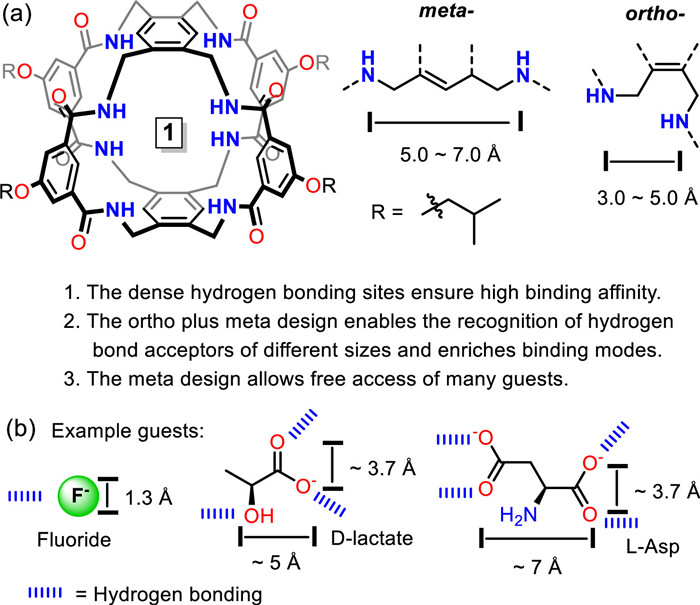
Figure 1.
(a) Molecule design. (b) The guests used in this study, i.e., fluoride, D-lactate and L-Asp. They were prepared as tetrabutylammonium (TBA) salts.
Synthesis and molecular recognition characteristics of a tetrapodal benzene cage
Caihong Mao , Yanfeng He , Xiaohan Wang , Yan Cai , Xiaobo Hu

Cage molecules play an important role in host-guest chemistry due to their unique structural features, remarkable molecular recognition characteristics and rich applications [1-5]. In order to obtain novel molecular recognition patterns and properties, tremendous efforts have been devoted to create new synthetic cages [6-30]. For instance, various oligopyrrolic cages for small molecular recognition [6-9], hexapodal cages for anion recognition [10-12], temple shaped cages for carbohydrate recognition [13-15], porous cages for gas sorption [16,17], tetrahedral cages for white phosphorus [18] and other guest molecular recognition [19]. Thanks to the confined structure and settled binding sites, one main merit of cage molecules is that they typically exhibit good selectivity towards guest molecules of appropriate size and functional groups. However, this could also limit the recognition scope of the cage to few types of molecules and narrow the object library when developing applications other than selectivity, such as molecular transport and catalysis [20,21]. Customizing novel cages for individual objects each time is obviously challenging and time-consuming. On the other hand, host molecules with multiple and well-designed binding sites are possible to compensate for the loss of binding affinity caused by object shrinkage or functional group changes, while still maintaining selectivity for objects with appropriate size and functional groups [22,23]. Here we present a new cage molecule (Fig. 1a) with dense and well-deployed hydrogen bond donor sites that can provide strong synergistic hydrogen bonding to bind multiple types of hydrogen bond acceptors. In this contribution, we show three distinct guests as examples (Fig. 1b), where F− represents the case of extremely small size, D-lactate represents the case of appropriate size, and L-Asp represents the case of branched shape. Through 1H NMR titration studies, the cage expressed good binding affinity to all the tested guests in a competitive solvent system (10%DMSO/THF), while exhibiting good selectivity for D-lactate versus F− and L-Asp.
The target cage comprised of two cap sections (symmetrical tetramethylbenzene) and four tether sections (isophthalamide) (Fig. 1a). Eight amide protons and four inner rim benzene protons provide dense hydrogen bond donor sites, allowing the cage to form strong synergistic hydrogen bonding with various hydrogen bond acceptors. The tethers are deployed in an ortho plus meta manner. The ortho region ensures high binding affinity to small hydrogen bond acceptors, such as F− and carboxyl group, while the meta region is suitable for larger hydrogen bond acceptors and allows free access of small or linear guest molecules. Moreover, the combination of ortho and meta regions could also enrich binding patterns of the cage. The side chains of the cage are modifiable, thus it would enable investigations for different purposes in different media, such as binding to bio-targets in aqueous media.
It is challenging to synthesize the target cage (1, whose caps are arranged in parallel), because it is easy to generate the structural isomer (caps are arranged in a cross pattern) that is extremely difficult to separate. The synthesis of 1 began with the preparation of the cap (5) and tether (3) precursors (Fig. 2a), where the production of 5 is the key to access the correct structural isomer of 1. Compound 3 could be conveniently prepared from the commercial compound 2 through substitution and saponification reactions with a good yield of 42%.
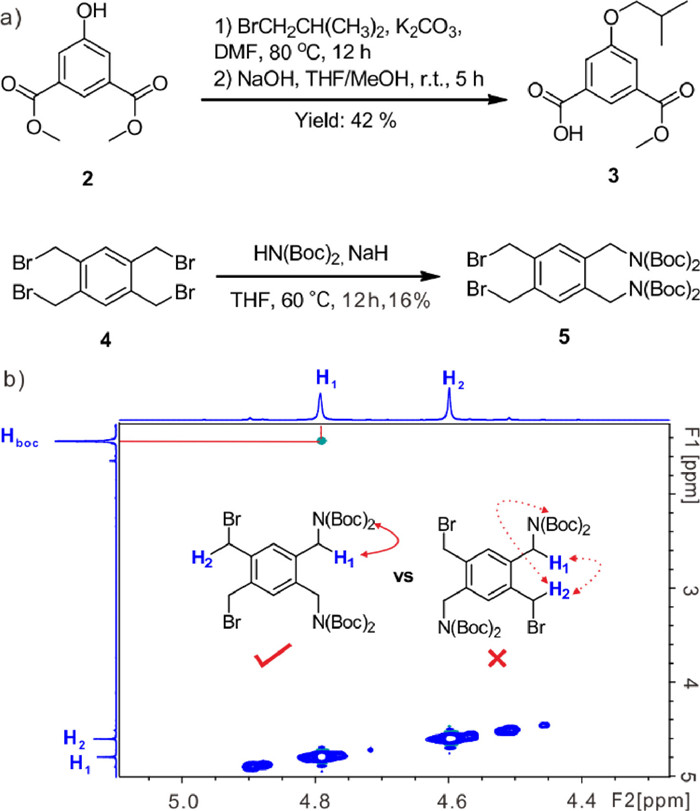
In contrast, the synthesis of 5 was not easy. After trying several synthesis routes, directly treating the commercial compound 4 with NaN(Boc)2 at 60 ℃ gave the desired structural isomer (5) with relatively high content (~30% by 1H NMR integration). The key to isolating 5 from the other two structural isomers was to perform fast column chromatography elution at an extremely shallow gradient, giving a yield of 16%. To verify the structure of 5 (ortho-type), 1H–1H NOESY NMR was performed. The spectrum (Fig. 2b) only shows the correlation of H1 ↔ HBoc, but not the correlations of H1 ↔ H2 and H2 ↔ HBoc, which excludes the para-type isomer (the meta-type isomer can be ruled out simply by 1H NMR). In addition, treating 5 with S3 (Scheme S1 in Supporting information) afforded a cyclic compound S2 (validated by 1H NMR and HRMS, Figs. S3 and S4 in Supporting information), which further confirms 5 is the desired structure.
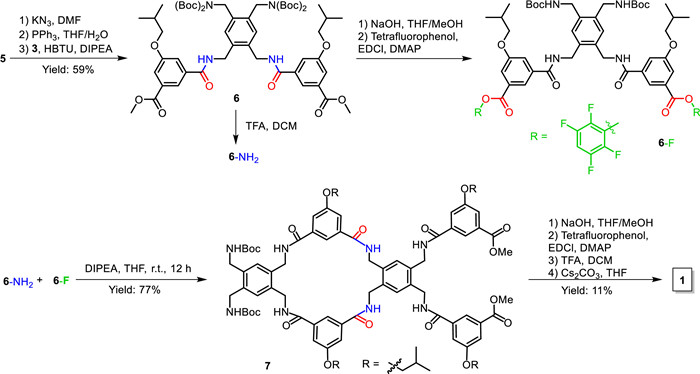
With the key precursor 5, the cage can be prepared through multi-step reactions (Scheme 1). Compound 5 was first converted to azide with KN3, then to amine with PPh3 and subsequently coupled with 3 by HBTU to afford the key intermediate 6 with a good overall yield of 59%. Afterwards, compound 6 was divided into two parts. One partwas treated with TFA to remove Boc groups to give the amine form of 6 (6-NH2), the other part was converted from methyl ester to more active tetrafluorophenyl ester (6-F). Compounds 6-NH2 and 6-F were then mixed together under high dilution condition (1 mmol/L) to afford the cyclic intermediate 7 with a high overall yield of 77%. Compound 7 was converted again to tetrafluorophenyl ester, followed by the removal of Boc groups, and then treated with Cs2CO3 under high dilution condition (0.25 mmol/L) to give the target cage 1 with an overall yield of 11%. This synthetic approach requires three times of column chromatography and allows 1 to be produced in one week.
Before molecular recognition studies, the key protons of 1 (H1–5, Fig. 3b and Fig. S5 in Supporting information) was firstly assigned with 1D and 2D NMR spectra of 1. When the cage structure is formed, the amide methylene (–NHCH2-, H5) protons exhibit two typical ABX signals at 4.78 and 4.48 ppm (doublet-doublet, Fig. S5) due to structural constraints (the two methylene protons have different orientations, one towards the ortho side while the other towards the meta side). By observing the key correlations of H2 ↔ H4, H1 ↔ H5A and H1 ↔ H5B (Fig. S7 in Supporting information), H1–5 can be assigned. Furthermore, the 1H–1H NOESY spectrum shows both H1↔ H2 and H1 ↔ H4 correlations (Fig. S9 in Supporting information), indicating that the amide protons are in the dynamic equilibrium between two conformations, i.e., H1 on the inner or outer rim of the cage. However, when 1 binds to guests, the conformation of H1 on the inner rim prevails (Figs. S16, S28 and S35 in Supporting information).
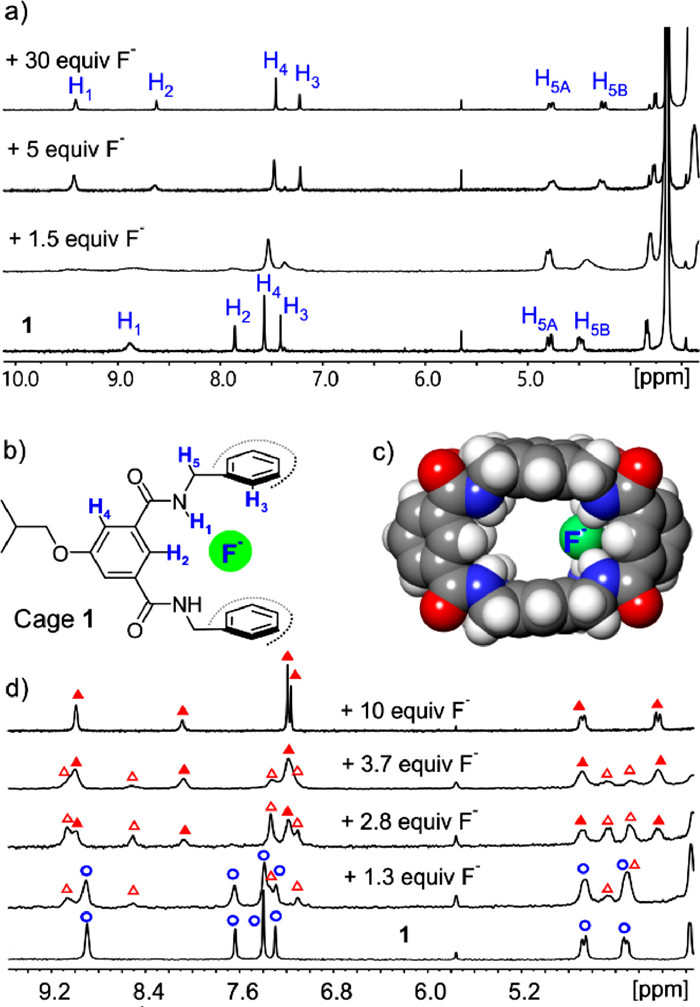
Cages that exhibit high affinity to larger-sized molecules normally show low or no binding affinity for much smaller-sized guests. In order to test whether 1 can compensate for this drawback, recognition studies were conducted on one of the smallest guests, F−. The recognition of fluoride anion [31,32] was investigated by adding F− (0.1-1.67 equiv.) to 1 in 10% DMSO/THF (Fig. 3a and Fig. S11 in Supporting information). The protons of 1 gradually disappeared with the concomitant appearance of some broad signals. The newly emerging signals should belong to F−⊂1 complexes, while the broadness of the spectrum is probably due to the coexistence of different binding forms and they undergo relatively slow exchange with each other. The CPK molecular model (Fig. 3c and Fig. S10 in Supporting information) of the 1:1 F−⊂1 complex clearly shows a large vacancy of the cavity of 1. Continuingly adding F− to 5 equiv. made the previously broad spectrum sharp. Further adding F− to 30 equiv. did not evoke significant spectral changes (Fig. 3a and Fig. S12 in Supporting information). This is consistent with the above assumption, and the sharp spectrum is due to the saturation of the binding sites of 1 by F−, resulting in the formation of a defined F−⊂1 complex. By comparing the chemical shifts of the protons of the defined F−⊂1 complex with those of the free 1, the amide (H1) and inner rim benzene (H2) protons of 1 undergo substantial downfield shifts (H1 from 8.88 ppm to 9.41 ppm and H2 from 7.86 ppm to 8.62 ppm), revealing that these protons directly involve in the interaction with F− through hydrogen bonding. Other protons of 1 also experience small to moderate shifts, implying 1 undergoes slight structural adjustments to accommodate F−.
The mass spectra of 1 + 2F− (2 equiv., Fig. S18 in Supporting information) and 1 + 20F− (20 equiv., Fig. S19 in Supporting information) only show 1:1 and 1:2 [1-F−] signals, respectively, indicating the 1:1 [1-F−] species are the prevalent ones in the NMR spectra before 2 equiv. F− while the 1:2 [1-F−] species dominate in the NMR spectra after 5 equiv. F−. NMR titration study of F− was also conducted in DMSO–d6, considering that the high polar medium may eliminate weakly bound host-guest complexes and lead to clear NMR spectra. Indeed, NMR spectra of 1 + F− in DMSO–d6 are relatively sharp (Fig. 3d and Fig. S17 in Supporting information). Thereby, spectral evolution from free 1 (blue circle) to 1:1 (red triangle) and further to 1:2 (red solid triangle) [1-F−] binding were observed. Due to the coexistence of different species, the exact binding constant (K) of [1-F−] binding cannot be determined. Nevertheless, by the competition study between F− and L-Asp (Fig. S40 in Supporting information, the recognition of L-Asp will be discussed later), an approximate value of K(F−) in 10% DMSO/THF can be obtained as ~106 L/mol, which is in sharp contrast with the K (4.4 × 102 L/mol) of [S1-F−] (S1 is a control cage with only meta-deployed tethers, Fig. S44 in Supporting information). This proves that the ortho design of cage 1 does work well for compensating for the loss of binding affinity caused by object shrinkage.
According to the molecular model (Fig. 4e and Fig. S20 in Supporting information), D-lactate should fit well to the cavity and most binding sites of 1. Thus, it was expected that 1 would exhibit high affinity to D-lactate. D-Lactate is considered harmful to human metabolism and can lead to decalcification and acidosis [33-35]. Therefore, developing artificial molecules capable of recognizing D-lactate is important [36,37]. By adding D-lactate (0.12-2.08 equiv.) to 1 in 10% DMSO/THF (Fig. 4a and Fig. S21 in Supporting information), the 1H NMR spectra first became broad with the concomitant appearance of some new signals and dramatic chemical shifts. When the amount of D-lactate approached 1 equiv., the spectra turned sharp, and the subsequent addition of D-lactate did not cause further noticeable changes. These observations indicate 1 undergoes substantial structural adjustments to adopt D-lactate, and they bind together through a high affinity 1:1 receptor-substrate mode. The 1:1 binding stoichiometry was also validated by Job’s plot and mass spectrum (Figs. S30 and S31 in Supporting information). By fitting the chemical shifts of a sharp signal (H3), the binding constant of D-lactate⊂1 was determined [38,39] as K(D-lactate) = (1.4 ± 0.5) × 107 L/mol (Fig. 4b). However, due to the limitations of the NMR titration technique, it is more appropriate to consider K(D-lactate) >> 105. Furthermore, the competition study between D-lactate and L-Asp (Fig. S38 in Supporting information, the recognition of L-Asp will be discussed later) gave K(D-lactate) ~107 L/mol (1.3 × 107 L/mol), which is in good agreement with the above value. The high binding affinity can be attributed to the appropriate size of D-lactate and the effective utilization of most binding sites of 1 by the carboxyl and hydroxyl groups of D-lactate.
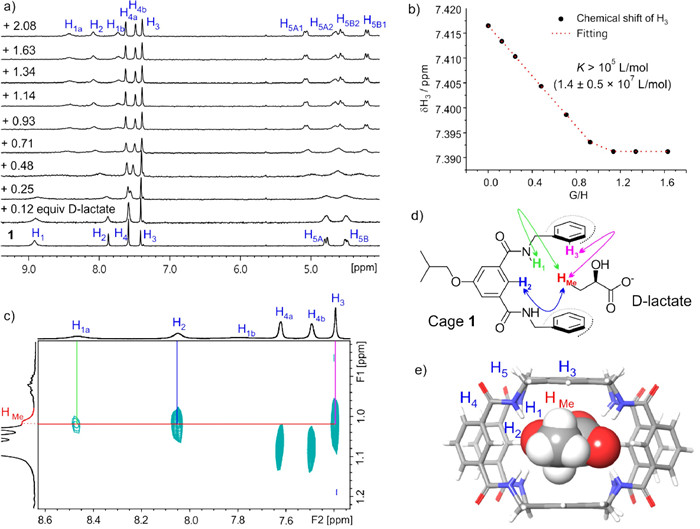
In order to assign the additional signals caused by the formation of D-lactate⊂1 complex as well as to understand the binding mode of 1 and D-lactate, 2D NMR studies (Figs. S23–S28 in Supporting information) were conducted. For instance, H5A1 and H5B1 are from the same amide methylene due to the existence of COSY correlation H5A1 ↔ H5B1 (Fig. S24); H4a and H4b are the outer rim benzene protons due to the observation of the COSY correlation H4a ↔ H4b (Fig. S25); H1a and H1b are amide protons conjunct with H5A/B1 and H5A/B2, respectively, due to the NOE correlations H1a ↔ H5A/B1 and H1b ↔ H5A/B2 (Fig. S27). Thanks to the observation of the key correlations (Figs. 4c and d) between the methyl group (HMe) of D-lactate and amide (H1a), inner rim benzene (H2) and cap benzene (H3) protons of 1, it further proves D-lactate is inside 1 (Fig. 4e and Fig. S20). Therefore, the origin of these new signals is probably because that the encapsulation of D-lactate makes 1 asymmetric. Unfortunately, the signal of H1b is too broad to observe the correlation H1b ↔ HMe. Similarly, the other protons of D-lactate are too weak to show NOE correlations with 1. Nevertheless, more recognition evidences can be found from NMR studies. For example, by increasing the amount of D-lactate after 1 equiv., the 1H NMR spectra show downfield shifts of the HMe of D-lactate (Fig. S22 in Supporting information), indicating that HMe is in the shielded area of the benzene rings of 1 and the downfield shifts of HMe is due to the signal averaging of the free and encapsulated D-lactate.
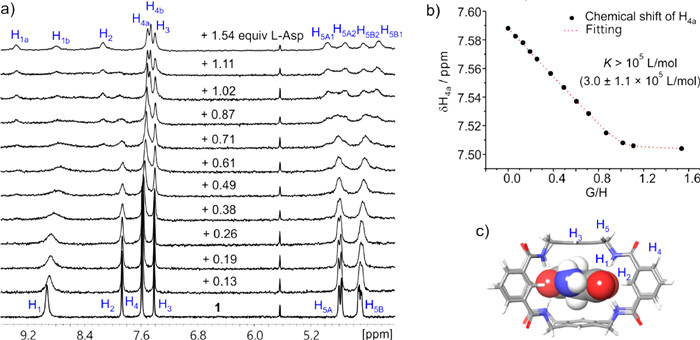
Encouraged by the success of D-lactate, we continued to carry out the recognition study of L-Asp [40,41], a branched molecule. Despite the steric hindrance of L-Asp, it was expected that 1 could still well bind L-Asp through fully utilizing the binding sites of 1 by the two carboxyl groups of L-Asp. Indeed, the NMR titration study (Fig. 5a and Fig. S33 in Supporting information) of L-Asp and 1 shows similar change as that of D-lactate and 1, i.e., the spectra also undergo significant changes and show additional peaks. These observations can also be explained by the encapsulation of L-Asp, which leads to the structural asymmetry of 1. Through Job’s plot and mass spectrum (Figs. S36 and S37 in Supporting information), the binding stoichiometry was determined to be 1:1. Thus, by fitting the chemical shifts of H4a with the 1:1 receptor-substrate binding model [38,39], the binding constant of L-Asp⊂1 was determined as K(L-Asp) ~105 L/mol [(3.0 ± 1.1) × 105 L/mol] (Fig. 5b). This high binding constant indicates 1 can efficiently recognize L-Asp in the competitive solvent medium (10% DMSO/THF). At last, the computational result implies L-Asp could be encapsulated by 1 in an expected binding mode (Fig. 5c and Fig. S32 in Supporting information).
In conclusion, we have developed a new cage with dense hydrogen bond donor sites. The key to successfully synthesizing the desired structural isomer of the cage is to obtain the correct structural isomer (ortho type) of the cap precursor 5. By molecular recognition studies of F− (extremely small size), D-lactate (appropriate size) and L-Asp (branched shape) in competitive solvent media (10% DMSO/THF), the cage exhibits a wide recognition scope without losing molecular selectivity (K(D-lactate) > K(F−) > K(L-Asp)). Remarkably, the much higher (four orders of magnitude) binding constant of [1-F−] versus [S1-F−] proves the importance of the ortho design in binding small-sized objects. Moreover, it was found in our lab that many other molecules can be recognized by 1, for instance 1,4-dinitrobenzene, isooctyl phosphate and aspirin (Figs. S41-S43 in Supporting information), further supporting the wide recognition scope feature of our cage. By varying the size of guest molecules, we also observed that the cage can express rich binding behavior. For instance, both 1:1 and 1:2 host-guest binding patterns can be observed with small-sized anions, while high selectivity is shown with appropriate-sized ones. In line with these properties, our lab is currently conducting binary and selective anion recognition studies. In addition, the binary binding property also allows for the envisioning of future applications of the cage and its analogues in catalysis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge financial support by the National Natural Science Foundation of China (No. 22101260) and Natural Science Foundation of Zhejiang Province (No. LQ22B020001).
Supplementary material associated with this article can be found, in the online version, at doi:
T.S. Koblenz, J. Wassenaar, J.N.H. Reek, Chem. Soc. Rev. 37 (2008) 247–262. doi: 10.1039/B614961H
G. Zhang, M. Mastalerz, Chem. Soc. Rev. 43 (2014) 1934–1947. doi: 10.1039/C3CS60358J
A.P. Davis, Chem. Soc. Rev. 49 (2020) 2531–2545. doi: 10.1039/c9cs00391f
H. Wang, Y. Jin, N. Sun, W. Zhang, J. Jiang, Chem. Soc. Rev. 50 (2021) 8874–8886. doi: 10.1039/d0cs01142h
D. Zhang, T.K. Ronson, Y.Q. Zou, J.R. Nitschke, Nat. Rev. Chem. 5 (2021) 168–182. doi: 10.1038/s41570-020-00246-1
F. Wang, C. Bucher, Q. He, A. Jana, J.L. Sessler, Acc. Chem. Res. 55 (2022) 1646–1658. doi: 10.1021/acs.accounts.2c00120
F. Wang, E. Sikma, Z. Duan, et al., Chem. Commun. 55 (2019) 6185–6188. doi: 10.1039/c9cc02490e
J.H. Oh, J.H. Kim, D.S. Kim, et al., Org. Lett. 21 (2019) 4336–4339. doi: 10.1021/acs.orglett.9b01515
J.H. Oh, B.P. Hay, V.M. Lynch, et al., J. Am. Chem. Soc. 144 (2022) 16996–17009. doi: 10.1021/jacs.2c06284
H. Xie, T.J. Finnegan, V.W.L. Gunawardana, et al., J. Am. Chem. Soc. 143 (2021) 3874–3880. doi: 10.1021/jacs.0c12329
H. Xie, V.W.L. Gunawardana, T.J. Finnegan, W. Xie, J.D. Badjić, Angew. Chem. Int. Ed. 61 (2022) e202116518. doi: 10.1002/anie.202116518
W. Zhou, F. Wang, A. Li, et al., Cell Rep. Phys. Sci. 4 (2023) 101295. doi: 10.1016/j.xcrp.2023.101295
Y. Ferrand, M.P. Crump, A.P. Davis, Science 318 (2007) 619–622. doi: 10.1126/science.1148735
R.A. Tromans, T.S. Carter, L. Chabanne, et al., Nat. Chem. 11 (2019) 52–56. doi: 10.1038/s41557-018-0155-z
A.P. Davis, Chem. Soc. Rev. 49 (2020) 2531–2545. doi: 10.1039/c9cs00391f
M. Mastalerz, M.W. Schneider, I.M. Oppel, O. Presly, Angew. Chem. Int. Ed. 50 (2011) 1046–1051. doi: 10.1002/anie.201005301
M. Mastalerz, Acc. Chem. Res. 51 (2018) 2411–2422. doi: 10.1021/acs.accounts.8b00298
P. Mal, B. Breiner, K. Rissanen, J.R. Nitschke, Science 324 (2009) 1697–1699. doi: 10.1126/science.1175313
D. Zhang, T.K. Ronson, J.R. Nitschke, Acc. Chem. Res. 51 (2018) 2423–2436. doi: 10.1021/acs.accounts.8b00303
J. Rodr-íguez, J. Mosquera, J.R. Couceiro, et al., J. Am. Chem. Soc. 139 (2017) 55–58. doi: 10.1021/jacs.6b11103
N. Luo, Y.F. Ao, D.X. Wang, Q.Q. Wang, Chem. Asian J. 16 (2021) 3599–3603. doi: 10.1002/asia.202100920
S.O. Kang, J.M. Llinares, D. Powell, D. VanderVelde, K. Bowman-James, J. Am. Chem. Soc. 125 (2003) 10152–10153. doi: 10.1021/ja034969+
H. Valkenier, O. Akrawi, P. Jurček, et al., Chem 5 (2019) 429–444. doi: 10.1016/j.chempr.2018.11.008
Q.F. Sun, J. Iwasa, D. Ogawa, et al., Science 328 (2010) 1144–1147. doi: 10.1126/science.1188605
T. Jiao, L. Chen, D. Yang, et al., Angew Chem. Int. Ed. 56 (2017) 14545–14550. doi: 10.1002/anie.201708246
B. Htan, D. Luo, C. Ma, J. Zhang, Q. Gan, Cryst. Growth. Des. 19 (2019) 2862–2868. doi: 10.1021/acs.cgd.9b00096
H. Duan, Y. Li, Q. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 10101–10110. doi: 10.1002/anie.201912730
S. Hollstein, O. Shyshov, M. Hanževački, et al., Angew. Chem. Int. Ed. 61 (2022) e202201831. doi: 10.1002/anie.202201831
V.W.L. Gunawardana, T.J. Finnegan, C.E. Ward, C.E. Moore, J.D. Badjić, Angew. Chem. Int. Ed. 61 (2022) e202207418. doi: 10.1002/anie.202207418
C. Xu, Q. Lin, C. Shan, et al., Angew. Chem. Int. Ed. 61 (2022) e202203099. doi: 10.1002/anie.202203099
H.J. Han, J.H. Oh, J.L. Sessler, S.K. Kim, Chem. Commun. 55 (2019) 10876–10879. doi: 10.1039/c9cc05613k
S. Xiong, M.V.N. Kishore, W. Zhou, Q. He, Coord. Chem. Rev. 461 (2022) 214480. doi: 10.1016/j.ccr.2022.214480
R. Datta, S.P. Tsai, P. Bonsignore, S.H. Moon, J.R. Frank, FEMS. Microbiol. Rev. 16 (1995) 221–231. doi: 10.1111/j.1574-6976.1995.tb00168.x
Y.J. Wee, J.N. Kim, H.W. Ryu, Food. Technol. Biotechnol. 44 (2006) 163–172.
K. Ghosh, A. Majumdar, RSC Adv. 5 (2015) 24499–24506. doi: 10.1039/C5RA00017C
M. Almaraz, C. Raposo, M. Martín, M.C. Caballero, J.R. Morán, J. Am. Chem. Soc. 120 (1998) 3516–3517. doi: 10.1021/ja972537r
L. Zhang, A.F. Martins, P. Zhao, et al., J. Am. Chem. Soc. 139 (2017) 17431–17437. doi: 10.1021/jacs.7b08292
P. Thordarson, Chem. Soc. Rev. 40 (2011) 1305–1323. doi: 10.1039/C0CS00062K
D.B. Hibbert, P. Thordarson, Chem. Commun. 52 (2016) 12792–12805. doi: 10.1039/C6CC03888C
J. Tian, Y. Wang, Y. Chen, et al., Chem. Commun. 56 (2020) 15012–15015. doi: 10.1039/d0cc06736a
J.N. Martins, J.C. Lima, N. Basílio, Molecules 26 (2021) 106.

Figure 1 (a) Molecule design. (b) The guests used in this study, i.e., fluoride, D-lactate and L-Asp. They were prepared as tetrabutylammonium (TBA) salts.

Figure 2 (a) Synthesis of precursors. (b) The 1H–1H NOESY (400 MHz) spectrum of 5 (10 mmol/L) in CDCl3 at 298 K, showing the correlation between H1 and HBoc.

Figure 3 (a) 1H NMR spectra of 1 (1 mmol/L) and 1 + F− (1.5, 5 and 30 equiv., respectively) in 10% DMSO–d6/THF-d8. (b) A section of the structure of F−⊂1 complex. (c) Molecular model (by DFT B3LYP-D3, 6–31+G(d,p)) of F−⊂1 complex. Redundant side chains are omitted for clarity. (d) 1H NMR spectra of 1 (1 mmol/L) and 1 + F− (1.3, 2.8, 3.7, and 10 equiv., respectively) in DMSO–d6. Blue circle = free 1, red triangle = 1:1 F−⊂1 complex, and red solid triangle = 2:1 F−⊂1 complex.

Figure 4 (a) From bottom to top: 1 in 10% DMSO–d6/THF-d8 (0.8 mmol/L) titrated with D-lactate in 10% DMSO–d6/THF-d8 (10 mmol/L, with 0.8 mmol/L 1) from 0.12 to 2.08 equiv. (b) Binding analysis curve of the NMR titration study. The chemical shift of H3 was used for the analysis. (c) The 1H–1H NOESY (400 MHz) spectrum of 1 + D-lactate (2 mmol/L + 1.6 mmol/L) in 10% DMSO–d6/THF-d8 at 298 K and (d) a section of the structure of 1 + D-lactate, showing the spatial correlations between protons of 1 and D-lactate. (e) Molecular model (by DFT B3LYP-D3, 6–31+G(d,p)) of D-lactate⊂1 complex. Redundant side chains are omitted for clarity.

Figure 5 (a) From bottom to top: 1 in 10% DMSO–d6/THF-d8 (1 mmol/L) titrated with L-Asp in 10% DMSO–d6/THF-d8 (10 mmol/L, with 1 mmol/L 1) from 0.13 equiv. to 1.54 equiv. (b) Binding analysis curve of the NMR titration study. The chemical shift of H4a was used for the analysis. (c) Molecular model (by DFT B3LYP-D3, 6–31+G(d,p)) of L-Asp⊂1 complex. Redundant side chains are omitted for clarity.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: