl)-1H-1,2,4-triazol-3-yl)-1,10-phenanthroline (L4, Fig. 6) would show great An(Ⅲ)/Ln(Ⅲ) selectivity. To verify our expectation, the ESP map as well as the HOMO and LUMO orbitals for ligand L4 are presented in Fig. 6. The negative minimum value of ESP for L4 (−80.78 kcal/mol) is obviously more negative than that of L1 (−70.47 kcal/mol), which reflects that L4 has stronger complexation ability with metal ions than L1. Furthermore, the higher energy level of HOMO (−5.64 eV) with smaller HOMO-LUMO energy gap (4.23 eV) for L4 compared to that of L1 indicate that L4 is a better electron donor. These results are consistent with earlier findings that tetradentate ligand with the same side chain has a stronger complexation ability compared to the tridentate one [40]. In addition, the η value of L4 is 3.37 eV, which is 0.65 eV smaller than that of L1, suggesting that L4 is relatively softer than L1.
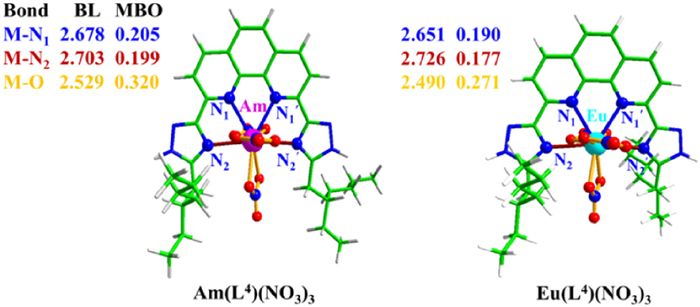
The optimized structures of AmL4(NO3)3 and EuL4(NO3)3 complexes with the bond lengths and MBOs of M-N and M-O bonds are shown in Fig. 7. The average M-N1 bonds in ML4(NO3)3 complex is shorter than that in ML1(NO3)3 due to the larger coordinated cavity of the former. By contrast, the M-N2 bond is longer in ML4(NO3)3 than that in ML1(NO3)3 thanks to the steric hindrance in the former. The Am-N1 and Am-O bonds in AmL4(NO3)3 are slightly longer than the Eu-N1 and Eu-O bonds in EuL4(NO3)3 complex, whereas the Am-N2 bond is shorter than the Eu-N2 bond, which suggests that L4 may have a higher Am(Ⅲ)/Eu(Ⅲ) selectivity than L1. The Am-N and Am-O MBOs in AmL4(NO3)3 are greater than the corresponding Eu-ones in EuL4(NO3)3, demonstrating that L4 has a complexation preference for Am(Ⅲ) ions. Fig. S6 (Supporting information) displayed the α-spin valence MOs of AmL4(NO3)3 and EuL4(NO3)3 complexes. It can be seen that ligand L4 has similar bonding interactions with Am(Ⅲ) and Eu(Ⅲ) ions, while the former has somewhat lower MO energy levels than the latter, suggesting the higher stability of AmL4(NO3)3. The values of ΔG and ΔΔG for the reactions with ligand L4 in Table 7 reflect significant complexing ability and high selectivity of Am(Ⅲ)/Eu(Ⅲ). For example, the ∆∆G value for reaction [M(NO3)(H2O)8]2+ aq + L4 org + 2NO3− aq = ML4(NO3)3 org + 8H2O aq in cyclohexanone/water solutions is −8.09 kcal/mol, which is more negative compared to that of L1 (−5.78 kcal/mol). Thus, we theoretically confirm that L4 has better Am(Ⅲ)/Eu(Ⅲ) selectivity than L1.
 DownLoad:
CSV
DownLoad:
CSV

|
In summary, we first investigated the effects of polyazole side chains on Am(Ⅲ)/Eu(Ⅲ) separation performance of three pyridine-derived ligands with side chains 1,2,4-triazole (L1), 1,2,3-triazole (L2) and pyrazole (L3) by scalar relativistic DFT methods. The results of M-L bond length and bonding nature suggest that the polyazole side chains play an important role in the complexation with Am(Ⅲ)/Eu(Ⅲ) ions and also reveal stronger complexation and higher covalency with Am(Ⅲ) than Eu(Ⅲ) ions of the three ligands. L1 with 1,2,4-triazole side chains has the strongest separation ability while the lowest complexation ability for Am(Ⅲ)/Eu(Ⅲ) among the three pyridine-derived polyazole ligands based on the ΔG and ΔΔG values for the reactions. Motivated by the above results, we suggested that the 1,2,4-triazole side chains could enhance the Am(Ⅲ)/Eu(Ⅲ) selectivity of ligand and designed a new ligand (L4) bearing 1,2,4-triazole side chains and a preorganized phenanthroline backbone. The results of ESP, molecular orbitals, bonding nature, and thermodynamic properties reflected that L4 has significant separation properties for Am(Ⅲ)/Eu(Ⅲ). We firstly clarify the effect of polyazole side chains on the complexation and selectivity ability for Am(Ⅲ)/Eu(Ⅲ) ions and then design a new ligand with potential Am(Ⅲ)/Eu(Ⅲ) separation performance. This work provides a new strategy for designing effective N-donor ligands with excellent actinides and lanthanides separation performance in the future.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. U2067212, 22076188, 11875058, U20B2019), the National Science Fund for Distinguished Young Scholars (No. 21925603).
Supplementary material associated with this article can be found, in the online version, at doi:
R.C. Ewing, Nat. Mater. 14 (2015) 252–257. doi: 10.1038/nmat4226
J. Veliscek-Carolan, J. Hazard. 318 (2016) 266–281. doi: 10.1016/j.jhazmat.2016.07.027
M. Salvatores, G. Palmiotti, Prog. Part. Nucl. Phys. 66 (2011) 144–166. doi: 10.1016/j.ppnp.2010.10.001
A. Salvatores, Nucl. Eng. Des. 235 (2005) 805–816. doi: 10.1016/j.nucengdes.2004.10.009
J.N. Mathur, M.S. Murali, K.L. Nash, Solvent Extr. Ion. Exch. 19 (2001) 357–390. doi: 10.1081/SEI-100103276
K.L. Nash, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1994, pp. 197–238.
A. Khayambashi, L. Chen, X. Dong, et al., Chin. Chem. Lett. 33 (2022) 3429–3434. doi: 10.1016/j.cclet.2022.02.011
L. Xu, N. Pu, G. Y, et al., Inorg. Chem. Front. 7 (2020) 1726–1740. doi: 10.1039/d0qi00200c
X. Yang, S.H. Wang, L. Xu, et al., Inorg. Chem. Front. 9 (2022) 4671–4684. doi: 10.1039/d2qi01153k
T.Y. Xiu, S.M. Zhang, P. Ren, et al., Chin. Chem. Lett. 34 (2023) 108440. doi: 10.1016/j.cclet.2023.108440
D.S. Tian, Y.Y. Liu, Y. Kang, et al., ACS Cent. Sci. 9 (2023) 1642–1649. doi: 10.1021/acscentsci.3c00504
R.G. Pearson, J. Am. Chem. Soc. 85 (1963) 3533–3539. doi: 10.1021/ja00905a001
A. Leoncini, J. Huskens, W. Verboom, Chem. Soc. Rev. 46 (2017) 7229–7273. doi: 10.1039/C7CS00574A
Z.R. Ye, Q.Y. Wu, C.Z. Wang, et al., Inorg. Chem. 60 (2021) 16409–16419. doi: 10.1021/acs.inorgchem.1c02256
A. Geist, P.J. Panak, Solvent Extr. Ion. Exch. 39 (2021) 128–151. doi: 10.1080/07366299.2020.1831235
C. Madic, M.J. Hudson, Report EUR 18038EN. Nuclear Science and Technology, 1998.
Z. Kolarik, U. Müllich, F. Gassner, Solvent Extr. Ion. Exch. 17 (1999) 23–32. doi: 10.1080/07360299908934598
F.W. Lewis, L.M. Harwood, M.J. Hudson, et al., J. Am. Chem. Soc. 133 (2011) 13093–13102. doi: 10.1021/ja203378m
M.G.B. Drew, M.J. Hudson, P.B. Iveson, et al., Dalton Trans. (1999) 2433–2440.
A. Bremer, C.M. Ruff, D. Girnt, et al., Inorg. Chem. 51 (2012) 5199–5207. doi: 10.1021/ic3000526
D.P. Su, Y. Liu, S.M. Li, et al., Eur. J. Inorg. Chem. (2017) 651–658 2017.
Y. Liu, X.Y. Yang, S.D. Ding, et al., Inorg. Chem. 57 (2018) 5782–5790. doi: 10.1021/acs.inorgchem.8b00074
J.R. Wang, D.P. Su, D.Q. Wang, et al., Inorg. Chem. 54 (2015) 10648–10655. doi: 10.1021/acs.inorgchem.5b01452
X.H. Kong, Q.Y. Wu, C.Z. Wang, et al., J. Phys. Chem. A 122 (2018) 4499–4507. doi: 10.1021/acs.jpca.8b00177
C. Kiefer, A.T. Wagner, B.B. Beele, et al., Inorg. Chem. 54 (2015) 7301–7308. doi: 10.1021/acs.inorgchem.5b00803
E. Macerata, E. Mossini, S. Scaravaggi, et al., J. Am. Chem. Soc. 138 (2016) 7232–7235. doi: 10.1021/jacs.6b03106
A.C. Edwards, P. Mocilac, A. Geist, et al., Chem. Commun. 53 (2017) 5001–5004. doi: 10.1039/C7CC01855J
A. Ossola, E. Macerata, E. Mossini, et al., J. Radioanal. Nucl. Ch. 318 (2018) 2013–2022. doi: 10.1007/s10967-018-6253-y
P. Weßling, M. Maag, G. Baruth, et al., Inorg. Chem. 61 (2022) 17719–17729. doi: 10.1021/acs.inorgchem.2c02871
F. Galluccio, E. Macerata, P. Weßling, et al., Inorg. Chem. 61 (2022) 18400–18411. doi: 10.1021/acs.inorgchem.2c02332
L.K. Liu, S.B. Xie, H.B. Lv, et al., Chin. Chem. Lett. 33 (2022) 3439–3443. doi: 10.1016/j.cclet.2022.04.001
Y.A. Ustynyuk, M.Y. Alyapyshev, V.A. Babain, et al., Russ. Chem. Rev. 85 (2016) 917–942. doi: 10.1070/RCR4588
X. Zhang, S.L. Adelman, B.T. Arko, et al., Inorg. Chem. 61 (2022) 11556–11570. doi: 10.1021/acs.inorgchem.2c00534
C. Ebenezer, R. Vijay Solomon, Inorg. Chem. Front. 8 (2021) 3012–3024. doi: 10.1039/d1qi00097g
R.C. Chapleski Jr., A.S. Ivanov, K.A. Peterson, et al., Phys. Chem. Chem. Phys. 23 (2021) 19558–19570. doi: 10.1039/d1cp02466c
J.P. Yu, K. Liu, Q.Y. Wu, et al., Chinese J. Chem. 39 (2021) 2125–2131. doi: 10.1002/cjoc.202100149
G.J.P. Deblonde, M.P. Kelley, J. Su, et al., Angew. Chem. Int. Ed. 57 (2018) 4521–4526. doi: 10.1002/anie.201709183
J.N. Cross, J. Su, E.R. Batista, et al., J. Am. Chem. Soc. 139 (2017) 8667–8677. doi: 10.1021/jacs.7b03755
J. Su, E.R. Batista, K.S. Boland, et al., J. Am. Chem. Soc. 140 (2018) 17977–17984. doi: 10.1021/jacs.8b09436
Q.Y. Wu, Y.T. Song, L. Ji, et al., Phys. Chem. Chem. Phys. 19 (2017) 26969–26979. doi: 10.1039/C7CP04625A
C. Wang, Q.Y. Wu, X.H. Kong, et al., Inorg. Chem. 58 (2019) 10047–10056. doi: 10.1021/acs.inorgchem.9b01200
X.H. Kong, Q.Y. Wu, J.H. Lan, et al., Inorg. Chem. 57 (2018) 14810–14820. doi: 10.1021/acs.inorgchem.8b02550
X.P. Lei, Q.Y. Wu, C.Z. Wang, et al., Dalton Trans. 51 (2022) 16659–16667. doi: 10.1039/d2dt02474h
X.P. Lei, Q.Y. Wu, C.Z. Wang, et al., Inorg. Chem. 62 (2023) 2705–2714. doi: 10.1021/acs.inorgchem.2c03823
Y.M. Chen, C.Z. Wang, Q.Y. Wu, et al., Inorg. Chem. 59 (2020) 3221–3231. doi: 10.1021/acs.inorgchem.9b03604
Z.R. Ye, Q.Y. Wu, C.Z. Wang, et al., Inorg. Chem. 61 (2022) 6110–6119. doi: 10.1021/acs.inorgchem.2c00232
Y. Zou, J.H. Lan, L.Y. Yuan, et al., Inorg. Chem. 62 (2023) 4581–4589. doi: 10.1021/acs.inorgchem.2c04476
Y. Zou, J.H. Lan, L.Y. Yuan, et al., Inorg. Chem. 61 (2022) 15423–15431. doi: 10.1021/acs.inorgchem.2c01952
A. Bhattacharyya, T. Gadly, A.S. Kanekar, et al., Inorg. Chem. 57 (2018) 5096–5107. doi: 10.1021/acs.inorgchem.8b00142
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785–789. doi: 10.1103/PhysRevB.37.785
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 16, Revision B, Gaussian, Inc., Wallingford, CT, 2016.
M. Dolg, H. Stoll, H. Preuss, J. Chem. Phys. 90 (1989) 1730–1734. doi: 10.1063/1.456066
W. Küchle, M. Dolg, H. Stoll, J. Chem. Phys. 100 (1994) 7535–7542. doi: 10.1063/1.466847
X.Y. Cao, M. Dolg, J. Mol. Struct. 581 (2002) 139–147. doi: 10.1016/S0166-1280(01)00751-5
X.Y. Cao, M. Dolg, H. Stoll, J. Chem. Phys. 118 (2002) 487–496.
X.Y. Cao, M. Dolg, J. Mol. Struct. 673 (2004) 203–209. doi: 10.1016/j.theochem.2003.12.015
G.A. Shamov, G. Schreckenbach, R.L. Martin, et al., Inorg. Chem. 47 (2008) 1465–1475. doi: 10.1021/ic7015403
G.A. Shamov, G. Schreckenbach, J. Phys. Chem. A 109 (2005) 10961–10974. doi: 10.1021/jp053522f
J. Narbutt, A. Wodyński, M. Pecul, Dalton Trans. 44 (2015) 2657–2666. doi: 10.1039/C4DT02657H
A. Zaiter, B. Amine, Y. Bouzidi, et al., Inorg. Chem. 53 (2014) 4687–4697. doi: 10.1021/ic500361b
C. Clavaguéra-Sarrio, V. Vallet, D. Maynau, et al., J. Chem. Phys. 121 (2004) 5312–5321. doi: 10.1063/1.1784412
M. Cossi, N. Rega, G. Scalmani, et al., J. Comput. Chem. 24 (2003) 669–681. doi: 10.1002/jcc.10189
V. Barone, M. Cossi, J. Phys. Chem. A 102 (1998) 1995–2001. doi: 10.1021/jp9716997
J. Ho, A. Klamt, M.L. Coote, J. Phys. Chem. A 114 (2010) 13442–13444. doi: 10.1021/jp107136j
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
I. Mayer, Chem. Phys. Lett. 97 (1983) 270–274. doi: 10.1016/0009-2614(83)80005-0
A.E. Reed, F. Weinhold, J. Chem. Phys. 78 (1983) 4066–4073. doi: 10.1063/1.445134
A.E. Reed, R.B. Weinstock, F. Weinhold, J. Chem. Phys. 83 (1985) 735–746. doi: 10.1063/1.449486
R.F.W. Bader, C.F. Matta, Inorg. Chem. 40 (2001) 5603–5611. doi: 10.1021/ic010165o
E. Espinosa, I. Alkorta, J. Elguero, et al., J. Chem. Phys. 117 (2002) 5529–5542. doi: 10.1063/1.1501133
T. Ziegler, A. Rauk, Theor. Chem. Acc. 46 (1977) 1–10. doi: 10.1007/BF02401406
G. te Velde, F.M. Bickelhaupt, E.J. Baerends, et al., J. Comput. Chem. 22 (2001) 931–967. doi: 10.1002/jcc.1056
C. Fonseca Guerra, J.G. Snijders, G. te Velde, et al., Theor. Chem. Acc. 99 (1998) 391–403.
T.Z.E.J. Baerends, J. Autschbach, D. Bashford, et al., ADF2022, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, URL:
R.G. Parr, R.G. Pearson, J. Am. Chem. Soc. 105 (1983) 7512–7516. doi: 10.1021/ja00364a005
R. Shannon, Acta Crystallogr. A 32 (1976) 751–767. doi: 10.1107/S0567739476001551
M.P. Jensen, A.H. Bond, J. Am. Chem. Soc. 124 (2002) 9870–9877. doi: 10.1021/ja0178620
D. Cremer, E. Kraka, J. Am. Chem. Soc. 107 (1985) 3811–3819. doi: 10.1021/ja00299a010
W.H. Tu, S.J. Zeng, Y. Bai, et al., Ind. Chem. Mater. 1 (2023) 262–270. doi: 10.1039/d2im00041e
P. Thakur, J.L. Conca, G.R. Choppin, J. Solution Chem. 41 (2012) 599–615. doi: 10.1007/s10953-012-9826-3

Figure 1 ESP maps and negative minimum ESP values (kcal/mol) for ligands L1, L2 and L3.

Figure 2 LUMOs and HOMOs for ligands and the corresponding MO energies (eV) as well as the HOMO-LUMO gap (eV) with the isosurface value of 0.02 a.u.

Figure 3 Structures of ML(NO3)3 complexes. C, H, O, N and metal atoms are denoted as green, white, red, blue and pink, respectively.

Figure 4 Energy level diagrams (eV) of MOs for M(L3)(NO3)3 with the isosurface value of 0.02 a.u.

Figure 5 The leading donor-acceptor interactions of ΔEorb1 in complexes. The metal composition in [M(NO3)3] acceptor orbital is also shown.

Figure 7 The optimized structures, bond length (BL) and MBOs of Am(L4)(NO3)3 and Eu(L4)(NO3)3.
Table 5. The Interaction energy (kcal/mol) between [L] and [M(NO3)3] (M = Am/Eu) fragments in complexes.
|
|
 下载: 导出CSV
下载: 导出CSV
Table 6. ΔG (kcal/mol) and ΔΔG (kcal/mol) for extraction reactions in cyclohexanone/water and nitrobenzene/water solutions using the B3LYP functional and 6–311G(d, p) basis set.a
|
|
下载: 导出CSV
Table 7. ΔG (kcal/mol) and ΔΔG (kcal/mol) for the reactions of ligand L4 with Am(Ⅲ) and Eu(Ⅲ) complexes in cyclohexanone/water and nitrobenzene/water solutions using the B3LYP functional and 6-311G(d, p) basis set.
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们







 下载:
下载: