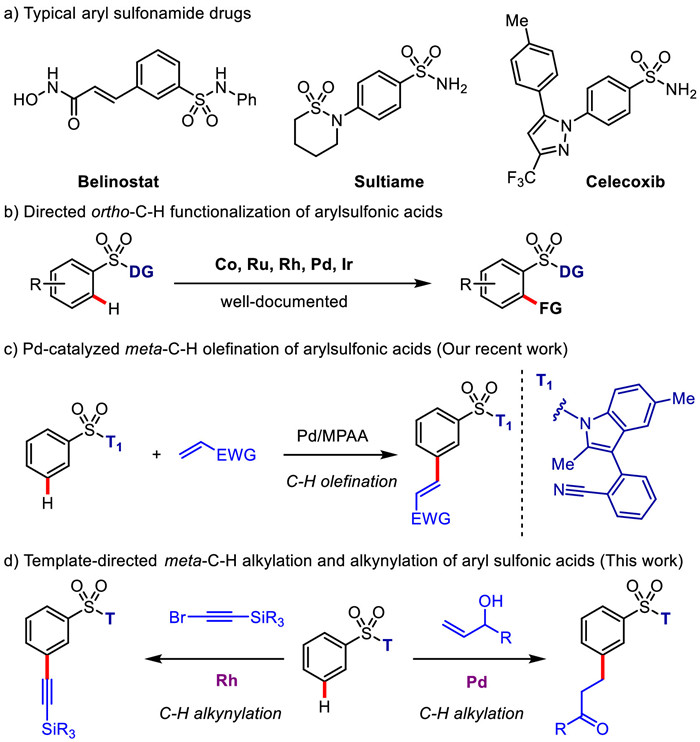
Figure 1.
Transition-metal-catalyzed sp2C—H functionalization of aryl sulfonic acids.
Transition-metal-catalyzed remote meta-C—H alkylation and alkynylation of aryl sulfonic acids enabled by an indolyl template
Pengfei Zhang , Qingxue Ma , Zhiwei Jiang , Xiaohua Xu , Zhong Jin

Transition-metal-catalyzed C—H functionalization has proved a powerful synthetic strategy that converts inert C—H bonds into a diverse range of C—C and C—heteroatom bonds [1]. One of the significant challenges for this field is to achieve the site selectivity of C—H activation, i.e., selective recognition of a target C—H bond where many enthalpically and entropically equivalent C—H bonds coexists in the molecules [2]. Directing group (DG)-assisted transition-metal-catalyzed C—H activation proved a successful vehicle for this process [3-15]. By means of proximity-driven cyclometalation process, activation of ortho-C—H bonds in the benzene rings via a conformationally rigid five- or six-membered metallacycle transition state (TS) was accomplished comprehensively [3,6,10,16-19]. In sharp contrast, transition-metal-catalyzed distal C—H bond functionalization in arenes still remains an enormous challenge [20,21-25]. The pioneering work by Yu provided a breakthrough solution in regard to this question [26]. Through using a nitrile or N-heterocycle, such as pyridine and pyrimidine, embedded in the template that serves as an end-on directing group, distal C—H bonds that locate at the meta or para position to the existing functional group in the arenes are successfully recognized by tuning the spatial and geometric parameters of directing templates [22,27-56]. These directing templates were commonly attached to the substrate by an amide or ester linkage, accommodating the substrate and catalyst to assemble a macrocyclophane (MCP) pre-transition state in the C—H activation step.
Despite these progresses, the remote meta-C—H functionalization of conjugated aryl sulfonic acids substantially suffers from the substrate-imposed challenges to date. The inherently directed ortho-C—H functionalization by the sulfonate group, e.g., the sulfonamide group, is shown to be the mainly competitive reaction with regard to the target meta-C—H activation. The electron-deficient characteristic of the phenyl ring further retards the sp2C—H metalation process significantly. Moreover, the contiguous sulfur(Ⅵ) center makes the whole molecule far more rigid than the related long-chain congeners, not beneficial to the assembly of MCP pre-transition state.
In light of the ubiquity of aryl sulfonic acids and the related sulfonamides as the key structural units in pharmaceuticals, agrochemicals and fine chemicals (Fig. 1a), synthesis and modification of aryl sulfonates by selective C—H functionalization appear particularly attractive. Different from transition-metal-catalyzed ortho-C—H functionalization of aryl sulfonate substrates (Fig. 1b) [57-73], direct meta-C—H bond functionalization of aryl sulfonate derivatives still remains rarely explored owing to the aforementioned limitations. Based on a recent statistical analysis [74] on the relationship between the site-selectivity and MCP-like pre-transition state, our research group, in collaborated with Yu’s group, have successfully developed a palladium-catalyzed inter- and intramolecular meta-selective C—H olefination reactions of aryl sulfonic acids using an indole-derived nitrile directing template (T1) very recently (Fig. 1c) [75].
To the best of our knowledge, the alkyl-substituted arenes as the core units have been widely applied for active pharmaceutical ingredients, fine chemicals, and material science. The alkylation reaction of arenes is an extremely important chemical transformation in organic synthesis. Direct alkylation reaction of aryl sulfonates, such as Friedel−Crafts reaction or radical reaction, often suffers from low reactivity, poor site selectivity, and limited substrate scope [76]. Therefore, we envisioned the incorporation of an alkyl side chain into aryl sulfonic acid via remote meta-selective C—H functionalization by using the aforementioned indolyl directing template. Herein, we disclosed a palladium-catalyzed meta-selective sp2C—H alkylation of aryl sulfonic acid using the allyl alcohols as an alkylation agents (Fig. 1d). In addition, with the assistance of this type of directing template, rhodium-catalyzed meta-C—H alkynylation of aryl sulfonic acids was also demonstrated to be viable. The introduced alkynyl groups were shown to be readily converted into the diverse substituents, for example, the alkyl, alkenyl, acyl groups.
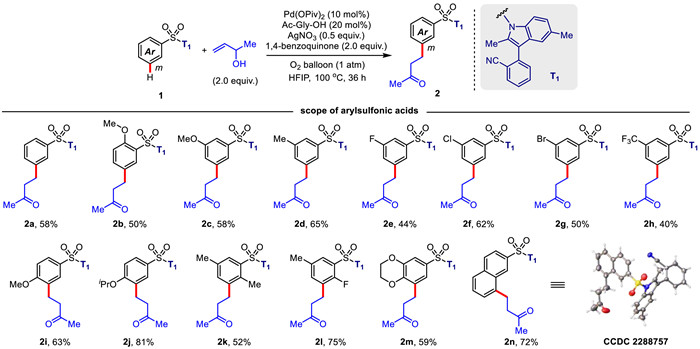
At outset, to achieve meta-C—H alkylation of aryl sulfonic acids using the established directing template, various reaction parameters including catalyst, oxidant, temperature, and time, etc., were thus optimized with but–3-en-2-ol as an alkylated reagent (Table 1). Palladium pivalate and silver nitrate were demonstrated to be the optimal catalyst and oxidant, respectively. AgNO3 was indispensable for this transformation and the catalytic process was almost halted when solely using 1,4-benzoquinone (2 equiv.) in the presence of O2 (1 atm) as the oxidant (Table 1, entry 16). However, excess silver nitrate (2.5 equiv.) caused the production of olefination product 2a′ (Table 1, entry 4), which were probably resulted from coupling with methyl vinyl ketones produced by oxidation of the allyl alcohols. Ultimately, with palladium pivalate as the catalyst, the use of a combination of silver nitrate (0.5 equiv.), 1,4-benzoquinone (2 equiv.) and molecular oxygen (1 atm) as the oxidant gave the highest yield (Table 1, entry 14). Notably, palladium-catalyzed C—H alkylation reaction gave the desired products 2a with exclusive meta selectivity (generally meta:others > 20:1) using the directing template (T1).
 DownLoad:
CSV
DownLoad:
CSV

|
The scope of aryl sulfonic acids in palladium-catalyzed meta-selective C—H alkylation reaction was subsequently surveyed under the optimal reaction conditions (Scheme 1). With but–3-en-2-ol as the coupling partner, electron-donating substituents including the methoxy, methyl, and isopropanoxyl groups in aryl sulfonates were generally conducive to the C—H alkylation reaction, providing the desired meta alkylated products (2c, 2i, 2d and 2j) in remarkably higher yields (58%–81%). Reaction with substrate bearing an o-OMe group delivered the product (2b) in slightly decreased yield (50%), probably due to the unfavored conformation caused by the steric repulsion between the methoxy group and tetrahedrally coordinated sulfonate group. The halo-substituted aryl sulfonate substrates were suitable for the protocol, giving the meta-alkylated products (2e–2g) in moderate yields (44%–62%). In particular, the product (2g) owning a bromo group was tolerated in this reaction, which is synthetically useful for further derivation of other functional groups. The presence of intrinsic sulfonic group makes the phenyl ring of aryl sulfonates highly electron-deficient. Incorporation of the additional electron-withdrawing functional groups into aryl sulfonates further augmented this trend, obviously not conducive to the C—H activation process. However, different from remote meta-C—H functionalization of benzoic acids [51,52], aryl sulfonates bearing a diverse set of electron-withdrawing groups, e.g., CF3 group, could still deliver the alkylated product (2h), albeit in lower yield (40%). Di-substituted aryl sulfonates in different substitution patterns were able to afford the meta-alkylated products (2k–2m) in 52%–75% yields. In addition, reaction with 2-naphthalenesulfonate is also feasible to afford the desired product in 72% yield, and C—H alkylation selectively occurred at the C8 position, which was comprehensively confirmed by X-ray diffraction analysis of product 2n. Notably, with the assistance of template T1, exclusive meta selectivity (generally meta:others > 20:1) were observed with all of the substrates outlined in Scheme 1.
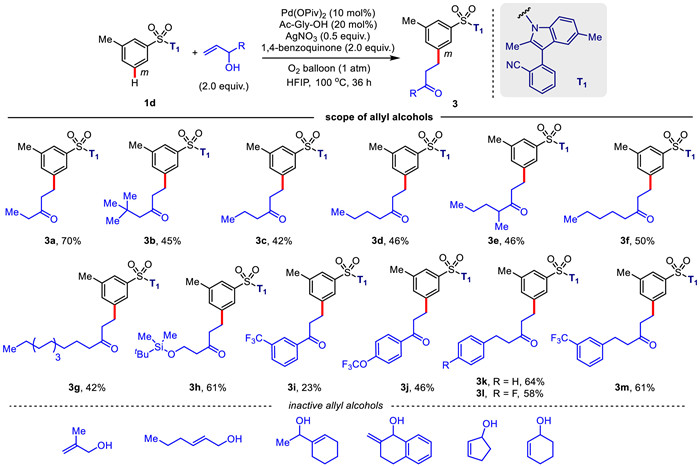
Next, we further investigated the compatibility of template-directed meta-C—H alkylation of aryl sulfonates with the diverse allyl alcohols (Scheme 2). The protocol revealed a widely synthetic utility, tolerating a range of allyl alcohols bearing various alkyl chains. Commonly, the desired products (3a–3g) were isolated in 42%–70% yields. Notably, allyl alcohol containing a terminal TBDMS-protected ether group was also suitable alkylated agent in this protocol, providing the β–hydroxy ketoneproduct (3h) in 61% yield. This allows for synthesis of selectively mono-protected 1,3-diol derivatives from product 3h. Moreover, meta-C—H alkylation with 1-phenylprop-2-en-3-ol is also feasible, successfully affording the aromatic ketones (3i and 3j). The terminal aryl groups in the allyl alcohol chains were well tolerated, successfully delivering the aliphatic ketones (3k–3m) in 58%–64% yields. As observed in Scheme 1, C—H alkylation of m-methyl benzenesulfonic acid-derived substrate 1d with the different allyl alcohols also gave the desired products with exclusive meta selectivity. Unfortunately, this protocol is incompatible with the cyclic allyl alcohols.
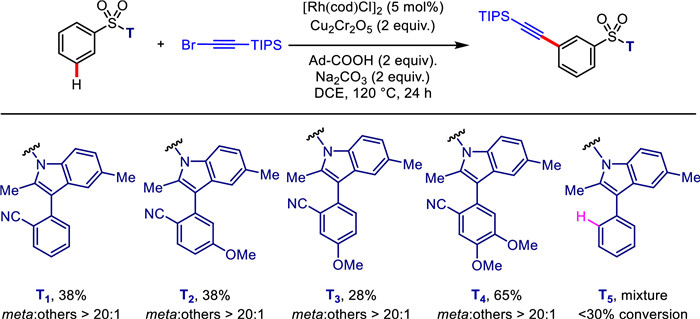
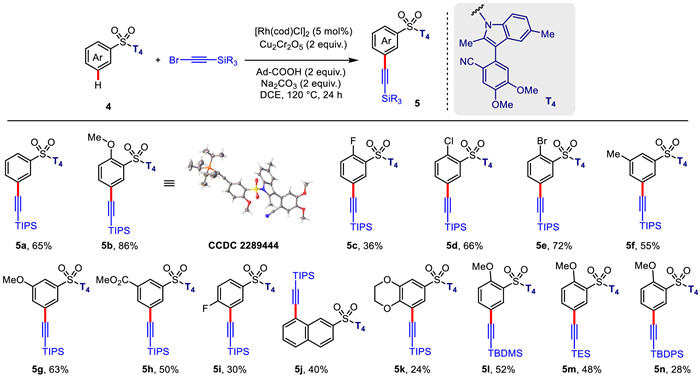
Alkynes are among the most useful motifs in bioactive and material molecules [61]. The alkynyl group is also one of the most valuable synthons in synthetic organic chemistry [21]. Encouraged by our success in template-directed meta-C—H alkylation, we further embarked on transition-metal-catalyzed meta-C—H alkynylation of aryl sulfonates. Through using (bromoethynyl)triisopropylsilane as the alkynylated reagent, reaction conditions of template-directed meta-selective C—H alkynylation of aryl sulfonic acids were extensively evaluated (For reaction condition optimization, see Supporting information for details). Pleasingly, with T1 as the directing template, rhodium(I)-catalyzed C—H alkynylation successfully afforded the desired products in 38% yield under the optimal conditions. Notably, the presence of 1-Ad-CO2H significantly accelerated the C—H activation/deprotonation process as observed previously by L. Ackermann [3,77]. Moreover, the electronic density of benzene ring significantly influences the coordination potential between the nitrile group and rhodium catalyst. As a consequence, with T4 as a directing template, the yield of alkynylation reaction drastically increased to 65% (Scheme 3).
With the optimal template (T4) in hand, we subsequently investigated the generality of rhodium-catalyzed meta-selective C—H alkynylation. It was found that substrates owning the methoxy and halo functional groups at ortho position were well tolerated under the present C—H alkynylation reaction conditions, providing the desired meta alkynylated products (5b–5e) in modest to good yields. The site-selectivity of C—H alkynylation was comprehensively confirmed by X-ray diffraction analysis of compound 5b. Aryl sulfonamides bearing a diverse set of electron-withdrawing or electron-donating substituents, such as methoxy, methyl, and ester group at meta position, are all compatible to provide the target products (5f–5h) in 50%–63% yields. Additionally, C—H alkynylation reaction with 2-naphthalenesulfonate-derived substrate is also feasible, occurring at C8-position (5j) selectively. Except for (bromoethynyl)triisopropylsilane, (bromoethynyl)(tert–butyl)dimethylsilane, (bromoethynyl)triethylsilane, and (bromoethynyl)(tert–butyl)diphenylsilane were tolerated under the present reaction conditions, producing the corresponding alkynylated products (5l–5n) in 28%–52% yields (Scheme 4). As validated by the aforementioned X-ray diffraction analysis, rhodium-catalyzed C—H alkynylation of benzenesulfonic acids also afforded the corresponding products 5a–5n in exclusive meta-selectivity (generally meta:others > 20:1).
As the vital drug moiety, 1-indanones were widely served as a type of intermediates to synthesize anti-inflammatory, anti-malarial, anti-tumor, anti-viral and other bioactive drugs, such as Indinavir and Paucifloral F(Ⅲ). Access to the substituted 1-indanone framework via intramolecular remote meta-C—H functionalization has not been achieved previously. To our delight, Pd-catalyzed intramolecular meta-C—H alkylation successfully afforded 5-sulfonate group-substituted 1-indanone 7 in 52% isolated yield with the present directing template T1 (Fig. 2a). Additionally, the introduced meta alkynyl groups in aryl sulfonates were conveniently converted into the various functional groups, further demonstrating the synthetic utility of the present remote meta-C—H functionalization strategy (Fig. 2b).
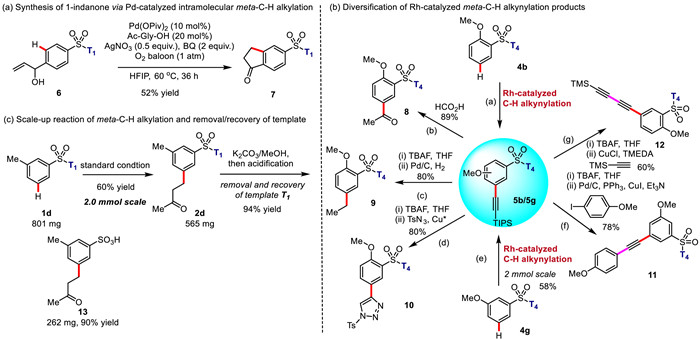
Finally, we successfully scaled up the meta-C—H reaction to 2.0 mmol scale (Fig. 2c). Palladium-catalyzed meta-C—H alkylation of aryl sulfonate 1d provided the target mono-product 2d in 60% isolated yield. The template T1 was easily removed under mild conditions (K2CO3, MeOH) and recovered in 94% yield, together with the desired m-alkylated arylsulfonic acid 13 in 90% yield.
In summary, we have successfully achieved transition-metal-catalyzed meta-selective C—H alkylation and alkynylation reactions of aryl sulfonic acids by means of a practical indolyl directing template developed by us very recently. Synthetic applications of the strategy were demonstrated in the synthesis of significant intermediate 1-indanone via a palladium-catalyzed intramolecular meta-C—H alkylation process. Further investigation on this template-assisted meta-C—H functionalization is underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from the National Natural Science Foundation of China (No. 22171145 to Z. Jin and 32072440 to X. Xu) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
H.J. Xu, Y. Lu, M.E. Farmer, et al., J. Am. Chem. Soc. 139 (2017) 2200–2203. doi: 10.1021/jacs.6b13269
Z. Huang, G. Dong, Acc. Chem. Res. 50 (2017) 465–471. doi: 10.1021/acs.accounts.6b00476
L. Ackermann, Acc. Chem. Res. 47 (2014) 281–295. doi: 10.1021/ar3002798
P.B. Arockiam, C. Bruneau, P.H. Dixneuf, Chem. Rev. 112 (2012) 5879–5918. doi: 10.1021/cr300153j
X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Angew. Chem. Int. Ed. 48 (2009) 5094–5115. doi: 10.1002/anie.200806273
O. Daugulis, H.Q. Do, D. Shabashov, Acc. Chem. Res. 42 (2009) 1074–1086. doi: 10.1021/ar9000058
K.M. Engle, J.Q. Yu, J. Org. Chem. 78 (2013) 8927–8955. doi: 10.1021/jo400159y
R. Giri, S. Thapa, A. Kafle, Adv. Synth. Catal. 356 (2014) 1395–1411. doi: 10.1002/adsc.201400105
Z. Huang, H.N. Lim, F. Mo, M.C. Young, G. Dong, Chem. Soc. Rev. 44 (2015) 7764–7786. doi: 10.1039/C5CS00272A
T.W. Lyons, M.S. Sanford, Chem. Rev. 110 (2010) 1147–1169. doi: 10.1021/cr900184e
S.R. Neufeldt, M.S. Sanford, Acc. Chem. Res. 45 (2012) 936–946. doi: 10.1021/ar300014f
M.P. Drapeau, L.J. Gooßen, Chem. Eur. J. 22 (2016) 18654–18677. doi: 10.1002/chem.201603263
A. Ros, R. Fernandez, J.M. Lassaletta, Chem. Soc. Rev. 43 (2014) 3229–3243. doi: 10.1039/C3CS60418G
T. Satoh, M. Miura, Synthesis 20 (2010) 3395–3409. doi: 10.1055/s-0030-1258225
G. Shi, Y. Zhang, Adv. Synth. Catal. 356 (2014) 1419–1442. doi: 10.1002/adsc.201400028
D.A. Colby, R.G. Bergman, J.A. Ellman, Chem. Rev. 110 (2010) 624–655. doi: 10.1021/cr900005n
D.A. Colby, A.S. Tsai, R.G. Bergman, J.A. Ellman, Acc. Chem. Res. 45 (2012) 814–825. doi: 10.1021/ar200190g
G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726–11743. doi: 10.1002/anie.201301451
K.M. Engle, T.S. Meit, M. Wasa, J.Q. Yu, Acc. Chem. Res. 45 (2012) 788–802. doi: 10.1021/ar200185g
H. Shi, P. Wang, S. Suzuki, M.E. Farmer, J.Q. Yu, J. Am. Chem. Soc. 138 (2016) 14876–14879. doi: 10.1021/jacs.6b11055
H.J. Xu, Y.S. Kang, H. Shi, et al., J. Am. Chem. Soc. 141 (2019) 76–79. doi: 10.1021/jacs.8b11038
S. Lee, H. Lee, K.L. Tan, J. Am. Chem. Soc. 135 (2013) 18778–18781. doi: 10.1021/ja4107034
B.E. Haines, Y. Saito, Y. Segawa, K. Itami, D.G. Musaev, ACS Catal. 6 (2016) 7536–7546. doi: 10.1021/acscatal.6b02317
M.E. Hoque, R. Bisht, C. Haldar, B. Chattopadhyay, J. Am. Chem. Soc. 139 (2017) 7745–7748. doi: 10.1021/jacs.7b04490
J.A. Leitch, C.L. McMullin, A.J. Paterson, et al., Angew. Chem. Int. Ed. 56 (2017) 15131–15135. doi: 10.1002/anie.201708961
D. Leow, G. Li, T.S. Mei, J.Q. Yu, Nature 486 (2012) 518–522. doi: 10.1038/nature11158
H.X. Dai, G. Li, X.G. Zhang, A.F. Stepan, J.Q. Yu, J. Am. Chem. Soc. 135 (2013) 7567–7571. doi: 10.1021/ja400659s
L. Wan, N. Dastbaravardey, G. Li, J.Q. Yu, J. Am. Chem. Soc. 135 (2013) 18056–18059. doi: 10.1021/ja410760f
R.Y. Tang, G. Li, J.Q. Yu, Nature 507 (2014) 215–220. doi: 10.1038/nature12963
G. Yang, P. Lindovska, D. Zhu, et al., J. Am. Chem. Soc. 136 (2014) 10807–10813. doi: 10.1021/ja505737x
L. Chu, M. Shang, K. Tanaka, et al., ACS Cent. Sci. 1 (2015) 394–399. doi: 10.1021/acscentsci.5b00312
Y. Deng, J.Q. Yu, Angew. Chem. Int. Ed. 54 (2015) 888–891. doi: 10.1002/anie.201409860
M. Bera, A. Maji, S.K. Sahoo, D. Maiti, Angew. Chem. Int. Ed. 54 (2015) 8515–8519. doi: 10.1002/anie.201503112
S. Bag, R. Jayarajan, R. Mondal, D. Maiti, Angew. Chem. Int. Ed. 56 (2017) 3182–3186. doi: 10.1002/anie.201611360
S. Bag, R. Jayarajan, U. Dutta, et al., Angew. Chem. Int. Ed. 56 (2017) 12538–12542. doi: 10.1002/anie.201706360
L. Zhang, C. Zhao, Y. Liu, et al., Angew. Chem. Int. Ed. 56 (2017) 12245–12249. doi: 10.1002/anie.201705495
Z. Zhang, K. Tanaka, J.Q. Yu, Nature 543 (2017) 538–542. doi: 10.1038/nature21418
T.K. Achar, X. Zhang, R. Mondal, et al., Angew. Chem. Int. Ed. 58 (2019) 10353–10360. doi: 10.1002/anie.201904608
S. Li, H. Wang, Y. Weng, G. Li, Angew. Chem. Int. Ed. 58 (2019) 18502–18507. doi: 10.1002/anie.201910691
S. Porey, X. Zhang, S. Bhowmick, et al., J. Am. Chem. Soc. 142 (2020) 3762–3774. doi: 10.1021/jacs.9b10646
A.F. Williams, A.J.P. White, A.C. Spivey, C.J. Cordier, Chem. Sci. 11 (2020) 3301–3306. doi: 10.1039/d0sc00230e
S. Bag, S. K, A. Mondal, J. Am. Chem. Soc. 142 (2020) 12453–12466. doi: 10.1021/jacs.0c05223
S. Bag, S. Jana, S. Pradhan, et al., Nat. Commun. 12 (2021) 1393. doi: 10.1038/s41467-021-21633-2
S. Bag, T. Patra, A. Modak, et al., J. Am. Chem. Soc. 137 (2015) 11888–11891. doi: 10.1021/jacs.5b06793
T. Patra, S. Bag, R. Kancherla, et al., Angew. Chem. Int. Ed. 55 (2016) 7751–7755. doi: 10.1002/anie.201601999
A. Maji, S. Guin, S. Feng, et al., Angew. Chem. Int. Ed. 56 (2017) 14903–14907. doi: 10.1002/anie.201708449
A. Maji, A. Dahiya, G. Lu, et al., Nat. Commun. 9 (2018) 3582. doi: 10.1038/s41467-018-06018-2
X. Chen, S. Fan, M. Zhang, et al., Chem. Sci. 12 (2021) 4126–4131. doi: 10.1039/d0sc07042d
G. Meng, N.Y.S. Lam, E.L. Lucas, et al., J. Am. Chem. Soc. 142 (2020) 10571–10591. doi: 10.1021/jacs.0c04074
Y.F. Yang, G.J. Cheng, P. Liu, et al., J. Am. Chem. Soc. 136 (2014) 344–355. doi: 10.1021/ja410485g
S. Li, L. Cai, H. Ji, L. Yang, G. Li, Nat. Commun. 7 (2016) 10443. doi: 10.1038/ncomms10443
L. Fang, T.G. Saint-Denis, B.L.H. Taylor, et al., J. Am. Chem. Soc. 139 (2017) 10702–10714. doi: 10.1021/jacs.7b03296
L. Cai, S. Li, C. Zhou, G. Li, Org. Lett. 22 (2020) 7791–7796. doi: 10.1021/acs.orglett.0c02528
S. Li, C. Zhang, L. Fu, et al., CCS Chem. 4 (2022) 1889–1900. doi: 10.31635/ccschem.021.202101156
M. Liu, L.J. Li, J. Zhang, H. Xu, H.X. Dai, Chin. Chem. Lett. 31 (2020) 1301–1304. doi: 10.1016/j.cclet.2019.09.057
W. Chang, Y. Wang, Y. Chen, J. Ma, Y. Liang, Chin. Chem. Lett. 34 (2023) 10879.
M.V. Pham, B. Ye, N. Cramer, Angew. Chem. Int. Ed. 51 (2012) 10610–10614. doi: 10.1002/anie.201206191
Y. Ran, Y. Yang, H. You, J. You, ACS Catal. 8 (2018) 1796–1801. doi: 10.1021/acscatal.7b04298
Y. Dong, X. Zhang, J. Chen, et al., Chem. Sci. 10 (2019) 8744–8751. doi: 10.1039/c9sc03691a
H.X. Dai, A.F. Stepan, M.S. Plummer, Y.H. Zhang, J.Q. Yu, J. Am. Chem. Soc. 133 (2011) 7222–7228. doi: 10.1021/ja201708f
G. Wu, W. Ouyang, Q. Chen, Y. Huo, X. Li, Org. Chem. Front. 6 (2019) 284–289. doi: 10.1039/c8qo01105b
T. Lan, L. Wang, Y. Rao, Org. Lett. 19 (2017) 972–975. doi: 10.1021/acs.orglett.6b03510
S. Rej, N. Chatani, Chem. Sci. 11 (2020) 389–395. doi: 10.1039/c9sc04308j
W. Liu, D. Wang, Y. Zhao, F. Yi, J. Chen, Adv. Synth. Catal. 358 (2016) 1968–1974. doi: 10.1002/adsc.201501104
S. Feng, S. Li, J. Li, J. Wei, Org. Chem. Front. 6 (2019) 517–522. doi: 10.1039/c8qo01311j
S. Ojha, N. Panda, Adv. Synth. Catal. 362 (2020) 561–571. doi: 10.1002/adsc.201900989
K. Parthasarathy, C. Bolm, Chem. Eur. J. 20 (2014) 4896–4900. doi: 10.1002/chem.201304925
W.J. Kerr, M. Reid, T. Tuttle, ACS Catal. 5 (2015) 402–410. doi: 10.1021/cs5015755
R. Vanjari, T. Guntreddi, K.N. Singh, Chem. Asian J. 11 (2016) 696–699. doi: 10.1002/asia.201501385
F. Li, T.X. Liu, G.W. Wang, Org. Lett. 14 (2012) 2176–2179. doi: 10.1021/ol3007452
Y. Dong, G. Liu, Chem. Commun. 49 (2013) 8066–8068. doi: 10.1039/c3cc44690e
Z. Qi, M. Wang, X. Li, Chem. Commun. 50 (2014) 9776–9778. doi: 10.1039/C4CC03627A
W. Ma, R. Mei, G. Tenti, L. Ackermann, Chem. Eur. J. 20 (2014) 15248–15251. doi: 10.1002/chem.201404604
N.Y.S. Lam, Z. Fan, K. Wu, et al., J. Am. Chem. Soc. 144 (2022) 2793–2803. doi: 10.1021/jacs.1c12654
P. Zhang, Z. Jiang, Z. Fan, et al., Chem. Sci. 14 (2023) 8279–8287. doi: 10.1039/d3sc01670f
S.B. Ankade, A.B. Shabade, V. Soni, B. Punji, ACS Catal. 11 (2021) 3268–3292. doi: 10.1021/acscatal.0c05580
L. Ackermann, Chem. Rev. 111 (2011) 1315–1345. doi: 10.1021/cr100412j

Scheme 1 Scope of aryl sulfonic acids for palladium-catalyzed meta-C—H alkylation. Reaction conditions: ArSO2-T1 (0.1 mmol), but–3-en-2-ol (0.2 mmol), Pd(OPiv)2 (10 mol%), Ac-Gly-OH (20 mol%), AgNO3 (0.05 mmol), 1,4-benzoquinone (0.2 mmol), O2 balloon (1 atm), HFIP (1.5 mL), 100 ℃, 36 h. Isolated yields were reported.

Scheme 2 Scope of allyl alcohols for palladium-catalyzed meta-C—H alkylation. Reaction conditions: ArSO2-T1 1d (0.1 mmol), allyl alcohol (0.2 mmol), Pd(OPiv)2 (10 mol%), Ac-Gly-OH (20 mol%), AgNO3 (0.05 mmol), 1,4-benzoquinone (0.2 mmol), O2 balloon (1 atm), HFIP (1.5 mL), 100 ℃, 36 h. Isolated yields were reported.

Scheme 3 Screening of directing template for rhodium-catalyzed meta-C—H alkynylation. Reaction conditions: ArSO2-T (0.1 mmol), (bromoethynyl)triisopropylsilane (0.6 mmol), [Rh(cod)Cl]2 (5 mol%), Cu2Cr2O5 (0.2 mmol), 1-Ad-COOH (0.2 mmol), Na2CO3 (0.2 mmol), DCE (1.5 mL), 120 ℃, 24 h. Isolated yields were reported.

Scheme 4 Substrate scope for rhodium-catalyzed meta-C—H alkynylation. Reaction conditions: ArSO2-T4 (0.1 mmol), bromoacetylene (0.6 mmol), [Rh(cod)Cl]2 (5 mol%), Cu2Cr2O5 (0.2 mmol), 1-Ad-COOH (0.2 mmol), Na2CO3 (0.2 mmol), DCE (1.5 mL), 120 ℃, 24 h. Isolated yields were reported.

Figure 2 Synthetic application of transition-metal-catalyzed meta-C—H functionalization of aryl sulfonic acids.
Table 1. Screening of reaction conditions for Pd-catalyzed meta-C—H alkylation of aryl sulfonic acids.a
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: