Table 1.
Main applications of PHI/PTI photocatalysts.
Citation:
Haojie Song, Laiyu Luo, Siyu Wang, Guo Zhang, Baojiang Jiang. Advances in poly(heptazine imide)/poly(triazine imide) photocatalyst[J]. Chinese Chemical Letters,
2024, 35(10): 109347.
doi:
10.1016/j.cclet.2023.109347

Advances in poly(heptazine imide)/poly(triazine imide) photocatalyst
English
Advances in poly(heptazine imide)/poly(triazine imide) photocatalyst
Abstract:
Polymeric carbon nitride (PCN) has garnered increasing attention as a metal-free photocatalyst with a suitable band gap. In efforts to enhance its photocatalytic performance, researchers have examined various PCN materials, including poly(heptazine imide) (PHI) and poly(triazine imide) (PTI), two isomers within the PCN family that exhibit distinct and superior photocatalytic activity compared to other forms. The challenge, however, lies in the common practice among researchers to categorize PHI and PTI along with other PCN types under the overarching term "g-C3N4, " which significantly impedes optimization efforts. The objective of this review is to provide comprehensive insights into the structural features, photoelectrochemical properties, and effective characterization methods employed for distinguishing between PHI and PTI materials. The review also summarizes various optimization strategies, such as crystallinity adjustments, defect engineering, morphology control, constructing heterojunction, and atomic-level metal loading dispersion, to elevate the photocatalytic activity of PHI and PTI, in addition to summarizing the history of carbon nitride development. Furthermore, this review highlights the primary applications of PHI and PTI, encompassing nitrogen fixation, biomass conversion, organic synthesis, CO2 reduction, pollutant degradation, H2O2 production, and photocatalytic water splitting. Lastly, the prospects and challenges associated with further advancing PHI and PTI are thoroughly examined.
-
Key words:
- Poly(heptazine imide)
- / Poly(triazine imide)
- / Modification
- / Photocatalytic
- / Application
-
1. Introduction
The worldwide energy crisis and environmental pollution pose grave threats to the sustainable development of human civilization [1]. To address these issues, the development of renewable energy sources has become an indispensable strategy. Solar energy, as a clean, green, and sustainable power source, delivers an estimated 120,000 TW of energy annually to the Earth [2,3]. To better harness solar energy, researchers have created various pathways for its conversion into other available resources such as photovoltaics [4,5], photothermal [6,7], and photochemistry [8,9]. Among these options, photocatalysis utilizing solar energy boasts immense benefits, and the choice of appropriate semiconductors as photocatalysts can induce numerous catalytic reactions such as hydrogen production from water splitting [10,11], CO2 reduction into hydrocarbon fuels [12,13], organic pollutant degradation [14,15], selective organic synthesis [16,17], and bacterial disinfection [18,19].
In 1972, Fujishima and Honda discovered that TiO2 could facilitate water splitting to produce H2 and O2 under UV light irradiation, consequently opening the door to photocatalysis in the energy field [20]. The development of highly efficient semiconductor photocatalysts has become a subject of significant interest in contemporary times. Several inorganic metal semiconductors, including TiO2 [21,22], Fe2O3 [23], SnO2 [24,25], ZrO2 [26,27], Ag3PO4 [28,29], Ag3VO4 [30,31], Bi2WO6 [32,33], BiVO4 [34,35], NiS [36,37], CdS [38,39] etc., have been reported for photocatalytic reactions. The practical application of metal-based photocatalysts is limited due to the rarity of nature and the leaching of toxic metals from photocorrosion [40,41]. On the other hand, organic semiconductors possess advantages like structural tunability and environmental friendliness compared to their inorganic counterparts, but conventional organic polymers display severe swelling and stability issues [42]. Therefore, there is a pressing need for a highly active and structurally durable organic photocatalyst.
Carbon nitride has recently emerged as a highly appealing organic polymer photocatalytic semiconductor, owing to its numerous outstanding characteristics [43,44]. The extraordinary chemical stability of g-C3N4, which does not decompose even in air at temperatures of up to 600 ℃, can be explained by the sturdy aromatic C—N heterocyclic ring [45,46]. It is also stable in most solvents such as water, alcohols, N, N-dimethylformamide, tetrahydrofuran, ether and toluene, as well as glacial acetic acid and aqueous NaOH solutions [47]. Additionally, the sp2 hybridization of nitrogen and carbon gives rise to a π-conjugated energy band structure with a low band gap (2.7–2.8 eV) and an excellent visible light response in the range up to 460 nm [48,49]. Furthermore, the distinctive properties of organic conjugated polymers allow carbon nitride to facilitate the construction of a variety of highly customizable composite photocatalysts featuring tunable composition, size, thickness, pore structure, size distribution and morphology [50–54]. Moreover, polymeric carbon nitride can be obtained by direct thermolysis of nitrogen-rich precursors like melamine [55,56], dicyandiamide [57,58], cyanamide [59,60], urea [61,62], thiourea [63,64] and ammonium thiocyanate [65,66]. These unique characteristics make it a very promising functional material for sustainable energy conversion and storage, as well as environmental remediation.
However, the application of carbon nitride in photocatalytic redox reactions is impeded by its lack of absorption of visible and near-infrared light and resultant photogenerated electron-hole pair complexation [67,68]. Researchers have employed various tactics to improve the practical applications of carbon nitride. These approaches include (1) selecting nitrogen-rich precursors and optimizing the reaction conditions to modulate the physicochemical properties of CN [69,70]; (2) deploying various synthetic techniques, such as soft-template [71,72], hard-template [73,74], supramolecular pre-organization [75,76], and exfoliation methods, to obtain CN with varying morphologies [77,78]; (3) modifying the electronic structure and energy gap of CN through copolymerization and doping strategies [79,80]; (4) establishment of different heterojunctions by coupling with other semiconductors and co-catalysts for extending the spectrum absorption range and promoting electron-hole separation [81,82] and (5) controlling structural defects [83,84]. Poly(heptazine imide) (PHI) and poly(triazine imide) (PTI) are two isomers in the carbon nitride family obtained by the molten salt method [70,85–87]. In contrast to the general characterization of g-C3N4 in the literature, PHI and PTI, as carbon nitride variants, stand out for their precisely-defined structures that set them apart from other graphite-phase carbon nitride species [88,89]. This modified carbon nitride exhibits enhanced photogenerated charge separation efficiency and a considerably expanded solar spectrum absorption range as a catalyst, demonstrating great promise in a variety of photocatalytic applications [90].
The distinctive properties of PHI and PTI, which differentiate them from graphitic-phase carbon nitride, have been the subject of extensive recent research and publication. However, it has been observed that certain studies have failed to acknowledge this differentiation, instead categorizing these materials under the general label of graphitic-phase or crystalline carbon nitride, hindering progress in investigating the unique structures and properties of PHI and PTI [91,92]. With consideration given to their advantageous features, this paper aims to enhance understanding of PHI and PTI through analysis of structural, characterization, optical, optimization aspects as well as their applications (Table 1).
Table 1
2. Development process of carbon nitride family
The historical development of carbon nitride (Fig. 1) and its precursors can be traced back to 1834 when Berzelius first synthesized a linear CN polymer called "melon, " which Liebig later named [93–95]. Melon was obtained as a yellow, amorphous, and insoluble product through the pyrolysis of ammonium chloride with potassium thiocyanate. Subsequently, in 1835, Gmelin discovered hydromelonate (K3C6N7NCN) by heating potassium ferricyanide and sulfur. However, due to the limited analytical tools available at the time, the precise structures of melon and K3C6N7NCN remained unknown [96]. In 1922, Franklin advanced our understanding of these compounds by investigating their structures in more depth. He proposed the concept of "carbonitrides" (C3N4) and suggested that C3N4 could be obtained by heat treatment of melon as the final deamination product of the ammoniacal series. Franklin also provided possible structural models for C3N4 [97]. Subsequently, in 1937, Pauling and Sturdivant conducted X-ray crystallographic studies, revealing a coplanar tri-s-triazine unit as the fundamental structural motif for these polymer derivatives [98].
Figure 1
 Figure 1. Historical development and milestones of carbon nitride. Reproduced and modified with permission [98]. Copyright 1937, National Academy of Sciences. Reproduced and modified with permission [102]. Copyright 1994, American Physical Society. Reproduced and modified with permission [104,87,109]. Copyright 2003, 2015 and 2019, the American Chemical Society. Reproduced and modified with permission [107]. Copyright 2009, the Royal Society of Chemistry. Reproduced and modified with permission [85,89,91,110]. Copyright 2008, 2011, 2021 and 2023, Wiley-VCH.
Figure 1. Historical development and milestones of carbon nitride. Reproduced and modified with permission [98]. Copyright 1937, National Academy of Sciences. Reproduced and modified with permission [102]. Copyright 1994, American Physical Society. Reproduced and modified with permission [104,87,109]. Copyright 2003, 2015 and 2019, the American Chemical Society. Reproduced and modified with permission [107]. Copyright 2009, the Royal Society of Chemistry. Reproduced and modified with permission [85,89,91,110]. Copyright 2008, 2011, 2021 and 2023, Wiley-VCH.In 1940, Redemann and Lucas observed formal similarities between graphite and melon, leading them to deduce that the 2,5,8-triamino-tris-s-triazine (C126H21N175) best matched Franklin's description of C3N4 [99]. These findings suggested that C3N4 may not possess a single crystalline structure but rather comprise a mixture of polymers with varying sizes and structures. After a prolonged period of relative disinterest, carbon nitride regained attention in the 1990s, predominantly focused on the pursuit of superhard material β-C3N4 [100,101]. In their theoretical computational work, Liu and Cohen presented the prediction of five stochastic phases of carbon nitride, one of which is the well-known β-C3N4 phase. They further predicted that β-C3N4 possesses a Young's modulus surpassing that of diamond. Additionally, their study identified a graphitic phase characterized by a two-dimensional binding arrangement and three-dimensional stacking resembling graphite. Subsequent theoretical investigations indicated g-C3N4 as the most thermodynamically stable phase under ambient conditions. The graphitic phase of carbon nitride was described as having a triazinyl structure during this specific period [102].
It was not until 2002 that Kroke et al. developed a synthetic route for trichloro-tri-s-triazine, successfully determining its crystal structure. Computational modeling studies showed the thermodynamic superiority of the heptazine unit over the triazolium unit [103]. In the subsequent year, Schnick's group independently reported the synthesis and the structure of melem, establishing it as an intermediate in the formation of g-C3N4 [104]. The potential application of g-C3N4 as an organic semiconductor in materials science and catalysis had been largely overlooked until 2006 when Goettmann et al. discovered its remarkable performance as a metal-free catalyst. By employing mesoporous g-C3N4, they achieved the activation of benzene and introduced carboxylic acids, alcohols, quaternary ammonium, and urea as electrophilic reagents for Friedel-Crafts reactions [105,106].
In 2009, Wang et al. made a groundbreaking discovery by identifying g-C3N4 as a metal-free conjugated semiconductor photocatalyst for hydrogen production, thus revolutionizing the research field [43]. This discovery propelled the exploration of polymer conjugated semiconductor photocatalysts, thereby shifting the research emphasis from inorganic conjugated semiconductors. Concurrently, PHI and PTI materials were successfully synthesized, with Tomas et al. achieving the preparation of highly crystalline carbon nitride in 2008 using a eutectic mixture of LiCl/KCl as a high-temperature solvent. Subsequent studies by Wirnhier et al. confirmed this material as PTI [85,89]. In 2009, Doblinger et al. successfully synthesized PHI through a combination of high temperature (630 ℃) and autogenous ammonia pressure to facilitate melon condensation [107]. In 2013, Ham et al. employed PTI/Li+Cl− co-catalysts, such as Pt or CoOx, for the photocatalytic generation of H2 or O2. They demonstrated that the precise positioning of the valence and conduction bands of PTI/Li+Cl− enabled efficient overall water splitting [108]. These findings significantly advanced the understanding and application of carbon nitride-based photocatalysis. Subsequently, in 2015, Dariya Dontsova et al. introduced the widely utilized molten salt method for obtaining PHI by treating triazole derivatives in LiCl/KCl molten salts [87]. In 2017, Chen et al. achieved the direct synthesis of photocatalytically active sodium, potassium, and cesium salts of PHI through a solid-phase reaction between melamine and the corresponding metal chlorides carried out at temperatures ranging from 550 ℃ to 600 ℃ [86]. In 2019, Schlomberg et al. resolved the structure of PHI in transmission electron microscopy and solid-state NMR spectroscopy supported by quantum chemical calculations, showing a planar imine-bridged heptazine backbone with triangular symmetry in both K-PHI and H-PHI [109]. In 2022–2023, Liu et al. prepared crystalline PTI photocatalyzed total water splitting reaction with an apparent quantum efficiency (AQE) reached 20.2% [90,110]. Nowadays, PHI/PTI has been extensively studied for photocatalysis and has been shown to have extraordinary photocatalytic activity.
3. Crystal phase structure and electronic structure of PHI/PTI
Carbon nitride, specifically polymeric carbon nitride (PCN), is typically prepared by direct thermal polymerization of nitrogen-rich precursors at temperatures ranging from 450 ℃ to 600 ℃ [111]. PCN primarily consists of one-dimensional melon (tri-triazine) units that are connected via secondary amines to form one-dimensional polymer chains. Interactions between different chains occur through hydrogen bonding, resulting in the formation of two-dimensional planes [91,92,112,113]. However, due to incomplete polymerization of the precursors, PCN exhibit moderate performance in photocatalytic reactions and often exist as amorphous or semi-crystalline phases [70,114].
In contrast, the synthesis of PHI and PTI involves the use of a molten salt as a high-temperature solvent, where the molten salt ions act as structural guides, promoting the formation of more ordered structures [115–120]. PHI exhibits a two-dimensional extended network with melon serving as the basic unit, wherein heptazine units are interconnected by -NH- bridges. The six connected heptazine units stack together, creating a large triangular gap capable of accommodating one melamine molecule (Fig. 2a) [109]. Similar to PHI, PTI also adopts a two-dimensional planar structure. It is somewhat similar to the recent star material, nitrogen-rich π-conjugated covalent triazine framework, both of which contain triazine structural units [121,122]. However, the basic unit in PTI is a planar network of triazines interconnected by -NH- functionalities, with ions filling the spaces between the layers. The triazine rings in PTI adopt an AB stacking pattern, generating uniform pore channels embedded with lithium halide ions. Consequently, these compounds are typically denoted as PTI/Li+X− (Fig. 2b) [89].
Figure 2
 Figure 2. Structural modeling of (a) PHI and (b) PTI. Reproduced and modified with permission [115]. Copyright 2020, Wiley-VCH.
Figure 2. Structural modeling of (a) PHI and (b) PTI. Reproduced and modified with permission [115]. Copyright 2020, Wiley-VCH.Carbon nitride exhibits an aromatic ring-like structure, characterized by sp2 hybridization of carbon and nitrogen atoms. The presence of a highly delocalized conjugated π system imparts notable photocatalytic properties to carbon nitride. Specifically, the lowest electron-unoccupied orbital (LUMO), corresponding to the semiconductor conduction band (CB), is constituted by C pZ and low coordination N pZ orbitals, while the highest electron-occupied orbital (HOMO), corresponding to the semiconductor valence band (VB), is predominantly composed of N pZ orbitals (Fig. 3a) [123].
Figure 3
 Figure 3. (a) Schematic diagram of catalyst light absorption and fluorescence emission. Optical properties of g-CN and K-PHI: (b) DRUV–vis spectra. (c) Photoluminescence spectroscopy. (d) VB and CB. Reproduced with permission [115]. Copyright 2018, Wiley-VCH. Optical properties of g-CN and PTI: (e) DRUV-vis spectra. (f) Photoluminescence spectroscopy. Reproduced with permission [130]. Copyright 2015, Wiley-VCH. (g) VB and CB. Reproduced with permission [124]. Copyright 2017, the Royal Society of Chemistry.
Figure 3. (a) Schematic diagram of catalyst light absorption and fluorescence emission. Optical properties of g-CN and K-PHI: (b) DRUV–vis spectra. (c) Photoluminescence spectroscopy. (d) VB and CB. Reproduced with permission [115]. Copyright 2018, Wiley-VCH. Optical properties of g-CN and PTI: (e) DRUV-vis spectra. (f) Photoluminescence spectroscopy. Reproduced with permission [130]. Copyright 2015, Wiley-VCH. (g) VB and CB. Reproduced with permission [124]. Copyright 2017, the Royal Society of Chemistry.Compared to PCN, PHI exhibits a more positive valence band potential, rendering photogenerated holes more oxidize the reactants (Fig. 3d) [115]. On the other hand, PTI possesses a band gap of approximately 2.8–3.2 eV, enabling it to effectively bridge the redox potential of water and manifest thermodynamic competency for photocatalytic overall water splitting (Fig. 3g) [124]. Generally, PHI, PTI, and PCN demonstrate light absorption bands at λ < 420–440 nm, arising from π→π* electron transitions within the conjugated aromatic rings (Figs. 3b and e) [125]. Notably, PHI exhibits an additional absorption band around λ = 520 nm, attributed to the n→π* electron transition involving nitrogen atoms at the heptazine ring's periphery (Fig. 3b) [115,126]. This phenomenon emerges due to the significant asymmetry in heptazine rings induced by the insertion of metal ions, thereby allowing the normally n →π* forbidden electron transition to become permissible [127]. Moreover, PHI and PTI form two-dimensional elongated π-conjugated systems, augmenting the rigidity of the conjugated polymer backbone and leading to significantly enhanced light absorption intensity compared to PCN. Additionally, the higher crystallinity and lower defectivity of PHI and PTI contribute to superior photogenerated carrier transfer capabilities and diminished rates of electron-hole (Figs. 3c and f) [128–130].
It is worth noting that redox reactions are typically driven by the presence of photogenerated electrons and holes, which often occur simultaneously. However, the behavior of photogenerated carriers in PHI (particularly K-PHI) exhibits distinct characteristics. In the presence of electron donors such as benzyl alcohol, PHI demonstrates the ability to transiently store photogenerated charges, with the potential for their conversion into long-lasting radicals. Notably, these photogenerated charges can facilitate electron release for "dark" reduction reactions even under dark conditions [113,131–133]. To illustrate, the utilization of long-lived radicals by Lotsch et al. resulted in a novel approach combining light harvesting and dark cycling techniques for proton reduction, subsequently leading to the liberation of molecular hydrogen (H2) [134]. Moreover, Zeng et al. harnessed long-lived radicals to generate hydrogen peroxide (H2O2) by introducing molecular oxygen (O2) in the absence of light [135]. Exploiting this unique characteristic of PHI, Ou et al. successfully applied it to achieve simultaneous alcohol dehydrogenation and H2O2 production [136].
On the other hand, alterations in the electronic configuration of PHI/PTI exhibit significant implications for its photocatalytic efficacy. Specifically, manipulation of the electronic framework can engender modifications in the energy band structure. To this effect, Xu et al. pursued the doping of PHI with C/K, resulting in the generation of PHI (RPCN) that boasts a narrow band gap (1.71 eV) and robust near-infrared light absorption (> 700 nm). Intriguingly, the introduction of carbon dopants induces an asymmetric architecture in RPCN, with density-functional theory (DFT) analyses demonstrating a substantially narrower band gap relative to PCN. Moreover, the incorporation of carbon species fosters enhanced delocalization of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbitals, thereby promoting improved spatial separation of the charge carriers. This, in turn, facilitates effective segregation of the redox sites from one another (Fig. 4a) [49]. In a similar vein, Bhunia et al. endeavored to incorporate 2,4,6-triaminopyrimidine (TAP) into PTI, culminating in the preparation of PTI with varying C/N ratios. Intricate DFT calculations revealed a negative shift in the valence band positioning subsequent to the substitution of N within the aryl ring by CH, along with a diminished band gap concomitant with escalated TAP/Mel ratios (Fig. 4b) [137].
Figure 4
 Figure 4. (a) DFT calculated HOMO and LUMO distributions and DOS of PCN and RPCN. Reproduced with permission [49]. Copyright 2021, Wiley-VCH. (b) DFT optimized PTI-TPA structure schematic diagram. Reproduced with permission [137]. Copyright 2015, American Chemical Society. (c) Quantum-chemical calculations of PHI and Mel-PHI, and band position determination of PHI and Mel-PHI. Reproduced with permission [120]. Copyright 2021, Wiley-VCH.
Figure 4. (a) DFT calculated HOMO and LUMO distributions and DOS of PCN and RPCN. Reproduced with permission [49]. Copyright 2021, Wiley-VCH. (b) DFT optimized PTI-TPA structure schematic diagram. Reproduced with permission [137]. Copyright 2015, American Chemical Society. (c) Quantum-chemical calculations of PHI and Mel-PHI, and band position determination of PHI and Mel-PHI. Reproduced with permission [120]. Copyright 2021, Wiley-VCH.As previously stated, the induction of planar asymmetry plays a pivotal role in facilitating n→π* electron transitions, ultimately augmenting spectral absorption. Consequently, there exists a plausible avenue for extending the near-infrared (NIR) optical absorption range by manipulating the polarization of the electronic structure, for instance, through the introduction of functional groups, defects, or heteroatoms. In a compelling study conducted by Zhao et al., NH-rich red PHI nanoparticles were synthesized, thereby yielding a substantial extension of the NIR optical absorption capabilities. The incorporation of NH groups elicits a reduction in π-deficiency, prompting an unbalanced electron density that instigates n→π* electron transitions, consequently expanding the optical response window to encompass 750 nm. This noteworthy finding underscores the potential of modulating the electronic structure to achieve desirable spectroscopic properties in materials [125].
Moreover, changes in the local electronic distribution have the potential to engender internal donor-acceptor (D-A) heterostructures, thereby facilitating efficient carrier separation. A noteworthy illustration of this phenomenon was showcased by Zhang et al., who conducted investigations on the impact of triazine-modified heptazine frameworks on the local electronic structure. By introducing melamine into PHI (Mel-PHI), internal heterojunctions were successfully established through functionalization of the extended π-aromatic architecture on the triazine surface. Notably, calculations pertaining to the localization of the lowest energy exciton state immediately post-excitation revealed a significant disparity in charge delocalization between the two model systems. In the absence of melamine binding (PHI model), excitons exhibited uniform delocalization across each heptazine unit. In contrast, the Mel-PHI sample exhibited strongly localized exciton states on the heptazine unit situated directly adjacent to the melamine moiety. The aggregation of charge carriers at the melamine "defect" site serves to enhance exciton separation (Fig. 4c) [120]. Therefore, additional modifications to the electronic structure of PHI/PTI present an effective means to optimize its redox and light absorption capabilities, thereby instigating a substantial enhancement in overall photocatalytic efficiency.
4. Characterization of PHI/PTI
Through material characterization, significant differences between the structures of PHI and PTI compared to PCN are revealed (Fig. 5). The X-ray diffraction (XRD) patterns of PHI and PCN exhibit similar characteristics, particularly in their two strongest diffraction peaks around 27°−28° (Figs. 5a and c), which correspond to the (002) crystal plane of graphite-like materials. However, due to the larger gap between the heptazine units, PHI displays a notable shift in the (100) crystal plane diffraction peak to approximately 8.0° (Fig. 5c), while conventional PCN exhibits this peak at around 13° (Fig. 5a) [138]. In contrast, PTI, possessing higher crystallinity than PHI, demonstrates numerous strong and sharp XRD diffraction peaks, most notably at 12°, 21°, 26°, 29°, and 32°, corresponding to the (100), (110), (002), (102), and (210) crystalline planes, respectively (Fig. 5b). SEM images of the more crystalline PTI exhibit a hexagonal morphology (Fig. 5e), while high-resolution transmission electron microscopy (HRTEM) images clearly reveal the presence of the (100) lattice stripe with a spacing of approximately 0.7 nm. In comparison, PHI displays a larger lattice stripe spacing of around 1 nm (Figs. 5h and i) [115].
Figure 5
 Figure 5. For PCN (a) XRD, (d) SEM and (g) TEM. For PTI (b) XRD, (e) SEM and (h) TEM. For PHI (c) XRD, (f) SEM and (i) TEM. Reproduced with permission [115]. Copyright 2020, Wiley-VCH.
Figure 5. For PCN (a) XRD, (d) SEM and (g) TEM. For PTI (b) XRD, (e) SEM and (h) TEM. For PHI (c) XRD, (f) SEM and (i) TEM. Reproduced with permission [115]. Copyright 2020, Wiley-VCH.Both the ideal heptazine-based graphite-phase carbon nitride and triazine-based graphite-phase carbon nitride consists of tertiary amines linking three heptazine or triazine rings [92,139]. In the case of PHI and PTI, however, secondary amines connect two heptazine or triazine rings. These bridging secondary amine groups are partially protonated, resulting in multiple signal splits or shifts in the carbon-nitrogen (C/N) nuclear magnetic resonance (NMR) spectra of PHI and PTI. Specifically, the C NMR spectrum of K-PHI demonstrates four distinct signals, namely the central carbon atom (C1) within the heptazine unit, the peripheral carbon atom adjacent to the NH group (C2), the carbon atom adjacent to the deprotonated imide bridge (C3), and the carbon atom (C4) of the heptazine unit connected to the terminal cyano group. Additionally, a weak signal is observed from a small quantity of carbon (C5) within the terminal NCN group (Fig. 6a). Conversely, the corresponding NMR spectra of the N atoms exhibit five distinct signals, including the terminal NH2 group (N1), the NH group of the bridged heptazine unit (N2), the central nitrogen atom of the heptazine unit (N3), the peripheral nitrogen atom of the heptazine unit (N4), and the deprotonated N-group of the bridged heptazine unit (N5) (Fig. 6b). Notably, H-PHI, obtained through proton exchange to compensate for protons, does not exhibit signal splitting in the C/N NMR spectra for C3 and N5 (Figs. 6c and d) [109].
Figure 6
 Figure 6. (a, c) 13C solid state NMR spectra and (b, d) 15N solid state NMR spectra of K-PHI and H-PHI. Reproduced with permission [109]. Copyright 2019, American Chemical Society. (e) FTIR spectra and (f) XPS survey profile of PHI/PTI. Reproduced with permission [142]. Copyright 2023, MDPI. (g) 1H, (h) 13C, (i) 15N, and (j) 7Li solid state NMR spectra of PTI. (k) Schematic diagram of the PTI/LiCl connection pattern. Reproduced with permission [140]. Copyright 2016, Wiley-VCH. (l) Schematic diagrams of the triazine nucleus ((left) and the heptazine nucleus (right). Reproduced with permission [89]. Copyright 2011, Wiley-VCH.
Figure 6. (a, c) 13C solid state NMR spectra and (b, d) 15N solid state NMR spectra of K-PHI and H-PHI. Reproduced with permission [109]. Copyright 2019, American Chemical Society. (e) FTIR spectra and (f) XPS survey profile of PHI/PTI. Reproduced with permission [142]. Copyright 2023, MDPI. (g) 1H, (h) 13C, (i) 15N, and (j) 7Li solid state NMR spectra of PTI. (k) Schematic diagram of the PTI/LiCl connection pattern. Reproduced with permission [140]. Copyright 2016, Wiley-VCH. (l) Schematic diagrams of the triazine nucleus ((left) and the heptazine nucleus (right). Reproduced with permission [89]. Copyright 2011, Wiley-VCH.PTI distinguishes itself from PHI by exhibiting a distinct signal from the central nitrogen atom ("Nc") of the heptazine unit (Fig. 6l). Consequently, PTI primarily showcases three different carbon signals. Notably, the chemical shifts observed at 158 ppm (C1) and 162 ppm (C2) primarily stem from carbon nuclei adjacent to protonated triazine nitrogen atoms, with only their Li+ environment differentiation, while carbon species neighboring non-protonated ring nitrogen atoms show resonances at 168 ppm (Figs. 6g-k). In the 15N CP MAS spectrum, three distinct nitrogen signals are identified: chemical shifts at −250 and −242 ppm correspond to the NH group bridging the triazine ring (N1H and N2H), with their splitting into two peaks also related to the Li+ position, and the signal at −180 ppm corresponds to the broad resonance of the tertiary nitrogen atom (N tert) located on the triazine ring (Figs. 6h and k) [140,141].
The distinguishing triazine and heptazine rings of PHI and PTI can be identified through their FTIR signals within the 800–850 cm−1 and 1400–1600 cm−1 regions. Furthermore, the secondary amine (-NH-) bridging the triazine/heptazine unit exhibits FTIR absorption primarily within the 1200–1400 cm−1 region. The symmetric and asymmetric vibrational absorptions of the metal-NC2 groups formed through the insertion of alkali metals in the molten salt are observed at 990 and 1065 cm−1. Contrarily, PTI displays a distinct absorption peak at 670 cm−1 (Fig. 6e). In PHI XPS spectra, the presence of sp3 hybridized nitrogen atoms is evident, as reflected in the N 1s spectrum, which showcases three prominent peaks corresponding to ring nitrogen (C—N=C), tertiary nitrogen (NC3), and terminal -NHx groups. In contrast, the triazinyl-based PTI lacks sp3 hybridized nitrogen atoms (Fig. 6f). The ratio of C—N=C/NC3 in PHI is approximately 6, while the absence of tertiary nitrogen (NC3) in PTI results in a ratio of C—N=C/NC3 around 2 [142].
5. Structure optimization of PHI/PTI
PHI is typically synthesized through post-treatment of pre-condensed covalent carbon nitrides or N-rich heterocycles (e.g., triazole [143,144], tetrazole [145], or melamine [86]) using alkali metal salts. In contrast, PTI is directly prepared from N-rich heterocycles in alkali metal molten salts containing lithium (Li) [146]. Notably, in comparison to the usage of a small amount of alkali metal salt for heating graphite carbon nitrides or cyanamide to obtain alkali metal-doped covalent carbon nitrides, the formation of PTI and PHI necessitates an excess of alkali metal salt [147]. In the pursuit of enhancing the photocatalytic efficiency of PHI and PTI, researchers have explored various optimization strategies. This section primarily focuses on structural optimization approaches for PHI and PTI, encompassing adjustments in crystallinity, defect engineering, modulation of morphology, construction of heterojunction junctions, and achieving atomic-level dispersion of metal loading.
5.1 Crystallinity adjustment
Owing to limitations in the reaction kinetics of thermal polymerization processes, polymer chains in coordination networks (PCN) often undergo incomplete condensation or polymerization. Furthermore, inadequate deamidation of precursor molecules can cause excessive hydrogen bonding in the carbon-nitrogen (CN) backbone, thereby significantly impeding the in-plane conduction of electrons and resulting in low electrical conductivity [148,149]. Moreover, the presence of numerous uncondensed amino groups on the PCN surface can serve as electron-hole pair complex centers. In contrast, highly crystalline CN structures possess a completer and more extended π-conjugation system, which can greatly enhance charge separation efficiency within the CN material [150,151].
Inspired by the successful utilization of molten salts in organic synthesis and electrochemistry [152], a crystalline form of CN, designated as CCN, was first synthesized using the ionothermal method with LiCl/KCl by Thomas et al. The high-temperature liquid reaction environment provided by the eutectic mixture of LiCl/KCl remarkably expedited the condensation process of CN. The resulting CCN exhibited distinct diffraction peaks in X-ray diffraction analysis (Figs. 7a and b), and lattice streaks were clearly observed in HRTEM images (Figs. 7c and d). In a related study, Liu et al. synthesized fully condensed PTI crystals by condensing in LiCl/NaCl molten salts within a sealed ampoule system. The highly crystalline PTI crystals adopted the form of regular hexagonal prisms (Figs. 8a and b). Analysis of the XRD spectrum indicated higher crystallinity of PTI-LiNa, as evidenced by increased peak height and narrower half-peak width (Fig. 8c). Additionally, the IR spectrum revealed weaker vibrational absorption bands corresponding to bridging/terminal N—H bonds, indicating a higher degree of polymerization in PTI-LiNa (Fig. 8d). The presence of an extended conjugated structure and reduced defects promoted charge separation, thereby enhancing the photocatalytic efficacy with a full hydrolysis apparent quantum yield (AQY) of 12% (λ = 365 nm). The researchers also investigated the correlation between crystallinity and various factors including molten salt composition ratio (Figs. 8e and f), percentage of molten salt (Figs. 8g and h), holding temperature (Figs. 8i and j), and holding time (Figs. 8k and l). Lower percentages of salt resulted in partial solubilization of the solid precursors, while an excess of salt hindered the nucleation and growth of PTI crystals. Heating temperatures above 550 ℃ caused significant depolymerization of triazine units, whereas temperatures below 550 ℃ did not induce depolymerization of heptazine units. The limited reaction durations of 4 h and 12 h did not yield well-formed crystals [90]. Furthermore, the choice of different precursors for thermal polymerization influenced the resulting crystallinity. For instance, Savateev et al. demonstrated that the utilization of more acidic tetrazole precursors yielded a more ordered structure of PHI through pyrolysis in LiCl/KCl salt melt. Interestingly, incorporating a small quantity of finely dispersed PHI products as crystalline species facilitated the enhancement of PHI crystallinity [145].
Figure 7
 Figure 7. (a) XRD spectra, (b) SEM images, (c, d) HRTEM images of CCN. Reproduced with permission [85]. Copyright 2008, Wiley-VCH.
Figure 7. (a) XRD spectra, (b) SEM images, (c, d) HRTEM images of CCN. Reproduced with permission [85]. Copyright 2008, Wiley-VCH.Figure 8
 Figure 8. (a, b) HRTEM images and HAADF-STEM images of PTI-LiNa. (c, d) XRD spectra and FTIR spectra of PTI synthesized at 550 ℃ for 24 h under different molten salt systems (e, f) XRD spectra and FTIR spectra of PTI-LiNa synthesized at 550 ℃ for 24 h at different LiCl/LiNa molar ratios. (g, h) XRD spectra and FTIR spectra of PTI-LiNa synthesized at 550 ℃ for 24 h at different DCDA to molten salt mass ratios. (i, j) XRD spectra and FTIR spectra of PTI-LiNa powders synthesized at 500, 550 and 570 ℃. (k, l) XRD spectra and FTIR spectra of PTI-LiNa powders synthesized at 550 ℃ for 4, 12, 24 and 48 h. Reproduced with permission [145]. Copyright 2021, Wiley-VCH.
Figure 8. (a, b) HRTEM images and HAADF-STEM images of PTI-LiNa. (c, d) XRD spectra and FTIR spectra of PTI synthesized at 550 ℃ for 24 h under different molten salt systems (e, f) XRD spectra and FTIR spectra of PTI-LiNa synthesized at 550 ℃ for 24 h at different LiCl/LiNa molar ratios. (g, h) XRD spectra and FTIR spectra of PTI-LiNa synthesized at 550 ℃ for 24 h at different DCDA to molten salt mass ratios. (i, j) XRD spectra and FTIR spectra of PTI-LiNa powders synthesized at 500, 550 and 570 ℃. (k, l) XRD spectra and FTIR spectra of PTI-LiNa powders synthesized at 550 ℃ for 4, 12, 24 and 48 h. Reproduced with permission [145]. Copyright 2021, Wiley-VCH.Liang et al. conducted a comprehensive investigation into the nucleation and crystallization processes of PTI at the molecular level, unveiling a mechanism for the thermal polymerization and crystal evolution of PTI. Through the use of sample quenching and water vapor separation techniques, they successfully characterized the polymerized structural units and the crystal precipitation interface at a molecular scale. Fig. 9 illustrates the proposed evolutionary mechanism, wherein the heptazine-based melon compound undergoes depolymerization into triazine structural units within the low melting point molten salt during the heating phase. This process leads to the formation of PTI molecules. During the subsequent cooling phase, precipitation of KCl cubes becomes the dominant phase condensation precipitation. Notably, the exposed (100) planes of the KCl cube serve as templates, guiding the layer-by-layer alignment of PTI molecular units on the KCl planes, resulting in the formation of vertically aligned PTI crystals. By adjusting the interface between PTI and the molten salt crystal precipitation, PTI crystals with improved dispersion and enhanced photocatalytic properties were successfully obtained [153]. Furthermore, the highly crystalline PTI/Li+Cl− system proves to be an ideal platform for investigating the reaction surface of conjugated polymers. Lin et al. conducted an in-depth analysis of the in situ photodeposition of PTI/Li+Cl− single crystals using co-catalysts. Intriguingly, their study demonstrated that the (101°) surface of the catalyst exhibited stronger photocatalytic activity compared to the (0001) surface. Consequently, enhancing the side area of PTI/Li+Cl− single crystals can effectively improve the efficiency of photocatalytic hydrogen precipitation by the catalyst [154].
Figure 9
 Figure 9. Schematic diagram of molecular conjugation and directional crystallization process of PTI polymer crystals in molten salt. Reproduced with permission [153]. Copyright 2023, Wiley-VCH.
Figure 9. Schematic diagram of molecular conjugation and directional crystallization process of PTI polymer crystals in molten salt. Reproduced with permission [153]. Copyright 2023, Wiley-VCH.5.2 Defective engineering
Defect engineering proves to be a highly effective strategy for tailoring the structural characteristics of carbon nitride (CN) frameworks [155]. The presence of material defects, arising from alterations in reaction conditions or additional processing steps, manifests as disruptions in the inherent atomic arrangement within a given material. These imperfections are commonly encountered across various materials and can significantly impact their performance across multiple applications. Fig. 10 shows the common defects in carbon nitride. Significantly, in the field of photocatalysis, defects can become carrier trapping centers and improving carrier separation efficiency [156]. Moreover, these defects can generate intermediate energy states and adjust the VB and CB of the semiconductor material, thus narrowing the band gap width and shift photoresponse range. Additionally, defects introduce additional active sites to improve the redox properties of photocatalytic materials. In terms of material preparation, the defects can enhance the stability and dispersion of the metal anchored on the supporting surface. Advanced comprehension of the underlying reaction mechanisms has unveiled the advantageous role played by diverse defects in augmenting the photocatalytic performance of materials [157,158].
Figure 10
 Figure 10. Schematic diagram of the typical locations of the (a) C defect, (b) N defect, (c) amino defects, and (d) cyano defect in CN. Reproduced with permission [156]. Copyright 2021, Elsevier.
Figure 10. Schematic diagram of the typical locations of the (a) C defect, (b) N defect, (c) amino defects, and (d) cyano defect in CN. Reproduced with permission [156]. Copyright 2021, Elsevier.One prevalent defect in carbon nitride is cyanamide, and Lau et al. skillfully designed a cyano-modified carbon nitride polymer employing a PHI-based model catalyst. Through their investigation, they identified a fraction of cyanamide as a photocatalytically relevant "defective" active site. In comparison to the PCN, the hydrogen production rate of 24.7 µmol/h and the apparent quantum efficiency (at 400 nm) of 9.3% were enhanced by a factor of 12 and 16, respectively. Two effects attributable to the presence of cyanamide defects were established: (1) Augmentation of the interaction between the co-catalyst and the catalyst, thereby promoting interfacial electron transfer to the Pt center; (2) Establishment of a built-in electric field conducive to improved charge separation for photoexcited species [159]. Furthermore, cyanamide defects also play a pivotal role in the generation of ultra-long-lived radicals within the polymerization network of cyanamide-functionalized heptazine units, as demonstrated in subsequent work by Lau et al. These long-lived radical species enable the subsequent release of trapped electrons in the dark, resulting in H2 production. Control experiments involving PCN lacking cyanamide introduction did not exhibit "dark" photocatalysis. This behavior can be attributed to the excellent electron absorption properties of the cyanide defect, with the excess electrons accumulating near the cyanide, thereby endowing the nitrogen within the cyanide with superior nucleophilic and radical reaction characteristics [134].
In some study, the authors investigated various strategies for introducing defects in the carbon nitride material. The introduction of defects can be achieved through different approaches such as heat treatment, chemical reduction, hydrothermal methods, and others [156]. For instance, Li et al. employed a molten salt polymerization technique using melamine with the addition of KOH to prepare carbon nitride with controlled defects. The FTIR spectrum revealed an increased absorption peak of cyano with an increasing content of KOH (Fig. 11a), indicating the controlled introduction of N vacancies. Moreover, 13C NMR analysis demonstrated the presence of bridging C atoms in cyano and adjacent C atoms (Fig. 11b), further supporting the controlled introduction of defects. The authors observed that the introduction of nitrogen vacancies led to a stronger binding ability of the catalyst to gas molecules, resulting in improved photocatalytic CO2 reduction performance compared to pristine carbon nitride and crystalline carbon nitride without defect modification (Figs. 11c and d) [160].
Figure 11
 Figure 11. (a) FTIR spectra of CN, CN-M-x. (b) 13C solid state NMR spectra of CN-M-0.01. (c) CO2 adsorption isotherms of CN. (d) CO2 TPD measurements of CN. Reproduced with permission [160]. Copyright 2020, the Royal Society of Chemistry.
Figure 11. (a) FTIR spectra of CN, CN-M-x. (b) 13C solid state NMR spectra of CN-M-0.01. (c) CO2 adsorption isotherms of CN. (d) CO2 TPD measurements of CN. Reproduced with permission [160]. Copyright 2020, the Royal Society of Chemistry.In a similar study, Chen et al. employed a solvothermal method to treat carbon nitride and obtain melon-PHI isotropic composites modified with carbon defects. 13C NMR measurements confirmed the presence of carbon defects in the melon-PHI carbon nitride isotropic composites. When combined with Pt co-catalyst and cavity scavenger, the carbon defect modified melon-PHI isotropic composites exhibited excellent photocatalytic activity for hydrogen precipitation. The melon-PHI composites achieved simultaneous hydrogen precipitation through the cleavage of water and selective oxidation of 5-hydroxymethylfuran to 2,5-dimethylfuran, with respective activities of 41 and 49 µmol h−1 g−1. The inherent defect in the carbon vacancy was found to enhance the photo-charge separation and generate additional active sites, thereby improving the performance of photocatalytic redox reactions [161].
These findings highlight the significance of defect engineering in carbon nitride materials, as controlled defect introduction can enhance their catalytic properties and improve their performance in photocatalytic reactions. The strategies employed in this study provide valuable insights for the development of efficient and sustainable photocatalytic systems.
5.3 Morphological control
The performance of PCN materials is significantly influenced by their structural characteristics, morphology, and specific surface area. Variation in PCN morphologies has a direct impact on the separation of photogenerated carriers, catalyst-reactant contact area, as well as reactant adsorption and desorption processes [162,163]. Hence, the manipulation of morphology plays a crucial role in enhancing catalytic activity. Particularly, ultrathin nanosheets facilitate efficient charge transfer from the inner regions to the surface and expose a greater number of active sites, leading to a substantial improvement in photocatalytic performance [164]. Notably, previous investigations have demonstrated that PHI/PTI possesses two-dimensional planar layers consisting of heptazine/triazine interconnected by imide, making it highly suitable for the production of high-quality ultrathin nanosheets. By subjecting PTI to sonication in an aqueous solution and subsequent centrifugation, Schwinghammer et al. successfully obtained highly crystalline PTI nanosheets at a concentration of up to 0.2 mg/mL, with thicknesses ranging from 1 nm to 2 nm (Fig. 12a). HRTEM analysis confirmed the retention of the hexagonal morphology observed in the microcrystals. Importantly, the PTI nanosheets exhibited an 18-fold increase in hydrogen generation from decomposed water compared to the unprocessed PTI material (Figs. 12b and c) [165].
Figure 12
 Figure 12. (a) Schematic diagram of the PTI exfoliation process. (b) AFM image of PTI nanosheets. (c) TEM image of PTI nanosheets. Reproduced with permission [165]. Copyright 2014, American Chemical Society.
Figure 12. (a) Schematic diagram of the PTI exfoliation process. (b) AFM image of PTI nanosheets. (c) TEM image of PTI nanosheets. Reproduced with permission [165]. Copyright 2014, American Chemical Society.In contrast to chemical or physical interventions, Miller et al. dissolved PTI-Li+Br− in a non-protonic polar solvent, resulting in the formation of several layers of thick PTI nanosheets (Fig. 13a). This dissolution process, driven by thermodynamics, effectively preserved the crystalline structure of the material, yielding a solution composed of defect-free hexagonal two-dimensional nanosheets with a well-defined size distribution. Fig. 13b illustrates the presence of complete lattice stripes and hexagonal shapes in the PTI-Li+Br− nanosheets [166]. Additionally, Jia et al. utilized NaNp/DMAc to charge reduce the PTI framework and stabilize the PTI layer by introducing C12H25Br (Fig. 13c), ultimately achieving highly exfoliated 2D few-layered PTI (FL-PTI) with an average thickness of 1.1 nm (Figs. 13d-f). The original PTI layer exhibited insolubility in water due to the strong interlayer interactions, however, the reduction process successfully mitigated these interactions and accelerated dissolution in DMAc. The resulting FL-PTI solution exhibited excellent stability, with a yield of 35 wt% [167].
Figure 13
 Figure 13. (a) Schematic diagram of spontaneous dissolution of PTI-LiBr. (b) TEM image of PTI nanosheet. Reproduced with permission [166]. Copyright 2017, American Chemical Society. (c) Schematic of charging and exfoliation of PTI. (d) HRTEM image of FL-PTI nanosheet. (e) AFM image of FL-PTI nanosheets. (f) PTI thickness histogram. Reproduced with permission [167]. Copyright 2018, Wiley-VCH.
Figure 13. (a) Schematic diagram of spontaneous dissolution of PTI-LiBr. (b) TEM image of PTI nanosheet. Reproduced with permission [166]. Copyright 2017, American Chemical Society. (c) Schematic of charging and exfoliation of PTI. (d) HRTEM image of FL-PTI nanosheet. (e) AFM image of FL-PTI nanosheets. (f) PTI thickness histogram. Reproduced with permission [167]. Copyright 2018, Wiley-VCH.An alternative approach to prepare ultrathin nanosheets involves the utilization of a bottom-up strategy. This technique involves the selection of an appropriate solid surface that enables the fusion and alignment of precursor materials. Through surface polymerization, ultrathin nanosheets with a high level of in-plane order can be synthesized. Wang et al. demonstrated this approach by inducing surface polymerization of melamine on NaCl crystal surfaces (Fig. 14a), successfully creating PHI flakes measuring approximately 4 nm in thickness and a few microns in lateral dimensions (Figs. 14b-d). This can be attributed to the relatively high surface energy of NaCl crystals, which facilitates the adsorption and activation of melamine on its surface. Moreover, the unique interaction between NaCl and melamine introduces a robust interfacial constraint, resulting in the organized assembly of two-dimensional nanosheets on the NaCl substrate [168].
Figure 14
 Figure 14. (a) Illustration for the synthesis process of PCNNs-IHO by on-surface polymerization. (b) High-resolution TEM image of PCNNs-IHO and SAED pattern of the (100) plane (inset). (c) AFM image and (d) corresponding thickness analysis taken around the white line in (c) of PCNNs-IHO. Reproduced with permission [168]. Copyright 2021, Wiley-VCH. (e) Schematic illustration of the formation process of TCNTs in the molten salt medium. (f)TEM image and (g) high-resolution TEM image of TCNTs. (h) N2 adsorption-desorption isotherms of B-CN and TCNTs. Reproduced with permission [169]. Copyright 2017, Elsevier.
Figure 14. (a) Illustration for the synthesis process of PCNNs-IHO by on-surface polymerization. (b) High-resolution TEM image of PCNNs-IHO and SAED pattern of the (100) plane (inset). (c) AFM image and (d) corresponding thickness analysis taken around the white line in (c) of PCNNs-IHO. Reproduced with permission [168]. Copyright 2021, Wiley-VCH. (e) Schematic illustration of the formation process of TCNTs in the molten salt medium. (f)TEM image and (g) high-resolution TEM image of TCNTs. (h) N2 adsorption-desorption isotherms of B-CN and TCNTs. Reproduced with permission [169]. Copyright 2017, Elsevier.Furthermore, the orientation of morphology can also be controlled using a molten salt template. Tian et al. proposed a salt-assisted self-assembly mechanism for the in-situ synthesis of tetragonal hollow tube PTI using the molten salt method (Fig. 14e). The recrystallized salt serves as an in-situ template on which carbon nitride nanosheets assemble into a tubular structure with distinctive one-dimensional hollow characteristics (Figs. 14f and g) and a substantial specific surface area (Fig. 14h). Consequently, these PTI nanosheets exhibit enhanced photocatalytic activity and superior adsorption properties compared to B-CN [169].
5.4 Construction of heterojunction
The design of composite/heterojunction structures represents a viable strategy for improving the process of charge separation/transfer and enhancing the overall efficiency of photocatalysis. By closely associating a semiconductor possessing a suitable energy band structure with carbon nitride, it becomes possible to achieve spatial separation of photogenerated electron-hole pairs through charge transfer at the interface of these two semiconductors [170,171].
Mazzant et al. have successfully engineered a donor-acceptor composite by superimposing K-PHI nanoparticles onto flavin microcrystals. Notably, the frontier orbital energy levels of flavin and the conduction band/valence band energy levels of K-PHI are aligned in a manner that forms a Z-shaped heterojunction (Fig. 15a). As a result of selective charge complexation at the interface, the composite exhibits a near-complete burst of fluorescence, effectively inhibiting energy transfer. Moreover, the K-PHI shell facilitates the release of oxygen due to its positive highest occupied molecular orbital (HOMO) energy level, thereby accelerating the reaction and potentially influencing product distribution through kinetic effects [172]. Cheng et al. have introduced Co atom-doped CeO2 into highly crystalline PTI to establish an S-type heterojunction for efficient photoreduction of CO2 to CH4. The heterojunction displays dynamic charge fluctuations that promote electron transfer from PTI to the site-specific Co, thereby facilitating directed electron transfer during the photocatalytic process (Fig. 15b) [173].
Figure 15
 Figure 15. (a) Schematic diagram of photoreduction of CO2 by Z-scheme heterojunction constructed with K-PHI and flavin. Reproduced with permission [172]. Copyright 2020, Elsevier. (b) Schematic illustration of formation of S-scheme heterojunction of CeCo-PTI. Reproduced with permission [173]. Copyright 2022, Wiley-VCH.
Figure 15. (a) Schematic diagram of photoreduction of CO2 by Z-scheme heterojunction constructed with K-PHI and flavin. Reproduced with permission [172]. Copyright 2020, Elsevier. (b) Schematic illustration of formation of S-scheme heterojunction of CeCo-PTI. Reproduced with permission [173]. Copyright 2022, Wiley-VCH.The carbon nitride family comprises PHI/PTI isomers, where under certain conditions, the heptazine phase can transform into the triazine phase. This intrinsic property enables the synthesis of heptazine/triazine heterojunction via a straightforward process. Zhang et al. achieved the synthesis of PHI/PTI heterojunction utilizing a two-step molten salt method, with CaCO3 serving as the structural modifier and LiCl/KCl as the molten salt. Notably, the presence of a strong built-in electric field at the surface facilitates rapid exciton dissociation and carrier separation, resulting in a high apparent quantum yield (AQY = 64%) for visible-light-driven hydrogen production (Figs. 16a-c) [174]. Sun et al. demonstrated the one-step synthesis of triazine-heptazine heterojunction by condensing precursor molecules in molten salts through sequential molecular self-assembly (Figs. 16d-f). The staggered band gap inherent in the heterojunction drives electron transfer from the heptazine unit to the triazine unit (Fig. 16g). Furthermore, potassium ions serve as temporary bridges, enabling rapid electron migration and charge separation. In addition, the high crystallinity of the heterojunction significantly enhances carrier mobility along the in-plane direction. When utilized in photocatalytic processes such as hydrogen production under visible light or single-wavelength irradiation, MA-rod arrays exhibit remarkable photocatalytic activity and stability. This is evidenced by their high apparent quantum yield (AQY = 60.0%) and substantial milligram yields in aromatic oxidation, leading to the formation of aldehydes or ketones [175].
Figure 16
 Figure 16. (a) The synthetic process of hybrid PHI/PTI copolymer with simi-cohenrent interface. (b) HR-TEM image of PHI/PTI. (c) Schematic illustration of exciton dissociation and charge transfer in PHI/PTI under visible light irradiation. Reproduced with permission [174]. Copyright 2022, Wiley-VCH. (d) The comparison of array structure and particle/sheet structure. (e) The synthesis path of self-supported hierarchical crystalline GCN array. (f) SEM images of MA-rod array. (g) Schematic of the proposed photo-electron transfer path in the framework of MA-rod array. Reproduced with permission [175]. Copyright 2022, Elsevier.
Figure 16. (a) The synthetic process of hybrid PHI/PTI copolymer with simi-cohenrent interface. (b) HR-TEM image of PHI/PTI. (c) Schematic illustration of exciton dissociation and charge transfer in PHI/PTI under visible light irradiation. Reproduced with permission [174]. Copyright 2022, Wiley-VCH. (d) The comparison of array structure and particle/sheet structure. (e) The synthesis path of self-supported hierarchical crystalline GCN array. (f) SEM images of MA-rod array. (g) Schematic of the proposed photo-electron transfer path in the framework of MA-rod array. Reproduced with permission [175]. Copyright 2022, Elsevier.5.5 Atomically dispersed metal loading
Compared with metal nanoparticles, single-atom catalysts (SACs) have garnered considerable attention from researchers due to their unique properties. This can be attributed to the low coordination environment of metal single atoms, which allows for high atomic utilization and improved strong metal support interactions [176]. Consequently, SACs demonstrate exceptional catalytic activity, selectivity, and stability across various reactions, including oxidation, water-gas conversion, and hydrogenation [177]. However, it is worth noting that achieving atomic-level dispersion of metals on substrates such as metal, metal oxide, or carbon is thermodynamically challenging, as they tend to aggregate into clusters or nanoparticles, particularly at elevated temperatures [178].
To overcome this challenge, carbon nitride has emerged as a promising candidate for anchoring metal atoms, thanks to the presence of nitrogen-rich macro heterocycles within its lattice structure. Chen et al. conducted a study investigating different carbon nitride structures' ability to act as individual atomic substrates by utilizing microwave-assisted deposition to load Pd onto these structures. They observed that PTI and PHI exhibited higher Pd loading compared to other carbon nitride carbons. Furthermore, the Pd distribution in PTI and PHI was more uniform and thermally stable in comparison to GCN. The larger cavity size and the presence of chloride ions in the PTI and PHI structures enhanced the metal-carrier interaction, thereby facilitating the Pd loading process [179].
Various methods, such as photodeposition [180], atomic layer deposition [181], impregnation [182], and heat treatment [183], are commonly employed to introduce single atoms on carbon nitride. Among these methods, cation exchange on PHI substrates offers relative ease compared to others. This is attributed to the presence of compensating charges of Na+ or K+ between the negatively charged layers of PHI, allowing for facile exchange of cations [184]. Teixeira et al. applied a simple cation exchange method to synthesize Fe-PHI single-atom catalysts on Na-PHI materials in a one-step process (Fig. 17a). This involved adding Na-PHI to a solution of FeCl3·6H2O and sonicating the mixture. By accurately controlling the FeCl3 concentration during the cation exchange process, the authors achieved varying iron concentrations ranging from 5 wt% to 0.02 wt% in PHI. The Fe-PHI single-atom catalysts exhibited excellent selectivity and conversion rates in C—H bond oxidation reactions (Figs. 17b and c) [185].
Figure 17
 Figure 17. (a) Schematic representation of the controlled non-thermal cation exchange method used to replace the Na+ cations in the structure of PHI by Fe3+ cations. (b) Proposed mechanism of formation of the active Fe(Ⅳ)/Fe(Ⅴ) sites. (c) Schematic representation of the e−/h+ separation promoting effect on the formation of active high oxidation state iron centers. Reproduced with permission [185]. Copyright 2021, Elsevier.
Figure 17. (a) Schematic representation of the controlled non-thermal cation exchange method used to replace the Na+ cations in the structure of PHI by Fe3+ cations. (b) Proposed mechanism of formation of the active Fe(Ⅳ)/Fe(Ⅴ) sites. (c) Schematic representation of the e−/h+ separation promoting effect on the formation of active high oxidation state iron centers. Reproduced with permission [185]. Copyright 2021, Elsevier.In conventional practice, the synthesis of PHI/PTI typically occurs in eutectic molten salts, affording the convenient on-site integration of select metal single atoms through the ionothermal methodology. In their study, Shen et al. employed a molten salt-assisted ionothermal approach to synthesize CCN/PTI heterojunctions with single-atom Co coordination (Fig. 18a). This was accomplished by introducing CoCl2·6H2O into a mixture of KCl and LiCl·H2O. Through high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), the presence of numerous bright spots at the atomic scale was observed, confirming the successful incorporation of Co single atoms (Figs. 18b and c). The Co-K-edge extended X-ray absorption fine structure (EXAFS) spectra indicated that the valence state of Co in Co-CCNPTI was near +2, further revealing the coordination of Co with carbon nitride through the formation of Co-N bonds, rather than Co-O or Co-Co bonds (Fig. 18d). The introduction of CoCl2·6H2O facilitated the phase transition from PHI to PTI, resulting in the formation of a type Ⅱ heterojunction, as well as the formation of Co-N coordinated single-atom Co within the CCN/PTI heterojunction. Notably, the presence of single-atom Co coordination enhanced charge transport and expedited the surface oxidation of organic compounds. Remarkably, under visible light, the Co-CCN/PTI catalyst displayed a record-high hydrogen yield of 3538 µmol h−1 g−1, accompanied by an impressive apparent quantum yield (AQY) of 20.88% at 425 nm [186].
Figure 18
 Figure 18. (a) Schematic mechanisms of the Co-CCN/PTI heterojunction for photocatalytic hydrogen production. (b) HRTEM image of Co-CCN/PTI-4. (c) HAADF-STEM image of Co-CCN/PTI-4. (d) FT k3-weighted Co K-edge EXAFS spectra of Co-CCN/PTI-4, Co-CCN/PTI-40 and the references. Reproduced with permission [186]. Copyright 2021, Science China Press.
Figure 18. (a) Schematic mechanisms of the Co-CCN/PTI heterojunction for photocatalytic hydrogen production. (b) HRTEM image of Co-CCN/PTI-4. (c) HAADF-STEM image of Co-CCN/PTI-4. (d) FT k3-weighted Co K-edge EXAFS spectra of Co-CCN/PTI-4, Co-CCN/PTI-40 and the references. Reproduced with permission [186]. Copyright 2021, Science China Press.6. PHI/PTI photocatalytic applications
6.1 Photocatalytic water decomposition
Hydrogen is regarded as a clean, high-energy density (143 MJ/kg) chemical fuel. The utilization of photocatalytic water decomposition for hydrogen production presents a promising and sustainable solution to address energy and environmental challenges [187,188]. Notably, substantial progress has been made in the development of highly efficient photocatalysts for water decomposition and subsequent hydrogen production, as reported by Wang et al. One notable example is the construction of a novel heterostructure through the coupling of K-PHI with ferroelectric BaxSr1-xTiO3 (BxST), achieved via a simple electrostatic self-assembly technique (Fig. 19a). The favorable ionic properties of K-PHI enable strong electrostatic interactions with ferroelectric materials, facilitating close interfacial contacts (Fig. 19b). The energy band structure analysis of 5% K-PHI/B0.8ST (Fig. 19c) reveals a characteristic type Ⅰ heterojunction that suppresses charge separation. Consequently, the remarkable enhancement in charge transfer and separation efficiency observed in 5% K-PHI/B0.8ST primarily stems from the polarized electric field contribution of B0.8ST. The K+-rich surface of K-PHI tends to bind with the negatively charged surface of B0.8ST, while the electronegative surface of K-PHI attracts the positively charged side of B0.8ST (Fig. 19c). In this configuration, the K+ ions within the heptazine unit serve as bridges for the rapid migration of photogenerated charges [189]. Furthermore, the strong internal electric field between K-PHI and B0.8ST supplies the necessary driving force to guide the separation of photogenerated carriers while limiting their recombination. Detailed investigation of the photocatalytic performance reveals that K-PHI/B0.8ST exhibits a remarkable H2-evolution rate of 1087.4 µmol h−1 g−1, with an apparent quantum yield (AQY) of 8.05% at 420 nm (Figs. 19d and e) [189]. In a separate study, Cheng et al. successfully obtained amorphous-crystalline carbon nitride isotype heterojunctions (BCN—NaK) with a high cyano content through a post-treatment strategy utilizing eutectic salts (Fig. 19f). The abundance of cyanide species facilitates efficient electron trapping and promotes the effective separation of photoexcited carriers. Moreover, the tight type Ⅱ isotype heterojunction formed between amorphous PCN and crystalline PHI significantly enhances the separation and migration of photoexcited electrons and holes. BCN-NaK demonstrates the highest photocatalytic hydrogen production rate at 26,900 µmol h−1 g−1. Interestingly, the addition of K2HPO4 to optimize the photocatalytic system further enhances the photocatalytic H2 production activity of BCN-NaK, reaching 48,656 µmol h−1 g−1 along with an impressive 68.9% AQYs at 405 nm (Figs. 19g and h) [190].
Figure 19
 Figure 19. (a) Fabrication process of K-PHI/BxST materials. (b) Illustration of the alkali metal cations as charge-transfer bridge for polarization promoted photocatalysts. (c) Schematic diagram of migration and separation of photogenerated charge carriers. (d) The results of cycling experiments on 5 % K-PHI/B0.8ST in H2 evolution reaction. (e) The DR UV–vis spectrum over K-PHI and the determined AQY at different wavelengths over 5 % K-PHI/B0.8ST. Reproduced with permission [198]. Copyright 2022, Wiley-VCH. (f) Schematic mechanism of BCN—NaK for photocatalytic H2 production. (g) Photocatalytic H2-production activities for BCN-T550, BCN-LiK, BCN—NaK and BCN—NaK + K2HPO4 under visible-light irradiation (λ > 400 nm). (h) Wavelength-dependence AQYs for BCN—NaK + K2HPO4. Reproduced with permission [190]. Copyright 2023, Elsevier.
Figure 19. (a) Fabrication process of K-PHI/BxST materials. (b) Illustration of the alkali metal cations as charge-transfer bridge for polarization promoted photocatalysts. (c) Schematic diagram of migration and separation of photogenerated charge carriers. (d) The results of cycling experiments on 5 % K-PHI/B0.8ST in H2 evolution reaction. (e) The DR UV–vis spectrum over K-PHI and the determined AQY at different wavelengths over 5 % K-PHI/B0.8ST. Reproduced with permission [198]. Copyright 2022, Wiley-VCH. (f) Schematic mechanism of BCN—NaK for photocatalytic H2 production. (g) Photocatalytic H2-production activities for BCN-T550, BCN-LiK, BCN—NaK and BCN—NaK + K2HPO4 under visible-light irradiation (λ > 400 nm). (h) Wavelength-dependence AQYs for BCN—NaK + K2HPO4. Reproduced with permission [190]. Copyright 2023, Elsevier.The thermodynamic capability of PTI to achieve integral water splitting (OWS) is conferred by its band gap, which intersects with the redox potential of water. To promote the condensation of carbon nitride, Liu et al. selected LiCl/NaCl as the molten salt with a melting point (Tm) of 552 ℃ (Figs. 20a and b). The utilization of LiCl/NaCl resulted in mild reaction conditions that effectively restrained the depolymerization of the s-triazine unit, enabling the formation of PTI/Li+Cl− crystals with complete condensation. This crystalline structure exhibits an extended planar conjugated system and a reduced number of defects, which in turn facilitate charge separation, thereby enhancing the photocatalytic efficiency. In the process of photocatalytic OWS experiments, PTI-LiNa demonstrated an apparent quantum yield (AQY) of 12% at a wavelength of 365 nm [90]. Encouraged by the impressive OWS performance of highly crystalline PTI, Liu et al. further refined the quantum efficiency of PTI in subsequent research by judiciously controlling the composition, structure, and morphology of the co-catalyst. Specifically, a stepwise photodeposition technique was employed to sequentially load CoOx, Rh, and Cr2O3 co-catalysts onto the surface of PTI, resulting in the formation of CoOx and Rh-Cr2O3 co-catalysts spatially segregated from each other (Figs. 20c and d). The deposition of Cr2O3 onto the Rh surface created a core-shell structure, effectively suppressing the inverse reaction, while the dual co-catalysts synergistically facilitated charge extraction from the bulk to the surface. Consequently, the AQY for the photocatalytic hydrolysis reaction was further increased to 20.2% at a wavelength of 365 nm [110].
Figure 20
 Figure 20. (a) Proposed salt melt synthetic processes of PTI/Li+Cl− catalysts by using different binary alkali metal chlorides with different melting points. Red circle represents nitrogen defect). (b) Schematic of PTI (Pt-CoOx) overall water splitting. Reproduced with permission [90]. Copyright 2022, Wiley-VCH. (c) Schematic of PTI (Rh/Cr2O3-CoOx) overall water splitting. (d) Proposed schematic process for step-by-step deposition of CoOx and Rh/Cr2O3 on poly(triazine imide) as H2 and O2 evolution cocatalysts, respectively. Reproduced with permission [110]. Copyright 2023, Wiley-VCH.
Figure 20. (a) Proposed salt melt synthetic processes of PTI/Li+Cl− catalysts by using different binary alkali metal chlorides with different melting points. Red circle represents nitrogen defect). (b) Schematic of PTI (Pt-CoOx) overall water splitting. Reproduced with permission [90]. Copyright 2022, Wiley-VCH. (c) Schematic of PTI (Rh/Cr2O3-CoOx) overall water splitting. (d) Proposed schematic process for step-by-step deposition of CoOx and Rh/Cr2O3 on poly(triazine imide) as H2 and O2 evolution cocatalysts, respectively. Reproduced with permission [110]. Copyright 2023, Wiley-VCH.6.2 Photocatalytic CO2 reduction
With the exponential growth in the utilization of fossil fuels, there has been a consequential surge in CO2 emissions, thereby exacerbating concerns regarding both fuel scarcity and global warming. Drawing inspiration from the concept of photosynthesis in green plants, scientists have been increasingly interested in harnessing a photocatalytic process to convert CO2 into valuable chemical fuels, with the aim of addressing these aforementioned challenges [191,192]. To this end, Cheng et al. demonstrated the efficacy of integrating single-atom Co-doped CeO2 into PTI, resulting in a synergistic interplay between the bimetallic CeCo co-catalyst and PTI, thereby achieving a remarkably selective reduction of CO2 to CH4 (yield: 181.7 µmol/g; selectivity: 88.3%). The implementation of single-atom Co not only modifies the electronic properties and local crystallinity of CeO2, but also induces dynamically interconnected electronic properties through Co-O bonding, thereby engendering structures with heightened surface free energy and coordination unsaturation, which, in turn, accelerates electron transfer from the PTI donor to the Co center (Fig. 21a). DRIFTS analysis further indicates the robust protonation capability of CeCo-PTI, wherein preferential protonation of CO* to hydride species occurs rather than the dissociation of CO* intermediates from the binding surface [173]. As another significant contribution to this field, Mazzanti et al. devised a bionic core-shell architecture incorporating flavin crystals modified by K-PHI nanoparticles. This innovative construction enabled the photocatalytic reduction of CO2 to yield a diversity of products (CH4, MeOH, EtOH, and CO) by utilizing water vapor as an electron and proton donor. Enhanced redox activity was achieved through the formation of Z-type heterojunctions between K-PHI and flavin. The K-PHI shell layer expeditiously facilitated the liberation of oxygen due to its positively charged HOMO, thereby expediting the reaction rate and altering the product distribution through kinetic mechanisms [172]. In order to improve the CO2 binding capacity of photocatalytic materials, Xia et al. adopted an ionothermal methodology to synthesize crystalline PHI nanorods on graphene, resulting in the formation of one-dimensional (1D) nano-array structures termed CNNA/rGO (Fig. 21b). These heterogeneous configurations exhibited a substantially increased CO2 adsorption capacity and demonstrated remarkable selectivity for CO2 adsorption. The superior crystallinity of CNNA coupled with the excellent electrical conductivity of rGO facilitated efficient charge migration and separation at the surface or interface. Remarkably, employing a reaction environment consisting of water vapor-saturated wet CO2 gas, the CO2 conversion rate of CNNA/rGO was recorded as 12.63 µmol h−1 g−1. Moreover, even in an atmosphere characterized by low CO2 concentration and high N2 concentration (90 vol%), the CO2 conversion rate remained significantly high, reaching 9.24 µmol h−1 g−1 [193].
Figure 21
 Figure 21. (a) Schematic for well-defined photocatalytic platform by integrating Co-doped CeO2 nanoparticles with highly crystalline PTI. Reproduced with permission [173]. Copyright 2022, Wiley-VCH. (b) Schematic diagram of CNNA/rGO photocatalytic CO2 reduction. Reproduced with permission [193]. Copyright 2019, Elsevier.
Figure 21. (a) Schematic for well-defined photocatalytic platform by integrating Co-doped CeO2 nanoparticles with highly crystalline PTI. Reproduced with permission [173]. Copyright 2022, Wiley-VCH. (b) Schematic diagram of CNNA/rGO photocatalytic CO2 reduction. Reproduced with permission [193]. Copyright 2019, Elsevier.6.3 Photocatalytic organic synthesis
Due to its cost-effectiveness, facile synthesis and remarkable selectivity towards reaction intermediates, the PHI/PTI photocatalyst has assumed a significant role in multiphase organic transformations. The K-PHI displays a valence band (VB) potential of +2.54 V (vs. RHE) that exceeds that of g-CN, thereby endowing PHI with pronounced oxidative capabilities suited for various photooxidation reactions involving organic compounds. For instance, Mazzanti et al. employed K-PHI nanoparticles to convert chloroform into dichloromethyl radicals, expanding the alkenone backbone through C1 extension. This methodology proved applicable to diverse enone reactions, yielding γ,γ-dichloroketones in the range of 18%−89%. Notably, the synthesis efficiency of γ,γ-dichloroketone was enhanced by 19-fold when the reaction occurred within a quasi-homogeneous flow photoreactor (Fig. 22a). A schematic representation of the synthesis mechanism is depicted in Fig. 22b. Initially, K-PHI undergoes excitation (to K-PHI*) upon blue light stimulation. Subsequently, in the presence of TEOA, K-PHI* accepts an electron, giving rise to the long-lived radical anion K-PHI•− characterized by its distinctive blue-green color (indicative of a reductive burst of photocatalyst). Subsequently, K-PHI•− donates a single charge to chloroform, leading to the formation of the chloroform radical anion and the elimination of the chloride anion, ultimately resulting in the generation of the dichloromethyl radical. The addition of the dichloromethyl radical to the β-carbon atom of the enone yields the intermediate product i-1. Finally, TEOA acts as a hydrogen donor to transfer hydrogen to the intermediate product i-1, generating the desired product γ,γ-dichloroketone [194].
Figure 22
 Figure 22. (a) Schematic representation of the reactor setup and reaction parameters. (b) Proposed mechanism of the generation of dichloromethyl radicals and their addition to enones. Reproduced with permission [194]. Copyright 2020, Nature Springer.
Figure 22. (a) Schematic representation of the reactor setup and reaction parameters. (b) Proposed mechanism of the generation of dichloromethyl radicals and their addition to enones. Reproduced with permission [194]. Copyright 2020, Nature Springer.Markushyna et al. utilized the enduring radicals engendered by K-PHI and CO2 as a weak Lewis acidic reaction medium to convert benzylamine into a series of imines, attaining yields between 72% and 96% (Fig. 23a). When CO2 served as the reaction medium, it reacted with benzylamine to produce benzylcarbamic acid. Under light irradiation, K-PHI was excited to K-PHI*, and the carbamic acid carefully acted as an electron donor by transferring an electron to the excited photocatalyst, thereby generating the long-lived surface radical anion K-PHI•−. In its radical cationic form, the electron-deficient phenylcarbamic acid interacted with another molecule of carbamic acid, liberating CO2 and two protons. Ultimately, the imine product was formed with the release of a CO2 molecule and NH3 (the mechanism is portrayed in Fig. 23b) [195]. In subsequent investigations, K-PHI was employed to selectively produce three distinct products (sulfonyl chloride, arylchloride, and diaryldisulfide) from S-arylthioacetate. Notably, the tunability of K-PHI's ability to sensitize both singlet oxygen and multi-electron transfer permits the selective conversion of thioacetates simply by adjusting the wavelength of the excitation light. Specifically, upon irradiation with low-energy light at 535 nm, only "IBS-CB" electron transfer occurs, resulting in a relatively low oxidation capacity within the IBS and enabling energy transfer solely to oxygen to form 1O2. In the presence of singlet oxygen (1O2), the transformation of aryl thioacetate to diaryldisulfide occurs via deacetylation, followed by coupling of the thiol radical to the disulfide moiety. Upon irradiation of K-PHI with higher energy photons (410 or 465 nm), electron excitation from the valence band to the conduction band takes place, resulting in the single-electron oxidation of thioacetate to form the radical-anion of K-PHI and the radical-cation of thioacetate. Subsequently, the radical-cation decomposes, producing the corresponding alkylthiyl radical that couples with chlorine to yield sulfenyl chloride. The K-PHI radical-anion is further oxidized by molecular oxygen (O2) to generate peroxide, which subsequently reacts with hydrogen chloride (HCl) to produce chlorine gas (Cl2). Under 465 nm irradiation, the one-electron oxidation of thioacetate predominates, whereas the use of 365 nm irradiation favors a two-electron process that enables the oxidation of chloride ions (Cl−). Consequently, the 465 nm radiation promotes the activation of O2 via energy and electron transfer processes, leading to the oxidation of sulfonyl chloride to produce reactive sulfonyl chloride. Conversely, 365 or 410 nm radiation facilitates the two-electron oxidation of chloride ions by K-PHI*. When an excess of chlorine is present, overchlorination of sulfonyl chloride occurs, resulting in the formation of unstable arylsulfur trichlorides, which rapidly decompose to afford arylchlorides (the mechanism is portrayed in Fig. 24) [196].
Figure 23
 Figure 23. (a) Scope of substituted benzylamines investigated. (b) Proposed mechanism for photocatalytic oxidative coupling of benzylamines to N-benzylidene benzylamine in the presence of CO2 and H2O. Reproduced with permission [195]. Copyright 2020, American Chemical Society.
Figure 23. (a) Scope of substituted benzylamines investigated. (b) Proposed mechanism for photocatalytic oxidative coupling of benzylamines to N-benzylidene benzylamine in the presence of CO2 and H2O. Reproduced with permission [195]. Copyright 2020, American Chemical Society.Figure 24
 Figure 24. The mechanism of chromoselective conversion of thio-derivatives to sulfonyl chlorides, arylchlorides, and disulfides. Reproduced with permission [196]. Copyright 2022, Wiley-VCH.
Figure 24. The mechanism of chromoselective conversion of thio-derivatives to sulfonyl chlorides, arylchlorides, and disulfides. Reproduced with permission [196]. Copyright 2022, Wiley-VCH.Furthermore, K-PHI exhibits versatile applications in various organic reactions, including photocatalytic oxidative halogenation of electron-rich aromatic compounds, oxidative thiolation of methyl aromatics, oxidation of benzyl alcohol, and synthesis of 1,4-dihydropyridines and 1,3,4-oxadiazoles. These notable reactions have been extensively discussed in several review articles authored by Aleksandr Savateev and Markus Antoniett, exemplifying the remarkable advantages offered by PHI-mediated organic transformations [115,147,197]. The performance of these reactions is comparable to that of transition metal catalysts under similar conditions, while providing a cost-effective alternative.
6.4 Photocatalytic biomass conversion
Photocatalytic reactions play a crucial role in facilitating the conversion of biomass and are integral to the promotion of a low carbon economy and sustainable energy systems [198,199]. In their recent study, Nimbalkar et al. conducted ionothermal treatment of various isoforms of PCN in KSCN, revealing that K-PHI-450 derived from melem exhibited exceptional activity in cellulose conversion (Fig. 25a). This superior performance can be attributed to K-PHI-450 possessing optimal crystallite size, minimal defective states, and the highest cyanamide content among the tested isoforms. These structural characteristics contribute to enhanced charge separation and electron trapping, promoting efficient catalyst and reactant interactions. Under visible light and 1 solar irradiation, K-PHI-450 demonstrates stable conversion of cellulose into H2 (34 µmol/h) and a diverse range of chemicals (λ = 420 nm, AQY of 10.4%). Specifically, the conversions of cellulose reforming into hydrogen were found to be 6.64% and 13.1%, respectively. K-PHI-450 follows a sequential process for photocatalytic reforming of cellulose-based carboxylic acids and aldoses/aldehydes/alcohols, involving hydrolysis and photocatalytic oxidation reactions. Ultimately, this photocatalytic process leads to the production of carbonates and o-C1 as the final products. The resulting reformed chemicals encompass cellulose-derived carboxylic acids (C6~C1) and aldehydes/aldehydes/alcohols (r-C5~r-C1). Further investigation into the mechanism of photocatalytic reforming elucidated that photogenerated holes are directly transferred to cellulose-derived oxygenates, while photogenerated electrons are reduced by Pt co-catalysts to produce H2 [200]. In contrast, conventional PCN materials suffer from low catalytic efficiency due to their limited surface area, impeding access to the active site. To address this issue, Krivtsov et al. developed water-soluble K, Na-PHI through low-temperature thermal treatment of melamine with molten salts (KOH/NaOH). The water-soluble K, Na-PHI catalyst exhibits excellent selectivity, reducing lignocellulosic compounds such as 4-methoxybenzyl alcohol (4MBA) to 4-methoxybenzaldehyde (4MBAL) while simultaneously reducing O2 to H2O2. Moreover, the use of benzyl alcohol and lignocellulosic feedstock derived from sources such as ethanol, glycerol, and glucose further enhance the rate of H2O2 generation. The water solubility of K, Na-PHI is attributed to the introduction of polar cyamelurate moieties through the substitution of terminal NHx-groups with KOH/NaOH. These negatively charged surface functional groups preferentially interact with water, forming an effective hydration layer on the surface of K, Na-PHI nanoparticles. Consequently, the formation of hydrogen bonds between heptazine units is inhibited, preventing coalescence and precipitation of K, Na-PHI. Notably, the water-soluble K, Na-PHI photocatalyst can be easily recovered by increasing the ionic strength of the solution, facilitating coagulation and precipitation by introducing NaCl (Fig. 25b) [201].
Figure 25
 Figure 25. (a) Schematic diagram of photocatalytic reforming of cellulose to H2 and biochemicals under visible light by K-PHI. Reproduced with permission [200]. Copyright 2022, Elsevier. (b) Schematic representation of the synthesis procedure resulting in a highly active water-soluble and recoverable PCN photocatalyst. Reproduced with permission [201]. Copyright 2019, Wiley-VCH.
Figure 25. (a) Schematic diagram of photocatalytic reforming of cellulose to H2 and biochemicals under visible light by K-PHI. Reproduced with permission [200]. Copyright 2022, Elsevier. (b) Schematic representation of the synthesis procedure resulting in a highly active water-soluble and recoverable PCN photocatalyst. Reproduced with permission [201]. Copyright 2019, Wiley-VCH.6.5 Photocatalytic pollutant degradation
Carbon nitride is a widely employed catalyst for the degradation of pollutants owing to its non-toxic nature and remarkable stability. The utilization of PCN and reactants leads to the generation of reactive oxygen species, namely singlet oxygen (1O2), hydroxyl radicals (•OH), and superoxide radicals (•O2−), which effectively eliminate NOx pollutants [138]. In a recent study by Zhou et al., a novel variant of carbon nitride, namely CN—NaLi, was successfully synthesized through the use of simple molten salts, resulting in expanded interlayer distances (Fig. 26a). Under visible light irradiation, CN—NaLi exhibited exceptional efficiency in NO removal, as evidenced by the attainment of an 81.1% removal rate within a 30-min irradiation period (Fig. 26b). Further investigations revealed that CN—NaLi possesses a high desorption temperature for NO, indicating its favorable diffusion properties and low activation barriers for NO. Notably, the presence of Na+ ions in CN—NaLi serves to modulate the energy band structure of carbon nitride, promoting the direct oxidation of NO to NO3− and facilitating the reduction of oxygen to reactive oxygen species such as superoxide radicals and hydrogen peroxide. Experimental results from species trapping techniques identified electron, hole, and superoxide radicals as the primary reactive species involved in the photocatalytic processes of CN—NaLi (Figs. 26d and e). Additionally, electron spin resonance (ESR) spectroscopic observations demonstrated a substantial increase in the production of superoxide radicals (•O2 −) on CN—NaLi, thereby ensuring a continuous supply of reactive species and enhancing charge dynamics (Fig. 26c). Furthermore, the presence of additional holes facilitated the conversion of NO to NO+, thereby contributing to an augmented photocatalytic performance [202]. It is noteworthy that the treatment of pollutants such as pharmaceuticals, pesticides, and organic dyes represents a significant challenge due to their toxic nature and difficult management. In this regard, Chen et al. successfully polymerized melamine with various alkali metal chlorides, resulting in the formation of different derivatives, including polycrystalline Na-CN through the reaction with sodium chloride. Na-CN exhibited a degradation rate for Rhodamine B that was 18 times higher than that of g-CN, with holes and oxygen molecules identified as the primary active substances responsible for Rhodamine B degradation [86]. Yang et al. have successfully incorporated a minute quantity of 1-methyl-1H-tetrazole-5-thiol (MTT) into PHI in order to facilitate the degradation of pharmaceutical contaminants and the simultaneous generation of H2O2 (Fig. 27a). The inclusion of MTT has resulted in a reduction in the layer-stacked structure size, an enhancement in the polymerization of the heptazine planes, and an improvement in the surface electron transfer process. Furthermore, there has been an observed negative shift in the conduction band position, which has consequently amplified the reduction capability of the photocatalyst and promoted the occurrence of the O2 reduction reaction. During the examination of photocatalytic degradation of pharmaceutical pollutants, K-PHI-2 effectively eliminated 20 ppm carbamazepine in just 90 min and concurrently produced H2O2 (Figs. 27b and c). Moreover, the presence of 5 ppm O3 in the reaction system facilitated the rapid elimination of ciprofloxacin, which had initially been present at a concentration of 100 ppm, via the peroxynone reaction (Fig. 27d) [203].
Figure 26
 Figure 26. (a) Schematic of NO removal for melon-CN and CN—NaLi. (b) NO removal performance of CN and CN—NaLi. (c) ESR spectra of the spin-reactive •O2− generated by CN and CN—NaLi with visible light. Comparison of photocatalytic activities of (d) CN and (e) CN—NaLi in different capture agents under visible-light-irradiation (λ > 420 nm). Reproduced with permission [202]. Copyright 2022, Elsevier.
Figure 26. (a) Schematic of NO removal for melon-CN and CN—NaLi. (b) NO removal performance of CN and CN—NaLi. (c) ESR spectra of the spin-reactive •O2− generated by CN and CN—NaLi with visible light. Comparison of photocatalytic activities of (d) CN and (e) CN—NaLi in different capture agents under visible-light-irradiation (λ > 420 nm). Reproduced with permission [202]. Copyright 2022, Elsevier.Figure 27
 Figure 27. (a) Schematic diagram of K-PHI photocatalytic pollutant degradation and H2O2 production. (b) K-PHI photocatalytic H2O2 production performance. (c) Photocatalytic H2O2 production with the degradation of carbamazepine (CBZ). (d) Photocatalytic degradation of ciprofloxacin in the presence of O3. Reproduced with permission [203]. Copyright 2022, American Chemical Society.
Figure 27. (a) Schematic diagram of K-PHI photocatalytic pollutant degradation and H2O2 production. (b) K-PHI photocatalytic H2O2 production performance. (c) Photocatalytic H2O2 production with the degradation of carbamazepine (CBZ). (d) Photocatalytic degradation of ciprofloxacin in the presence of O3. Reproduced with permission [203]. Copyright 2022, American Chemical Society.6.6 Photocatalytic preparation of H2O2
Hydrogen peroxide (H2O2), a valuable chemical feedstock with a wide range of applications including environmental remediation, textile whitening, paper bleaching, and chemical synthesis, is recognized for its environmentally friendly, active, and safe properties [204]. The conventional anthraquinone process, known for its high energy consumption, is being challenged by the sustainable and straightforward pathway of producing H2O2 from atmospheric O2 through photocatalytic oxygen reduction reactions [205,206]. As an illustration, Rogolino et al. conducted a study on the synthesis of metal-doped PHIs (Fe-PHIs, Ni-PHIs, Co-PHIs, Ru-PHIs) and protonated PHIs through cation-exchange using NaPHI as a precursor. Cation substitution led to modifications in the layer spacing and enhanced visible light absorption of the PHIs. The conduction bands of all catalysts, except Co-PHIs, exhibited potentials more positive than the O2/HOO− reduction potential, thereby suppressing the theoretical 1e− ORR and favoring the generation of H2O2 via the 2e− ORR. By utilizing glycerol as a sacrificial electron donor and 410 nm irradiation, the protonated H-PHIs achieved O2 to H2O2 conversion rates reaching up to 1556 mmol L−1 h−1 g−1 (Figs. 28a and b). The improved catalytic performance of the H-PHIs can be attributed to their higher proton content, which facilitates the release of H2O2 [207]. To address challenges related to the decomposition and disproportionation of photocatalytically produced H2O2 on the catalyst surface, Krivtsov et al. employed hydrophobic PHI in a biphasic system for photocatalysis. This approach effectively mitigated H2O2 decomposition on the catalyst surface, resulting in a high concentration of H2O2 in the aqueous phase (~0.12 mol/L). The surface modification of suspended particles with dispersed silica through a sol-gel process, followed by silylation, rendered the composites hydrophobic. Under visible light irradiation, the hydrophobic PHI-based photocatalyst utilized fatty alcohol as an electron donor, leading to the reduction of oxygen to H2O2, which was promptly extracted into the aqueous phase. Consequently, the continuously separated H2O2 effectively prevented its decomposition on the photocatalyst surface. Furthermore, hydrophobic PHI exhibited a higher protonation efficiency, thereby promoting H2O2 generation in the biphasic system (Fig. 28c) [208].
Figure 28
 Figure 28. (a) Proposed general mechanism of photocatalysed ORR on carbon nitrides. (b) Comparison of different M-PHIs in photocatalytic H2O2 production. Reproduced with permission [207]. Copyright 2022, American Chemical Society. (c) Concept of biphasic photocatalysis using hydrophobized PHI. Reproduced with permission [208]. Copyright 2023, the Royal Society of Chemistry.
Figure 28. (a) Proposed general mechanism of photocatalysed ORR on carbon nitrides. (b) Comparison of different M-PHIs in photocatalytic H2O2 production. Reproduced with permission [207]. Copyright 2022, American Chemical Society. (c) Concept of biphasic photocatalysis using hydrophobized PHI. Reproduced with permission [208]. Copyright 2023, the Royal Society of Chemistry.6.7 Photocatalytic N2 reduction reactions
Nitrogen reduction for the synthesis of ammonia is a highly significant and challenging chemical conversion process with wide applications in modern industry and agriculture. In recent years, the use of photocatalytic N2 reduction reactions (PCNRR) has emerged as a promising, sustainable, and cost-effective strategy for direct NH3 production under mild conditions [209]. Carbon nitride-based materials, particularly those containing highly activated pyridine-like N sites, have shown tremendous potential for PCNRR [210]. Among them, PHI/PTI, a popular material in the PCN family, has attracted significant attention due to its exceptional photocatalytic nitrogen fixation performance.
In a comparative study by Li et al., various alkali metal-containing crystalline carbon nitrides were systematically prepared using different combinations of alkali metal chlorides (Fig. 29). Crystalline carbon nitride, especially the PHI-phase LK-HTCN, exhibited significantly higher ammonia synthesis rates compared to conventional thermally polymerized BCN. This enhanced performance can be attributed to the presence of a substantial number of cyano groups in the KN—HCN structure and the loading mode of potassium and sodium ions, which facilitate shorter photogenerated electron transfer paths and promote the migration and separation of photogenerated carriers for efficient photocatalytic nitrogen fixation (Fig. 30a) [211].
Figure 29
 Figure 29. The synthesis process of the different types of carbon nitride. Reproduced with permission [211]. Copyright 2022, the Royal Society of Chemistry.
Figure 29. The synthesis process of the different types of carbon nitride. Reproduced with permission [211]. Copyright 2022, the Royal Society of Chemistry.Figure 30
 Figure 30. (a) Photocatalytic N2 reduction mechanism schemes of KN—HCN. Reproduced with permission [211]. Copyright 2022, the Royal Society of Chemistry. (b) Schematic for the synthesis of PCON. (c) Mechanism for the photocatalytic N2 fixation over PCON-2. (d) Piezo-photocatalytic nitrogen reduction mechanism over DKCN. Reproduced with permission [127]. Copyright 2022, Elsevier.
Figure 30. (a) Photocatalytic N2 reduction mechanism schemes of KN—HCN. Reproduced with permission [211]. Copyright 2022, the Royal Society of Chemistry. (b) Schematic for the synthesis of PCON. (c) Mechanism for the photocatalytic N2 fixation over PCON-2. (d) Piezo-photocatalytic nitrogen reduction mechanism over DKCN. Reproduced with permission [127]. Copyright 2022, Elsevier.Further structural modification of PHI has been explored to enhance its photocatalytic nitrogen fixation performance. For instance, Yang et al. employed a monomer-directed co-polymerization strategy to prepare PHI (PCON) with C—N and N—C-O units connected to terminal melem units (Fig. 30b). The optimized PCON demonstrated excellent photocatalytic nitrogen fixation rates and near-infrared yield in pure water without the use of co-catalysts. The introduction of embedded C—N and N–C-O units in the photocatalyst enabled near-infrared optical response and provided channels for the separation of photogenerated carriers. The embed linker and heptazine unit also served as active sites for N2 and OH−, respectively, further improving the nitrogen fixation efficiency (Fig. 30c) [127].
Interestingly, the same research team utilized PHI as a piezoelectric photocatalyst for piezoelectric photocatalytic nitrogen hydrogenation. By encapsulating the benzene-1,4-dicarboxylic acid dihydrazide unit in PHI (DKCN) through a co-polymerization method, DKCN demonstrated excellent performance in piezoelectric photocatalytic reduction of N2 to NH3 in pure water without the need for co-catalysts or sacrificial agents. The introduction of linkages and the D-A unit redistributed charge centers and altered the bandgap, resulting in enhanced near-infrared light absorption, increased in-plane polarisation of PHI, and the provision of active sites. The piezoelectric potential generated by ultrasound-induced stress created a built-in electric field that polarized the charge and facilitated the directional migration of photogenerated carriers (Fig. 30d) [212].
These studies highlight the tremendous potential of carbon nitride-based materials, especially PHI/PTI, in promoting efficient and sustainable photocatalytic nitrogen fixation reactions. Further research and exploration of structural modifications hold significant promise for achieving even higher performance in this field.
7. Conclusion
In summary, PHI and PTI are introduced herein, focusing on their structural characteristics, characterization methods, optoelectronic properties, structure optimization, and applications. Both PHI and PTI demonstrate notable advantages over other carbon nitride variants. The well-defined structure and high crystallinity of PHI and PTI offer exemplary models for investigating the photocatalytic mechanisms of organic semiconductors. The 2D structure of PHI and PTI, with minimal defects, facilitates efficient photogenerated charge carrier transfer, lower rates of electron-hole recombination, and enhanced visible light absorption. Notably, the higher valence band potential of PHI and PTI enhances their ability to drive photocatalytic oxidation reactions effectively.
However, several challenges pertaining to PHI and PTI warrant attention: (1) In-depth investigations on the charge storage mechanism of PHI are lacking, as the underlying structure governing the kinetics of electron accumulation and transfer remains ambiguous. Moreover, there is a need to establish the upper limit capacity of stored charge and its relationship with the structural features. (2) The mechanism underlying the formation of PTI in molten salts, which differs from conventional thermal polymerization leading to PCN formation, remains poorly understood. (3) The preparation of PTI in molten salts without the inclusion of Li salt compositions has not been achieved, and the indispensability of Li salt in PTI synthesis requires clarification. (4) The synthesis of highly crystalline PTI mainly relies on sealed ampoules, which pose safety risks and operational complexities. Exploring alternative methods for the preparation of highly crystalline PTI is thus warranted. (5) Despite the thermodynamic stability of the heptazine phase and its larger π-conjugated system, there is a relative dearth of research on the photocatalytic properties of triazine-based PTI. Nevertheless, the well-resolved structure of PTI provides a favorable platform for exploring its photocatalytic mechanisms, offering potential avenues for future research and applications. In practical settings, the synthesis of PHI/PTI to meet industrial-scale production needs is hindered by the limitations of preparation via the molten salt approach. Furthermore, an important factor that must be taken into account is the stability of PHI/PTI, particularly concerning the loss of alkali metal ions within the structure during the photocatalytic process, which ultimately leads to a decline in performance. In addition to these challenges, the economic feasibility of PHI/PTI is a crucial consideration, and there is a need for further enhancements in the photocatalytic efficiency of PHI/PTI. The academic community is encouraged to address these issues to further our understanding and utilization of PHI and PTI as exceptional photocatalytic materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No. 52273264).
-
-
[1]
M. Farghali, A.I. Osman, I.M.A. Mohamed, et al., Environ. Chem. Lett. 21 (2023) 2003–2039. doi: 10.1007/s10311-023-01591-5
-
[2]
L. Zhou, H. Zhang, H. Sun, et al., Catal. Sci. Technol. 6 (2016) 7002–7023. doi: 10.1039/C6CY01195K
-
[3]
C. Zhang, Y.L. Xie., H.X. Zhang, Y.J. Gu, X.G. Zhang, Energy 262 (2023) 125453. doi: 10.1016/j.energy.2022.125453
-
[4]
N. Yan, C. Zhao, S. You, Y. Zhang, W. Li, Chin. Chem. Lett. 31 (2020) 643–653. doi: 10.1016/j.cclet.2019.08.022
-
[5]
L. Zhan, S. Li, Y. Li, et al., Adv. Energy Mater. 12 (2022) 2201076. doi: 10.1002/aenm.202201076
-
[6]
K. Wang, P. Yang, R. Guo, X. Yao, W. Yang, Chin. Chem. Lett. 30 (2019) 2013–2016. doi: 10.1016/j.cclet.2019.04.005
-
[7]
M. Chang, Z. Hou, M. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 12971–12979. doi: 10.1002/anie.202101924
-
[8]
C. Aydogan, G. Yilmaz, A. Shegiwal, D.M. Haddleton, Y. Yagci, Angew. Chem., Int. Ed. 61 (2022) e202117377. doi: 10.1002/anie.202117377
-
[9]
U. Raucci, H. Weir, C. Bannwarth, D.M. Sanchez, T.J. Martinez, Nat. Commun. 13 (2022) 2091. doi: 10.1038/s41467-022-29662-1
-
[10]
W. Wang, Y. Tao, J. Fan, et al., Adv. Funct. Mater. 32 (2022) 2201357. doi: 10.1002/adfm.202201357
-
[11]
X.P. Li, L.R. Zheng, S.J. Liu, et al., Chin. Chem. Lett. 33 (2022) 4761–4765. doi: 10.1016/j.cclet.2021.12.095
-
[12]
Q. Li, J.N. Chang, Z. Wang, et al., J. Am. Chem. Soc. 145 (2023) 23167–23175. doi: 10.1021/jacs.3c07471
-
[13]
L. Ran, Z. Li, B. Ran, et al., J. Am. Chem. Soc. 144 (2022) 17097–17109. doi: 10.1021/jacs.2c06920
-
[14]
L. Xu, L. Li, L. Yu, J.C. Yu, Chem. Eng. J. 431 (2022) 134241. doi: 10.1016/j.cej.2021.134241
-
[15]
T. Xia, Y. Lin, W. Li, M. Ju, Chin. Chem. Lett. 32 (2021) 2975–2984. doi: 10.1016/j.cclet.2021.02.058
-
[16]
A. Vijeta, C. Casadevall, E. Reisner, Angew. Chem. Int. Ed. 61 (2022) e202203176. doi: 10.1002/anie.202203176
-
[17]
T.Y. Yu, Q. Niu, Y. Chen, et al., J. Am. Chem. Soc. 145 (2023) 8860–8870. doi: 10.1021/jacs.2c12313
-
[18]
Y. Jiang, Z. Xiong, J. Huang, et al., Chin. Chem. Lett. 33 (2022) 415–423. doi: 10.1016/j.cclet.2021.06.058
-
[19]
Z. Liang, H. Wang, K. Zhang, et al., Chem. Eng. J. 428 (2022) 131349. doi: 10.1016/j.cej.2021.131349
-
[20]
A. Fujishima, K. Honda, Nature 238 (1972) 37–38. doi: 10.1038/238037a0
-
[21]
F. Wang, S.X. Min, Chin. Chem. Lett. 18 (2007) 1273–1277. doi: 10.1016/j.cclet.2007.08.010
-
[22]
Y. Zhang, J. Zhao, H. Wang, et al., Nat. Commun. 13 (2022) 58. doi: 10.1038/s41467-021-27698-3
-
[23]
S. Huang, B.F. Zheng, Z.Y. Tang, et al., Chem. Eng. J. 422 (2021) 130086. doi: 10.1016/j.cej.2021.130086
-
[24]
H. Wang, J. Liu, X. Xiao, et al., Chin. Chem. Lett. 34 (2023) 107125. doi: 10.1016/j.cclet.2022.01.018
-
[25]
L. Hao, H. Huang, Y. Zhang, T. Ma, Adv. Funct. Mater. 31 (2021) 2100919. doi: 10.1002/adfm.202100919
-
[26]
N. Zhang, H. Zuo, C. Xu, et al., Chin. Chem. Lett. 31 (2020) 337–340. doi: 10.1016/j.cclet.2019.06.008
-
[27]
H. Zhang, T. Itoi, T. Konishi, Y. Izumi, Angew. Chem. Int. Ed. 60 (2021) 9045–9054. doi: 10.1002/anie.202016346
-
[28]
S. Zhang, Z. Zhang, B. Li, et al., J. Colloid Interface Sci. 586 (2021) 708–718. doi: 10.1016/j.jcis.2020.10.140
-
[29]
K. Saravanakumar, C.M. Park, Chem. Eng. J. 423 (2021) 130076. doi: 10.1016/j.cej.2021.130076
-
[30]
I. Mahboob, I. Shafiq, S. Shafique, et al., Chem. Eng. J. 441 (2022) 136063. doi: 10.1016/j.cej.2022.136063
-
[31]
L. Liu, T. Hu, K. Dai, J. Zhang, C. Liang, Chin. J. Catal. 42 (2021) 46–55. doi: 10.1117/12.2604945
-
[32]
Y. Zhang, Y. Zhao, Z. Xiong, et al., Appl. Catal. B 282 (2021) 119534. doi: 10.1016/j.apcatb.2020.119534
-
[33]
H. Nie, K. Wei, Y. Li, et al., Chin. Chem. Lett. 32 (2021) 2283–2286. doi: 10.1016/j.cclet.2021.01.041
-
[34]
Y. Li, Y. Liu, D. Xing, et al., Appl. Catal. B 285 (2021) 119855. doi: 10.1016/j.apcatb.2020.119855
-
[35]
D. Dai, X. Liang, B. Zhang, et al., Adv. Sci. 9 (2022) 2105299. doi: 10.1002/advs.202105299
-
[36]
W. Zhong, X. Wu, Y. Liu, et al., Appl. Catal. B 280 (2021) 119455. doi: 10.1016/j.apcatb.2020.119455
-
[37]
M. Wang, J. Cheng, X. Wang, et al., Chin. J. Catal. 42 (2021) 37–45. doi: 10.1016/S1872-2067(20)63633-6
-
[38]
J. Li, Z. Zhao, Z. Li, et al., Chin. Chem. Lett. 33 (2022) 3705–3708. doi: 10.1016/j.cclet.2021.10.080
-
[39]
Y. Liu, W. Yang, Q. Chen, et al., J. Am. Chem. Soc. 144 (2022) 2705–2715. doi: 10.1021/jacs.1c11745
-
[40]
C. Tang, M. Cheng, C. Lai, et al., Coord. Chem. Rev. 474 (2023) 214846. doi: 10.1016/j.ccr.2022.214846
-
[41]
J. Xue, S. Ma, Y. Zhou, Z. Zhang, X. Liu, RSC Adv. 5 (2015) 58738–58745. doi: 10.1039/C5RA09978A
-
[42]
Y. Wang, X. Wang, M. Antonietti, Angew. Chem. Int. Ed. 51 (2012) 68–89. doi: 10.1002/anie.201101182
-
[43]
S. Zhang, Y. Yang, Y. Zhai, et al., Chin. Chem. Lett. 34 (2023) 107652. doi: 10.1016/j.cclet.2022.06.075
-
[44]
M. Yang, R. Lian, X. Zhang, et al., Nat. Commun. 13 (2022) 4900. doi: 10.1038/s41467-022-32623-3
-
[45]
E.G. Gillan, Chem. Mater. 12 (2000) 3906–3912. doi: 10.1021/cm000570y
-
[46]
Y. Wang, L. Liu, T. Ma, Y. Zhang, H. Huang, Adv. Funct. Mater. 31 (2021) 2102540. doi: 10.1002/adfm.202102540
-
[47]
S. Cao, J. Low, J. Yu, M. Jaroniec, Adv. Mater. 27 (2015) 2150–2176. doi: 10.1002/adma.201500033
-
[48]
J. Zander, J. Timm, M. Weiss, R. Marschall, Adv. Energy Mater. 12 (2022) 2202403. doi: 10.1002/aenm.202202403
-
[49]
Y. Xu, M. Fan, W. Yang, et al., Adv. Mater. 33 (2021) 2101455. doi: 10.1002/adma.202101455
-
[50]
Z. Zhou, Y. Zhang, Y. Shen, S. Liu, Y. Zhang, Chem. Soc. Rev. 47 (2018) 2298–2321. doi: 10.1039/c7cs00840f
-
[51]
B. Fan, L. Xing, Q. He, et al., Chem. Eng. J. 435 (2022) 135086. doi: 10.1016/j.cej.2022.135086
-
[52]
C. Cheng, J. Shi, L. Mao, et al., J. Colloid Interface Sci. 637 (2023) 271–282. doi: 10.1016/j.jcis.2023.01.098
-
[53]
Y. Li, D. Zhang, J. Fan, Q. Xiang, Chin. J. Catal. 42 (2021) 627–636. doi: 10.1016/S1872-2067(20)63684-1
-
[54]
J. Kroeger, A. Jimenez-Solano, G. Savasci, et al., Adv. Funct. Mater. 31 (2021) 2102468. doi: 10.1002/adfm.202102468
-
[55]
Y. Wang, F. He, L. Chen, et al., Chin. Chem. Lett. 31 (2020) 2668–2672. doi: 10.1016/j.cclet.2020.08.003
-
[56]
B. Qu, P. Li, L. Bai, et al., Adv. Mater. 35 (2023) 2211575. doi: 10.1002/adma.202211575
-
[57]
X. Zhang, X.R. Zhang, P. Yang, H.S. Chen, S.P. Jiang, Chem. Eng. J. 450 (2022) 138030. doi: 10.1016/j.cej.2022.138030
-
[58]
Y. Gu, T. Xu, X. Chen, W. Chen, W. Lu, Chem. Eng. J. 427 (2022) 131973. doi: 10.1016/j.cej.2021.131973
-
[59]
Y. Shiraishi, S. Kanazawa, Y. Sugano, et al., ACS Catal. 4 (2014) 774–780. doi: 10.1021/cs401208c
-
[60]
Y. Shiraishi, Y. Kofuji, H. Sakamoto, et al., ACS Catal. 5 (2015) 3058–3066. doi: 10.1021/acscatal.5b00408
-
[61]
X. Zong, L. Niu, W. Jiang, et al., Appl. Catal. B 291 (2021) 120099. doi: 10.1016/j.apcatb.2021.120099
-
[62]
Y. Ma, F. Liu, Y. Liu, et al., Chem. Eng. J. 414 (2021) 128802. doi: 10.1016/j.cej.2021.128802
-
[63]
M. Ran, P. Chen, J. Li, et al., Chin. Chem. Lett. 30 (2019) 875–880. doi: 10.1016/j.cclet.2019.03.016
-
[64]
J. Xu, Y. Chen, M. Chen, J. Wang, L. Wang, Chem. Eng. J. 442 (2022) 136208. doi: 10.1016/j.cej.2022.136208
-
[65]
L. Duan, G. Li, S. Zhang, et al., Chem. Eng. J. 411 (2021) 128551. doi: 10.1016/j.cej.2021.128551
-
[66]
X. Yu, T. Fan, W. Chen, et al., Carbon 144 (2019) 649–658. doi: 10.1016/j.carbon.2018.12.104
-
[67]
C. Xu, H. Liu, D. Wang, et al., Appl. Catal. B 334 (2023) 122835. doi: 10.1016/j.apcatb.2023.122835
-
[68]
X. Wu, R. Zhong, X. Lv, et al., Appl. Catal. B 330 (2023) 122666. doi: 10.1016/j.apcatb.2023.122666
-
[69]
Y. Zhang, S. Zong, C. Cheng, et al. Appl. Catal. B 233 (2018) 80–87. doi: 10.1016/j.apcatb.2018.03.104
-
[70]
L. Lin, Z. Yu, X. Wang, Angew. Chem. Int. Ed. 58 (2019) 6164–6175. doi: 10.1002/anie.201809897
-
[71]
S. Zhao, Y. Zhang, Y. Zhou, et al., Carbon 126 (2018) 247–256. doi: 10.1016/j.carbon.2017.10.033
-
[72]
Y. Yu, W. Xu, J. Fang, et al., Appl. Catal. B 268 (2020) 118751. doi: 10.1016/j.apcatb.2020.118751
-
[73]
X. Li, D. Chen, N. Li, et al., Appl. Catal. B 229 (2018) 155–162. doi: 10.1016/j.apcatb.2018.02.028
-
[74]
S.U. Lee, Y.S. Jun, E.Z. Lee, et al., Carbon 95 (2015) 58–64. doi: 10.5050/KSNVE.2015.25.1.058
-
[75]
B. Wu, L. Zhang, B. Jiang, et al., Angew. Chem. Int. Ed. 60 (2021) 4815–4822. doi: 10.1002/anie.202013753
-
[76]
S. Guo, Z. Deng, M. Li, et al., Angew. Chem. Int. Ed. 55 (2016) 1830–1834. doi: 10.1002/anie.201508505
-
[77]
C. Zhang, P. Ni, B. Wang, et al., Chin. Chem. Lett. 33 (2022) 757–761. doi: 10.1016/j.cclet.2021.08.017
-
[78]
Y. Xiao, G. Tian, W. Li, et al., J. Am. Chem. Soc. 141 (2019) 2508–2515. doi: 10.1021/jacs.8b12428
-
[79]
X.H. Jiang, F. Yu, D.S. Wu, et al., Chin. Chem. Lett. 32 (2021) 2782–2786. doi: 10.1016/j.cclet.2021.01.011
-
[80]
Z. Wang, E. Almatrafi, H. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202202338. doi: 10.1002/anie.202202338
-
[81]
F. Bi, Y. Su, Y. Zhang, et al., Appl. Catal. B 306 (2022) 121109. doi: 10.1016/j.apcatb.2022.121109
-
[82]
H. Li, Q. Qing, L. Zheng, et al., Chin. Chem. Lett. 33 (2022) 3573–3576. doi: 10.1016/j.cclet.2022.01.050
-
[83]
X. Wang, J. Meng, X. Zhang, et al., Adv. Funct. Mater. 31 (2021) 2010763. doi: 10.1002/adfm.202010763
-
[84]
L. Chen, Y. Wang, S. Cheng, et al., Appl. Catal. B 303 (2022) 120932. doi: 10.1016/j.apcatb.2021.120932
-
[85]
M.J. Bojdys, J.O. Mueller, M. Antonietti, A. Thomas, Chem. Eur. J. 14 (2008) 8177–8182. doi: 10.1002/chem.200800190
-
[86]
Z. Chen, A. Savateev, S. Pronkin, et al., Adv. Mater. 29 (2017) 1700555. doi: 10.1002/adma.201700555
-
[87]
D. Dontsova, S. Pronkin, M. Wehle, et al., Chem. Mater. 27 (2015) 5170–5179. doi: 10.1021/acs.chemmater.5b00812
-
[88]
Z. Liao, C. Li, Z. Shu, et al., Int. J. Hydrogen Energy 46 (2021) 26318–26328. doi: 10.1016/j.ijhydene.2021.05.138
-
[89]
E. Wirnhier, M. Doeblinger, D. Gunzelmann, et al., Chem. Eur. J. 17 (2011) 3213–3221. doi: 10.1002/chem.201002462
-
[90]
M. Liu, C. Wei, H. Zhuzhang, et al., Angew. Chem., Int. Ed. 61 (2022) e202113389. doi: 10.1002/anie.202113389
-
[91]
V.W.h. Lau, B.V. Lotsch, Adv. Energy Mater. 12 (2022) 2101078. doi: 10.1002/aenm.202101078
-
[92]
F.K. Kessler, Y. Zheng, D. Schwarz, et al., Nat. Rev. Mater. 2 (2017) 17030. doi: 10.1038/natrevmats.2017.30
-
[93]
J. Lieblg, Ann. Pharm. 10 (1834) 1–47. doi: 10.1002/jlac.18340100102
-
[94]
J. Liebig, Justus Liebigs Ann. Chem. 53 (1845) 330–348. doi: 10.1002/jlac.18450530303
-
[95]
J. Liebig, Justus Liebigs Ann. Chem. 95 (1855) 257–282. doi: 10.1002/jlac.18550950302
-
[96]
L. Gmelin, Ann. Pharm. 15 (1835) 252–258. doi: 10.1002/jlac.18350150306
-
[97]
E.C. Franklin, J. Am. Chem. Soc. 44 (1922) 486–509. doi: 10.1021/ja01424a007
-
[98]
L. Pauling, J.H. Sturdivant, Proc. Natl. Acad. Sci. U. S. A. 23 (1937) 615–620. doi: 10.1073/pnas.23.12.615
-
[99]
C.E. Redemann, H.J. Lucas, J. Am. Chem. Soc. 62 (1940) 842–846. doi: 10.1021/ja01861a038
-
[100]
M.L. Cohen, Science 261 (1993) 307–308. doi: 10.1126/science.261.5119.307
-
[101]
D.M. Teter, R.J. Hemley, Science 271 (1996) 53–55. doi: 10.1126/science.271.5245.53
-
[102]
A.Y. Liu, R.M. Wentzcovitch, Phys. Rev. B 50 (1994) 10362–10365. doi: 10.1103/PhysRevB.50.10362
-
[103]
E. Kroke, M. Schwarz, E. Horath-Bordon, et al., New J. Chem. 26 (2002) 508–512. doi: 10.1039/b111062b
-
[104]
B. Jurgens, E. Irran, J. Senker, et al., J. Am. Chem. Soc. 125 (2003) 10288–10300. doi: 10.1021/ja0357689
-
[105]
F. Goettmann, A. Fischer, M. Antonietti, A. Thomas, Angew. Chem. Int. Ed. 45 (2006) 4467–4471. doi: 10.1002/anie.200600412
-
[106]
F. Goettmann, A. Fischer, M. Antonietti, A. Thomas, Chem. Commun. (2006) 4530–4532.
-
[107]
M. Doblinger, B.V. Lotsch, J. Wack, et al., Chem. Commun. (2009) 1541–1543. doi: 10.1039/b820032g
-
[108]
Y. Ham, K. Maeda, D. Cha, K. Takanabe, K. Domen, Chem. Asian J. 8 (2013) 218–224. doi: 10.1002/asia.201200781
-
[109]
H. Schlomberg, J. Kroger, G. Savasci, et al., Chem. Mater. 31 (2019) 7478–7486. doi: 10.1021/acs.chemmater.9b02199
-
[110]
M. Liu, G. Zhang, X. Liang, et al., Angew. Chem. Int. Ed. 62 (2023) e202304694. doi: 10.1002/anie.202304694
-
[111]
J. Wang, S. Wang, Coord. Chem. Rev. 453 (2022) 214338. doi: 10.1016/j.ccr.2021.214338
-
[112]
B.V. Lotsch, M. Döblinger, J. Sehnert, et al., Chem. Eur. J. 13 (2007) 4969–4980. doi: 10.1002/chem.200601759
-
[113]
P.S. Wu, T.J. Lin, S.S. Hou, et al., J. Mater. Chem. A 10 (2022) 7728–7738. doi: 10.1039/d1ta10091b
-
[114]
M. Chang, Z. Pan, D. Zheng, et al., ChemSusChem 16 (2023) e202202255. doi: 10.1002/cssc.202202255
-
[115]
S. Mazzanti, A. Savateev, Chempluschem 85 (2020) 2499–2517. doi: 10.1002/cplu.202000606
-
[116]
K. Schwinghammer, B. Tuffy, M.B. Mesch, et al., Angew. Chem. Int. Ed. 52 (2013) 2435–2439. doi: 10.1002/anie.201206817
-
[117]
J.N. Burrow, R.A. Ciufo, L.A. Smith, et al., ACS Nano 16 (2022) 5393–5403. doi: 10.1021/acsnano.1c08912
-
[118]
J. Kröger, A. Jiménez-Solano, G. Savasci, et al., Adv. Energy Mater. 11 (2020) 2003016. doi: 10.1002/aenm.202003016
-
[119]
D. Burmeister, H.A. Tran, J. Muller, et al., Angew. Chem. Int. Ed. 61 (2022) e202111749. doi: 10.1002/anie.202111749
-
[120]
J. Kröger, A. Jiménez-Solano, G. Savasci, et al., Adv. Energy Mater. 11 (2021) 2003016. doi: 10.1002/aenm.202003016
-
[121]
R. Xu, D.H. Si, S.S. Zhao, et al., J. Am. Chem. Soc. 145 (2023) 8261–8270. doi: 10.1021/jacs.3c02370
-
[122]
M. Zhang, P. Huang, J.P. Liao, et al., Angew. Chem. Int. Ed. 62 (2023) e202311999. doi: 10.1002/anie.202311999
-
[123]
W. Wang, C. Zhou, Y. Yang, et al., Chem. Eng. J. 404 (2021) 126540. doi: 10.1016/j.cej.2020.126540
-
[124]
L. Lin, C. Wang, W. Ren, et al., Chem. Sci. 8 (2017) 5506–5511. doi: 10.1039/C7SC00900C
-
[125]
Z. Zhao, Z. Shu, J. Zhou, et al., J. Mater. Chem. A 10 (2022) 17668–17679. doi: 10.1039/d2ta04213d
-
[126]
W. Wang, Z. Shu, J. Zhou, et al., ACS Appl. Mater. Interfaces 14 (2022) 41131–41140. doi: 10.1021/acsami.2c12959
-
[127]
S. Yang, X. Deng, P. Chen, et al., Appl. Catal. B 311 (2022) 121370. doi: 10.1016/j.apcatb.2022.121370
-
[128]
W. Wang, Z. Shu, Z. Liao, et al., Chem. Eng. J. 424 (2021) 130332. doi: 10.1016/j.cej.2021.130332
-
[129]
J. Zhang, G. Ye, C. Zhang, et al., ChemSusChem 15 (2022) e202201616. doi: 10.1002/cssc.202201616
-
[130]
Q. Liang, Z.H. Huang, F. Kang, Q.H. Yang, ChemCatChem 7 (2015) 2897–2902. doi: 10.1002/cctc.201500076
-
[131]
C. Adler, S. Selim, I. Krivtsov, et al., Adv. Funct. Mater. 31 (2021) 2105369. doi: 10.1002/adfm.202105369
-
[132]
F. Podjaski, B.V. Lotsch, Adv. Energy Mater. 11 (2020) 2003049. doi: 10.1002/aenm.202003049
-
[133]
L. Luo, S. Wang, L. Zhang, et al., Appl. Catal., B, 343 (2024) 123475. doi: 10.1016/j.apcatb.2023.123475
-
[134]
V.W.h. Lau, D. Klose, H. Kasap, et al., Angew. Chem. Int. Ed. 56 (2017) 510–514. doi: 10.1002/anie.201608553
-
[135]
Z. Zeng, X. Quan, H. Yu, et al., Appl. Catal. B 236 (2018) 99–106. doi: 10.1016/j.apcatb.2018.05.003
-
[136]
H. Ou, C. Tang, X. Chen, M. Zhou, X. Wang, ACS Catal. 9 (2019) 2949–2955. doi: 10.1021/acscatal.9b00314
-
[137]
M.K. Bhunia, S. Melissen, M.R. Parida, et al., Chem. Mater. 27 (2015) 8237–8247. doi: 10.1021/acs.chemmater.5b02974
-
[138]
L.S. Zhang, X.H. Jiang, Z.A. Zhong, et al., Angew. Chem. Int. Ed. 60 (2021) 21751–21755. doi: 10.1002/anie.202109488
-
[139]
G. Algara-Siller, N. Severin, S.Y. Chong, et al., Angew. Chem. Int. Ed. 53 (2014) 7450–7455. doi: 10.1002/anie.201402191
-
[140]
M.B. Mesch, K. Bärwinkel, Y. Krysiak, et al., Chem. Eur. J. 22 (2016) 16878–16890. doi: 10.1002/chem.201603726
-
[141]
C.Z. Liao, V.W.h. Lau, M. Su, et al., Inorg. Chem. 58 (2019) 15880–15888. doi: 10.1021/acs.inorgchem.9b02287
-
[142]
K. Zhang, C. Liu, Q. Liu, Z. Mo, D. Zhang, Catalysts 13(4) (2023) 717. doi: 10.3390/catal13040717
-
[143]
A. Savateev, S. Pronkin, J.D. Epping, et al., J. Mater. Chem. A 5 (2017) 8394–8401. doi: 10.1039/C7TA01714F
-
[144]
N.A. Rodriguez, A. Savateev, M.A. Grela, D. Dontsova, ACS Appl. Mater. Interfaces 9 (2017) 22941–22949. doi: 10.1021/acsami.7b04745
-
[145]
A. Savateev, S. Pronkin, J.D. Epping, et al., ChemCatChem 9 (2017) 167–174. doi: 10.1002/cctc.201601165
-
[146]
G. Zhang, G. Li, T. Heil, et al., Angew. Chem., Int. Ed. 58 (2019) 3433–3437. doi: 10.1002/anie.201811938
-
[147]
A. Savateev, M. Antonietti, ChemCatChem 11 (2019) 6166–6176. doi: 10.1002/cctc.201901076
-
[148]
Y. Li, D. Zhang, X. Feng, Q. Xiang, Chin. J. Catal. 41 (2020) 21–30. doi: 10.1016/S1872-2067(19)63427-3
-
[149]
L. Lin, W. Ren, C. Wang, et al., Appl. Catal., B 231 (2018) 234–241. doi: 10.1016/j.apcatb.2018.03.009
-
[150]
C.C. Chen, J.J. Wu, ACS Appl. Energy Mater. 5 (2022) 9733–9741. doi: 10.1021/acsaem.2c01412
-
[151]
B. Zhai, H. Li, G. Gao, et al., Adv. Funct. Mater. 32 (2022) 2207375. doi: 10.1002/adfm.202207375
-
[152]
G. Zhang, L. Lin, G. Li, et al., Angew. Chem., Int. Ed. 57 (2018) 9372–9376. doi: 10.1002/anie.201804702
-
[153]
X. Liang, S. Xue, C. Yang, et al., Angew. Chem. Int. Ed. 62 (2023) e202216434. doi: 10.1002/anie.202216434
-
[154]
L. Lin, Z. Lin, J. Zhang, et al., Nat. Catal. 3 (2020) 649–655. doi: 10.1038/s41929-020-0476-3
-
[155]
X. Zhang, P. Ma, C. Wang, et al., Energy Environ. Sci. 15 (2022) 830–842. doi: 10.1039/d1ee02369a
-
[156]
L. Jiang, J. Yang, X. Yuan, et al., Adv. Colloid Interface Sci. 296 (2021) 102523. doi: 10.1016/j.cis.2021.102523
-
[157]
A. Meng, Z. Teng, Q. Zhang, C. Su, Chem. Eur. J. 15 (2020) 3405–3415. doi: 10.1002/asia.202000850
-
[158]
H. Kasap, C.A. Caputo, B.C. Martindale, et al., J. Am. Chem. Soc. 138 (2016) 9183–9192. doi: 10.1021/jacs.6b04325
-
[159]
V.W. Lau, I. Moudrakovski, T. Botari, et al., Nat. Commun. 7 (2016) 12165. doi: 10.1038/ncomms12165
-
[160]
H. Li, B. Zhu, S. Cao, J. Yu, Chem. Commun. 56 (2020) 5641–5644. doi: 10.1039/d0cc01338b
-
[161]
C.C. Chen, D.L. Tsai, H.T. Liu, J.J. Wu, ACS Sustain. Chem. Eng. 11 (2023) 6435–6444. doi: 10.1021/acssuschemeng.3c00363
-
[162]
B.X. Zhou, S.S. Ding, B.J. Zhang, et al., Appl. Catal. B 254 (2019) 321–328. doi: 10.1016/j.apcatb.2019.05.015
-
[163]
D. Liu, C. Li, C. Zhao, et al., Chem. Eng. J. 438 (2022) 135623. doi: 10.1016/j.cej.2022.135623
-
[164]
T. Huo, Q. Deng, F. Yu, et al., ACS Appl. Mater. Interfaces 14 (2022) 13419–13430. doi: 10.1021/acsami.2c01522
-
[165]
K. Schwinghammer, M.B. Mesch, V. Duppel, et al., J. Am. Chem. Soc. 136 (2014) 1730–1733. doi: 10.1021/ja411321s
-
[166]
T.S. Miller, T.M. Suter, A.M. Telford, et al., Nano. Lett. 17 (2017) 5891–5896. doi: 10.1021/acs.nanolett.7b01353
-
[167]
J. Jia, E.R. White, A.J. Clancy, et al., Angew. Chem. Int. Ed. 57 (2018) 12656–12660. doi: 10.1002/anie.201800875
-
[168]
F. Guo, B. Hu, C. Yang, et al., Adv. Mater. 33 (2021) e2101466. doi: 10.1002/adma.202101466
-
[169]
L. Tian, J. Li, F. Liang, et al., Appl. Catal. B 225 (2018) 307–313. doi: 10.1016/j.apcatb.2017.11.082
-
[170]
Y. Li, H. Wang, X. Zhang, et al., Angew. Chem. Int. Ed. 60 (2021) 12891–12896. doi: 10.1002/anie.202101090
-
[171]
Z. Zhang, Z. Pan, Y. Guo, et al., Appl. Catal. B 261 (2020) 118212. doi: 10.1016/j.apcatb.2019.118212
-
[172]
S. Mazzanti, S. Cao, K. Brummelhuis, et al., Appl. Catal. B 285 (2021) 119773. doi: 10.1016/j.apcatb.2020.119773
-
[173]
L. Cheng, X. Yue, J. Fan, Q. Xiang, Adv. Mater. 34 (2022) e2200929. doi: 10.1002/adma.202200929
-
[174]
J. Zhang, X. Liang, C. Zhang, et al., Angew. Chem. Int. Ed. 61 (2022) e202210849. doi: 10.1002/anie.202210849
-
[175]
Z. Sun, H. Dong, Q. Yuan, et al., Chem. Eng. J. 435 (2022) 134865. doi: 10.1016/j.cej.2022.134865
-
[176]
J.C. Liu, Y. Tang, Y.G. Wang, T. Zhang, J. Li, Natl. Sci. Rev. 5 (2018) 638–641. doi: 10.1093/nsr/nwy094
-
[177]
Y. Zhang, J. Yang, R. Ge, et al., Coord. Chem. Rev. 461 (2022) 214493. doi: 10.1016/j.ccr.2022.214493
-
[178]
S. Wei, A. Li, J.C. Liu, et al., Nat. Nanotechnol. 13 (2018) 856–861. doi: 10.1038/s41565-018-0197-9
-
[179]
Z. Chen, E. Vorobyeva, S. Mitchell, et al., Natl. Sci. Rev. 5 (2018) 642–652. doi: 10.1093/nsr/nwy048
-
[180]
M. Liu, X. Liu, D. Fu, et al., Appl. Catal. B 318 (2022) 121896. doi: 10.1016/j.apcatb.2022.121896
-
[181]
Y. Cao, S. Chen, Q. Luo, et al., Angew. Chem. Int. Ed. 56 (2017) 12191–12196. doi: 10.1002/anie.201706467
-
[182]
L. Hu, T. Wang, Q. Nie, et al., Carbon 200 (2022) 187–198. doi: 10.1016/j.carbon.2022.08.031
-
[183]
B. Kumru, D. Cruz, T. Heil, M. Antonietti, Chem. Mater. 32 (2020) 9435–9443. doi: 10.1021/acs.chemmater.0c03616
-
[184]
L. Xing, Q. Yang, C. Zhu, et al., Nat. Commun. 14 (2023) 1501-1501. doi: 10.1038/s41467-023-37113-8
-
[185]
M.A.R. da Silva, I.F. Silva, Q. Xue, et al., Appl. Catal. B 304 (2022) 120965. doi: 10.1016/j.apcatb.2021.120965
-
[186]
S. Shen, J. Chen, Y. Wang, et al., Sci. Bull. 67 (2022) 520–528. doi: 10.1016/j.scib.2021.11.024
-
[187]
S. Guo, X. Li, J. Li, B. Wei, Nat. Commun. 12 (2021) 1343. doi: 10.1038/s41467-021-21526-4
-
[188]
J. Yang, A. Acharjya, M.Y. Ye, et al., Angew. Chem. Int. Ed. 60 (2021) 19797–19803. doi: 10.1002/anie.202104870
-
[189]
M. Wang, Z. Zhang, Z. Chi, et al., Adv. Funct. Mater. 33 (2023) 2211565. doi: 10.1002/adfm.202211565
-
[190]
C. Cheng, L. Mao, X. Kang, et al., Appl. Catal. B 331 (2023) 122733. doi: 10.1016/j.apcatb.2023.122733
-
[191]
M. Lu, M. Zhang, J. Liu, et al., J. Am. Chem. Soc. 144 (2022) 1861–1871. doi: 10.1021/jacs.1c11987
-
[192]
J.J. Masana, J. Xiao, H. Zhang, et al., Appl. Catal. B 323 (2023) 122199. doi: 10.1016/j.apcatb.2022.122199
-
[193]
Y. Xia, Z. Tian, T. Heil, et al., Joule 3 (2019) 2792–2805. doi: 10.1016/j.joule.2019.08.011
-
[194]
S. Mazzanti, B. Kurpil, B. Pieber, M. Antonietti, A. Savateev, Nat. Commun. 11 (2020) 1387. doi: 10.1038/s41467-020-15131-0
-
[195]
Y. Markushyna, P. Lamagni, J. Catalano, et al., ACS Catal. 10 (2020) 7336–7342. doi: 10.1021/acscatal.0c02176
-
[196]
Y. Markushyna, C.M. Schusslbauer, T. Ullrich, et al., Angew. Chem., Int. Ed. 60 (2021) 20543–20550. doi: 10.1002/anie.202106183
-
[197]
A. Savateev, I. Ghosh, B. Konig, M. Antonietti, Angew. Chem. Int. Ed. 57 (2018) 15936–15947. doi: 10.1002/anie.201802472
-
[198]
D. Aboagye, R. Djellabi, F. Medina, S. Contreras, Angew. Chem. Int. Ed. (2023) e202301909.
-
[199]
M.F. Kuehnel, E. Reisner, Angew. Chem. Int. Ed. 57 (2018) 3290–3296. doi: 10.1002/anie.201710133
-
[200]
D.B. Nimbalkar, V.C. Nguyen, C.Y. Shih, H. Teng, Appl. Catal. B 316 (2022) 121601. doi: 10.1016/j.apcatb.2022.121601
-
[201]
I. Krivtsov, D. Mitoraj, C. Adler, et al., Angew. Chem. Int. Ed. 59 (2020) 487–495. doi: 10.1002/anie.201913331
-
[202]
M. Zhou, L. Zeng, R. Li, et al., Appl. Catal. B 317 (2022) 121719. doi: 10.1016/j.apcatb.2022.121719
-
[203]
Z. Yang, L. Li, J. Gao, et al., ACS ES&T Engin. 2 (2022) 2142–2149. doi: 10.1021/acsestengg.2c00178
-
[204]
C. Liang, X.M. Wang, W. Liu, et al., Chem. Eng. J. 466 (2023) 142931. doi: 10.1016/j.cej.2023.142931
-
[205]
J.N. Chang, Q. Li, J.W. Shi, et al., Angew. Chem. Int. Ed. 62 (2023) e202218868. doi: 10.1002/anie.202218868
-
[206]
Y.Z. Zhang, C. Liang, H.P. Feng, W. Liu, Chem. Eng. J. 446 (2022) 137379. doi: 10.1016/j.cej.2022.137379
-
[207]
A. Rogolino, I.F. Silva, N.V. Tarakina, et al., ACS Appl. Mater. Interfaces 14 (2022) 49820–49829. doi: 10.1021/acsami.2c14872
-
[208]
I. Krivtsov, A. Vazirani, D. Mitoraj, et al., J. Mater. Chem. A 11 (2023) 2314–2325. doi: 10.1039/d2ta08045a
-
[209]
D. Zhang, W. He, J. Ye, et al., Small 17 (2021) 2005149. doi: 10.1002/smll.202005149
-
[210]
B. Tian, D. Ho, J. Qin, et al., Prog. Mater. Sci. 133 (2023) 101056. doi: 10.1016/j.pmatsci.2022.101056
-
[211]
Y. Li, B. Wang, Q.J. Xiang, Q. Zhang, G. Chen, Dalton Trans. 51 (2022) 16527–16535. doi: 10.1039/d2dt02731c
-
[212]
M. Song, S. Yang, H. Peng, et al., Nano Energy 116 (2023) 108784. doi: 10.1016/j.nanoen.2023.108784
-
[1]
-
Figure 1 Historical development and milestones of carbon nitride. Reproduced and modified with permission [98]. Copyright 1937, National Academy of Sciences. Reproduced and modified with permission [102]. Copyright 1994, American Physical Society. Reproduced and modified with permission [104,87,109]. Copyright 2003, 2015 and 2019, the American Chemical Society. Reproduced and modified with permission [107]. Copyright 2009, the Royal Society of Chemistry. Reproduced and modified with permission [85,89,91,110]. Copyright 2008, 2011, 2021 and 2023, Wiley-VCH.
Figure 2 Structural modeling of (a) PHI and (b) PTI. Reproduced and modified with permission [115]. Copyright 2020, Wiley-VCH.
Figure 3 (a) Schematic diagram of catalyst light absorption and fluorescence emission. Optical properties of g-CN and K-PHI: (b) DRUV–vis spectra. (c) Photoluminescence spectroscopy. (d) VB and CB. Reproduced with permission [115]. Copyright 2018, Wiley-VCH. Optical properties of g-CN and PTI: (e) DRUV-vis spectra. (f) Photoluminescence spectroscopy. Reproduced with permission [130]. Copyright 2015, Wiley-VCH. (g) VB and CB. Reproduced with permission [124]. Copyright 2017, the Royal Society of Chemistry.
Figure 4 (a) DFT calculated HOMO and LUMO distributions and DOS of PCN and RPCN. Reproduced with permission [49]. Copyright 2021, Wiley-VCH. (b) DFT optimized PTI-TPA structure schematic diagram. Reproduced with permission [137]. Copyright 2015, American Chemical Society. (c) Quantum-chemical calculations of PHI and Mel-PHI, and band position determination of PHI and Mel-PHI. Reproduced with permission [120]. Copyright 2021, Wiley-VCH.
Figure 5 For PCN (a) XRD, (d) SEM and (g) TEM. For PTI (b) XRD, (e) SEM and (h) TEM. For PHI (c) XRD, (f) SEM and (i) TEM. Reproduced with permission [115]. Copyright 2020, Wiley-VCH.
Figure 6 (a, c) 13C solid state NMR spectra and (b, d) 15N solid state NMR spectra of K-PHI and H-PHI. Reproduced with permission [109]. Copyright 2019, American Chemical Society. (e) FTIR spectra and (f) XPS survey profile of PHI/PTI. Reproduced with permission [142]. Copyright 2023, MDPI. (g) 1H, (h) 13C, (i) 15N, and (j) 7Li solid state NMR spectra of PTI. (k) Schematic diagram of the PTI/LiCl connection pattern. Reproduced with permission [140]. Copyright 2016, Wiley-VCH. (l) Schematic diagrams of the triazine nucleus ((left) and the heptazine nucleus (right). Reproduced with permission [89]. Copyright 2011, Wiley-VCH.
Figure 7 (a) XRD spectra, (b) SEM images, (c, d) HRTEM images of CCN. Reproduced with permission [85]. Copyright 2008, Wiley-VCH.
Figure 8 (a, b) HRTEM images and HAADF-STEM images of PTI-LiNa. (c, d) XRD spectra and FTIR spectra of PTI synthesized at 550 ℃ for 24 h under different molten salt systems (e, f) XRD spectra and FTIR spectra of PTI-LiNa synthesized at 550 ℃ for 24 h at different LiCl/LiNa molar ratios. (g, h) XRD spectra and FTIR spectra of PTI-LiNa synthesized at 550 ℃ for 24 h at different DCDA to molten salt mass ratios. (i, j) XRD spectra and FTIR spectra of PTI-LiNa powders synthesized at 500, 550 and 570 ℃. (k, l) XRD spectra and FTIR spectra of PTI-LiNa powders synthesized at 550 ℃ for 4, 12, 24 and 48 h. Reproduced with permission [145]. Copyright 2021, Wiley-VCH.
Figure 9 Schematic diagram of molecular conjugation and directional crystallization process of PTI polymer crystals in molten salt. Reproduced with permission [153]. Copyright 2023, Wiley-VCH.
Figure 10 Schematic diagram of the typical locations of the (a) C defect, (b) N defect, (c) amino defects, and (d) cyano defect in CN. Reproduced with permission [156]. Copyright 2021, Elsevier.
Figure 11 (a) FTIR spectra of CN, CN-M-x. (b) 13C solid state NMR spectra of CN-M-0.01. (c) CO2 adsorption isotherms of CN. (d) CO2 TPD measurements of CN. Reproduced with permission [160]. Copyright 2020, the Royal Society of Chemistry.
Figure 12 (a) Schematic diagram of the PTI exfoliation process. (b) AFM image of PTI nanosheets. (c) TEM image of PTI nanosheets. Reproduced with permission [165]. Copyright 2014, American Chemical Society.
Figure 13 (a) Schematic diagram of spontaneous dissolution of PTI-LiBr. (b) TEM image of PTI nanosheet. Reproduced with permission [166]. Copyright 2017, American Chemical Society. (c) Schematic of charging and exfoliation of PTI. (d) HRTEM image of FL-PTI nanosheet. (e) AFM image of FL-PTI nanosheets. (f) PTI thickness histogram. Reproduced with permission [167]. Copyright 2018, Wiley-VCH.
Figure 14 (a) Illustration for the synthesis process of PCNNs-IHO by on-surface polymerization. (b) High-resolution TEM image of PCNNs-IHO and SAED pattern of the (100) plane (inset). (c) AFM image and (d) corresponding thickness analysis taken around the white line in (c) of PCNNs-IHO. Reproduced with permission [168]. Copyright 2021, Wiley-VCH. (e) Schematic illustration of the formation process of TCNTs in the molten salt medium. (f)TEM image and (g) high-resolution TEM image of TCNTs. (h) N2 adsorption-desorption isotherms of B-CN and TCNTs. Reproduced with permission [169]. Copyright 2017, Elsevier.
Figure 15 (a) Schematic diagram of photoreduction of CO2 by Z-scheme heterojunction constructed with K-PHI and flavin. Reproduced with permission [172]. Copyright 2020, Elsevier. (b) Schematic illustration of formation of S-scheme heterojunction of CeCo-PTI. Reproduced with permission [173]. Copyright 2022, Wiley-VCH.
Figure 16 (a) The synthetic process of hybrid PHI/PTI copolymer with simi-cohenrent interface. (b) HR-TEM image of PHI/PTI. (c) Schematic illustration of exciton dissociation and charge transfer in PHI/PTI under visible light irradiation. Reproduced with permission [174]. Copyright 2022, Wiley-VCH. (d) The comparison of array structure and particle/sheet structure. (e) The synthesis path of self-supported hierarchical crystalline GCN array. (f) SEM images of MA-rod array. (g) Schematic of the proposed photo-electron transfer path in the framework of MA-rod array. Reproduced with permission [175]. Copyright 2022, Elsevier.
Figure 17 (a) Schematic representation of the controlled non-thermal cation exchange method used to replace the Na+ cations in the structure of PHI by Fe3+ cations. (b) Proposed mechanism of formation of the active Fe(Ⅳ)/Fe(Ⅴ) sites. (c) Schematic representation of the e−/h+ separation promoting effect on the formation of active high oxidation state iron centers. Reproduced with permission [185]. Copyright 2021, Elsevier.
Figure 18 (a) Schematic mechanisms of the Co-CCN/PTI heterojunction for photocatalytic hydrogen production. (b) HRTEM image of Co-CCN/PTI-4. (c) HAADF-STEM image of Co-CCN/PTI-4. (d) FT k3-weighted Co K-edge EXAFS spectra of Co-CCN/PTI-4, Co-CCN/PTI-40 and the references. Reproduced with permission [186]. Copyright 2021, Science China Press.
Figure 19 (a) Fabrication process of K-PHI/BxST materials. (b) Illustration of the alkali metal cations as charge-transfer bridge for polarization promoted photocatalysts. (c) Schematic diagram of migration and separation of photogenerated charge carriers. (d) The results of cycling experiments on 5 % K-PHI/B0.8ST in H2 evolution reaction. (e) The DR UV–vis spectrum over K-PHI and the determined AQY at different wavelengths over 5 % K-PHI/B0.8ST. Reproduced with permission [198]. Copyright 2022, Wiley-VCH. (f) Schematic mechanism of BCN—NaK for photocatalytic H2 production. (g) Photocatalytic H2-production activities for BCN-T550, BCN-LiK, BCN—NaK and BCN—NaK + K2HPO4 under visible-light irradiation (λ > 400 nm). (h) Wavelength-dependence AQYs for BCN—NaK + K2HPO4. Reproduced with permission [190]. Copyright 2023, Elsevier.
Figure 20 (a) Proposed salt melt synthetic processes of PTI/Li+Cl− catalysts by using different binary alkali metal chlorides with different melting points. Red circle represents nitrogen defect). (b) Schematic of PTI (Pt-CoOx) overall water splitting. Reproduced with permission [90]. Copyright 2022, Wiley-VCH. (c) Schematic of PTI (Rh/Cr2O3-CoOx) overall water splitting. (d) Proposed schematic process for step-by-step deposition of CoOx and Rh/Cr2O3 on poly(triazine imide) as H2 and O2 evolution cocatalysts, respectively. Reproduced with permission [110]. Copyright 2023, Wiley-VCH.
Figure 21 (a) Schematic for well-defined photocatalytic platform by integrating Co-doped CeO2 nanoparticles with highly crystalline PTI. Reproduced with permission [173]. Copyright 2022, Wiley-VCH. (b) Schematic diagram of CNNA/rGO photocatalytic CO2 reduction. Reproduced with permission [193]. Copyright 2019, Elsevier.
Figure 22 (a) Schematic representation of the reactor setup and reaction parameters. (b) Proposed mechanism of the generation of dichloromethyl radicals and their addition to enones. Reproduced with permission [194]. Copyright 2020, Nature Springer.
Figure 23 (a) Scope of substituted benzylamines investigated. (b) Proposed mechanism for photocatalytic oxidative coupling of benzylamines to N-benzylidene benzylamine in the presence of CO2 and H2O. Reproduced with permission [195]. Copyright 2020, American Chemical Society.
Figure 24 The mechanism of chromoselective conversion of thio-derivatives to sulfonyl chlorides, arylchlorides, and disulfides. Reproduced with permission [196]. Copyright 2022, Wiley-VCH.
Figure 25 (a) Schematic diagram of photocatalytic reforming of cellulose to H2 and biochemicals under visible light by K-PHI. Reproduced with permission [200]. Copyright 2022, Elsevier. (b) Schematic representation of the synthesis procedure resulting in a highly active water-soluble and recoverable PCN photocatalyst. Reproduced with permission [201]. Copyright 2019, Wiley-VCH.
Figure 26 (a) Schematic of NO removal for melon-CN and CN—NaLi. (b) NO removal performance of CN and CN—NaLi. (c) ESR spectra of the spin-reactive •O2− generated by CN and CN—NaLi with visible light. Comparison of photocatalytic activities of (d) CN and (e) CN—NaLi in different capture agents under visible-light-irradiation (λ > 420 nm). Reproduced with permission [202]. Copyright 2022, Elsevier.
Figure 27 (a) Schematic diagram of K-PHI photocatalytic pollutant degradation and H2O2 production. (b) K-PHI photocatalytic H2O2 production performance. (c) Photocatalytic H2O2 production with the degradation of carbamazepine (CBZ). (d) Photocatalytic degradation of ciprofloxacin in the presence of O3. Reproduced with permission [203]. Copyright 2022, American Chemical Society.
Figure 28 (a) Proposed general mechanism of photocatalysed ORR on carbon nitrides. (b) Comparison of different M-PHIs in photocatalytic H2O2 production. Reproduced with permission [207]. Copyright 2022, American Chemical Society. (c) Concept of biphasic photocatalysis using hydrophobized PHI. Reproduced with permission [208]. Copyright 2023, the Royal Society of Chemistry.
Figure 29 The synthesis process of the different types of carbon nitride. Reproduced with permission [211]. Copyright 2022, the Royal Society of Chemistry.
Figure 30 (a) Photocatalytic N2 reduction mechanism schemes of KN—HCN. Reproduced with permission [211]. Copyright 2022, the Royal Society of Chemistry. (b) Schematic for the synthesis of PCON. (c) Mechanism for the photocatalytic N2 fixation over PCON-2. (d) Piezo-photocatalytic nitrogen reduction mechanism over DKCN. Reproduced with permission [127]. Copyright 2022, Elsevier.
-

 DownLoad:
DownLoad:

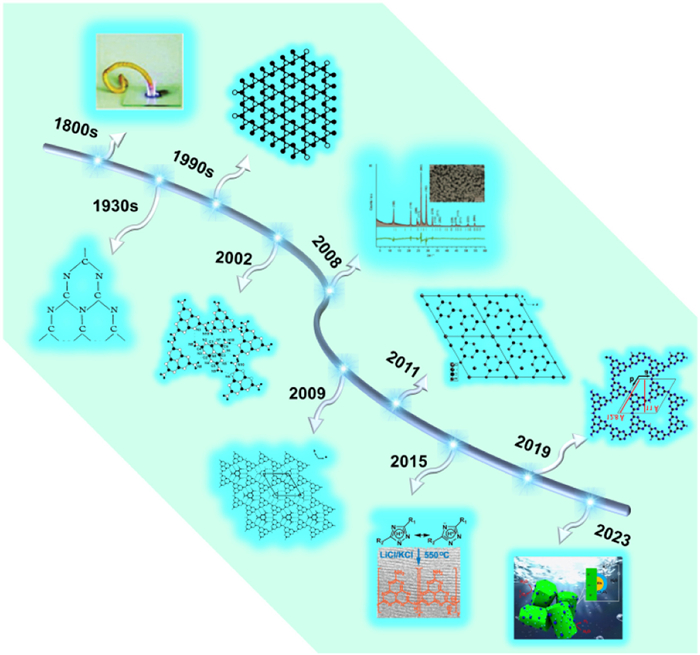
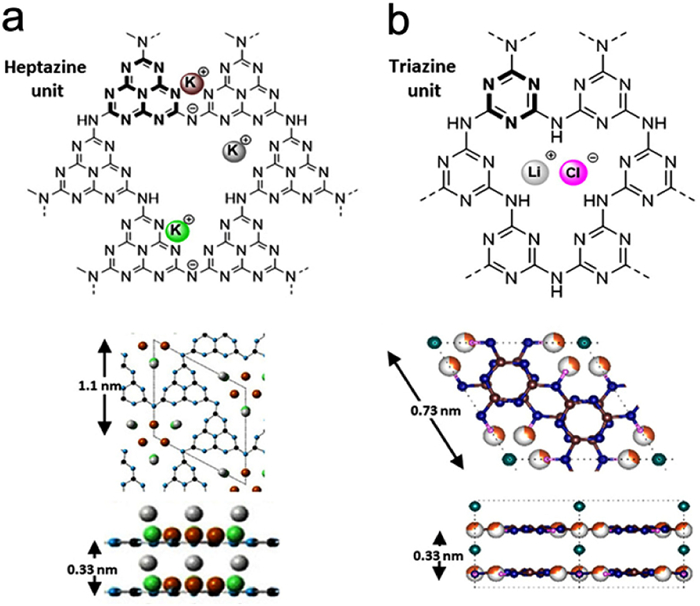
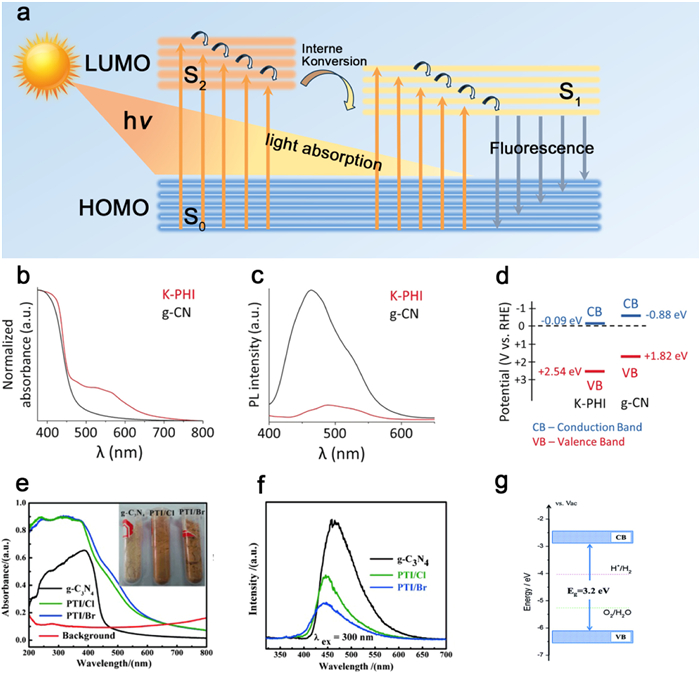
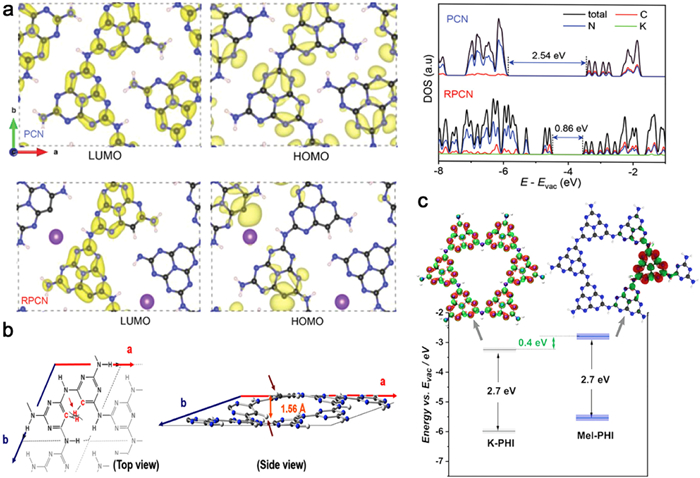
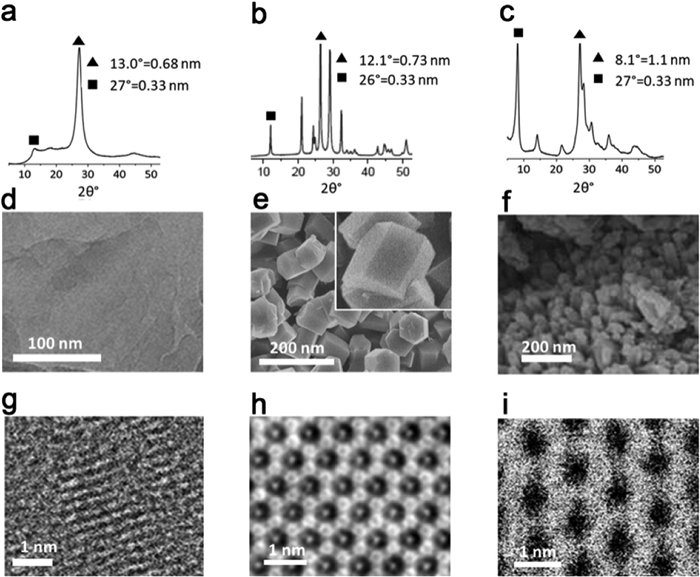
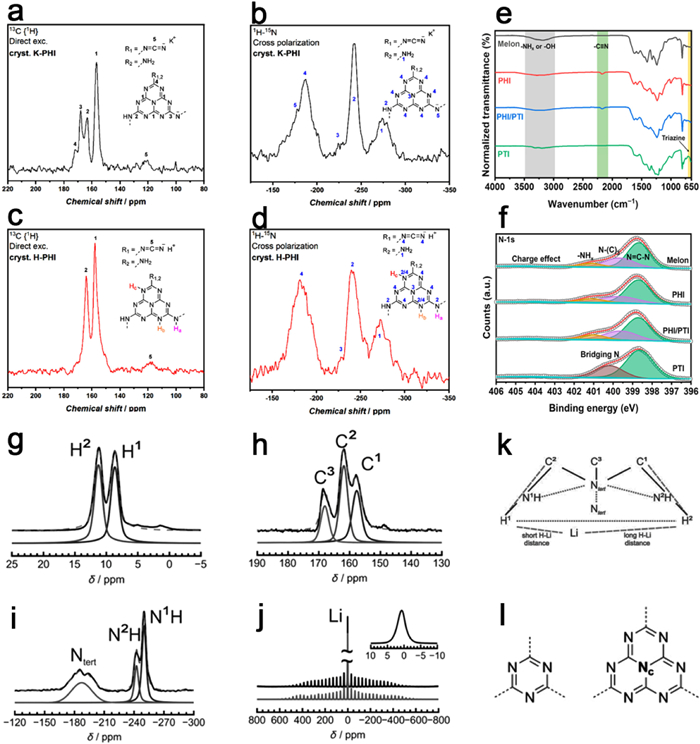
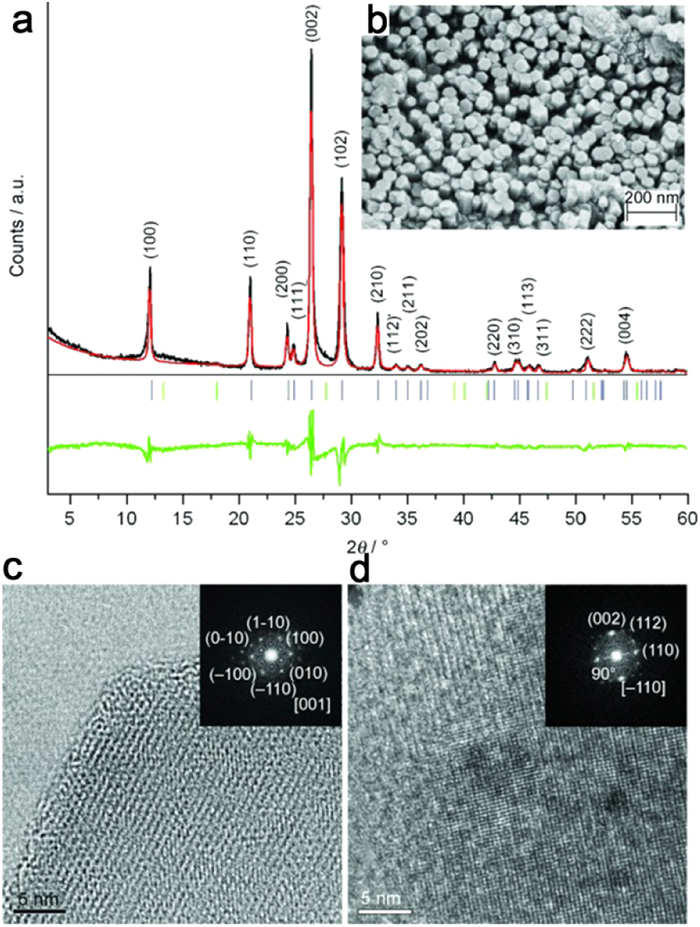
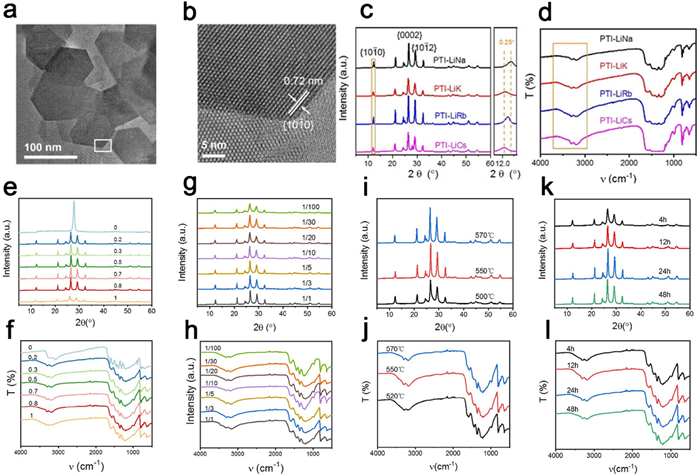
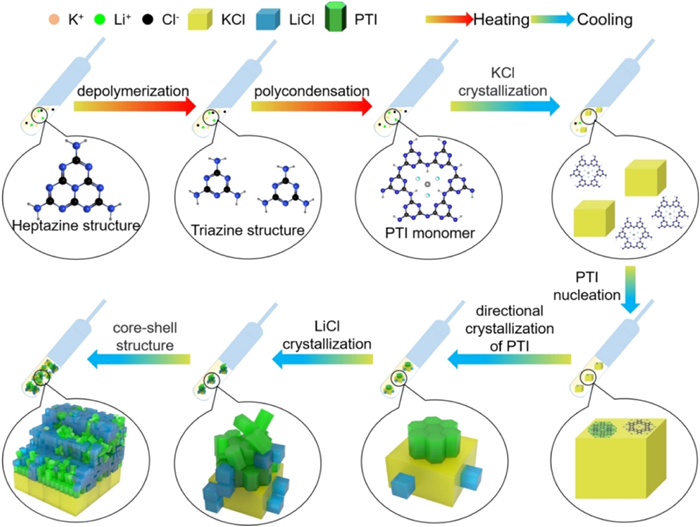
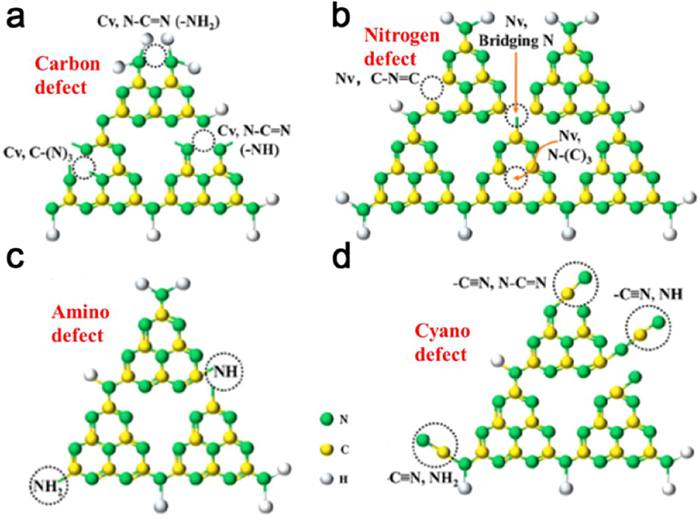
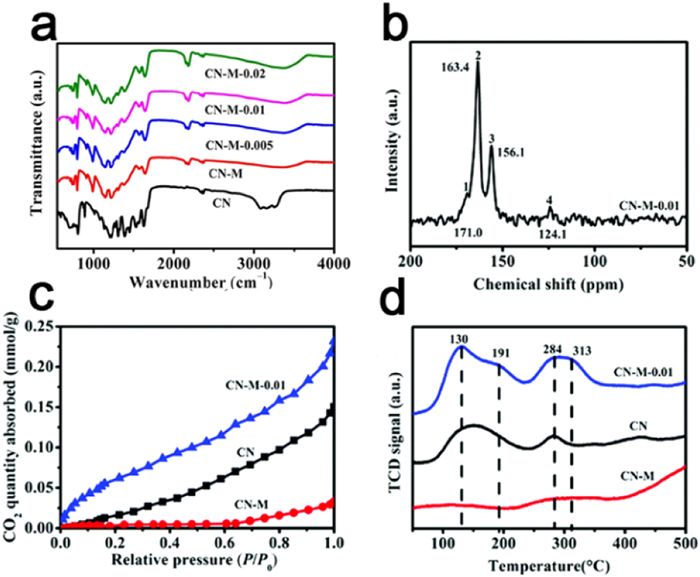
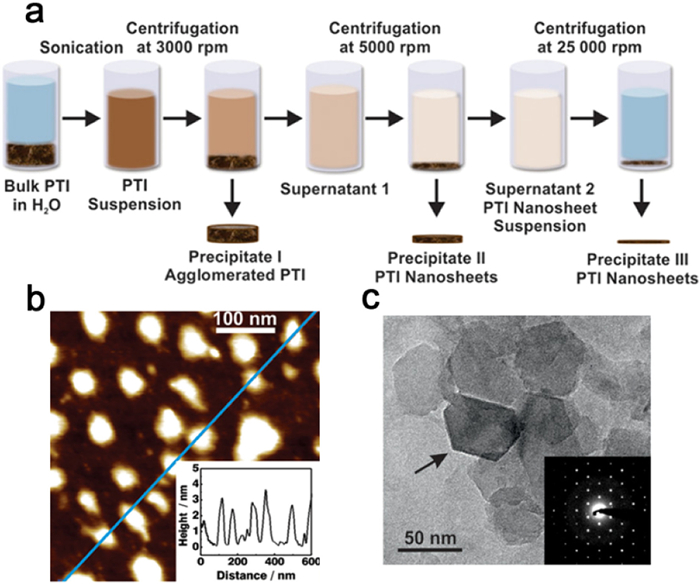
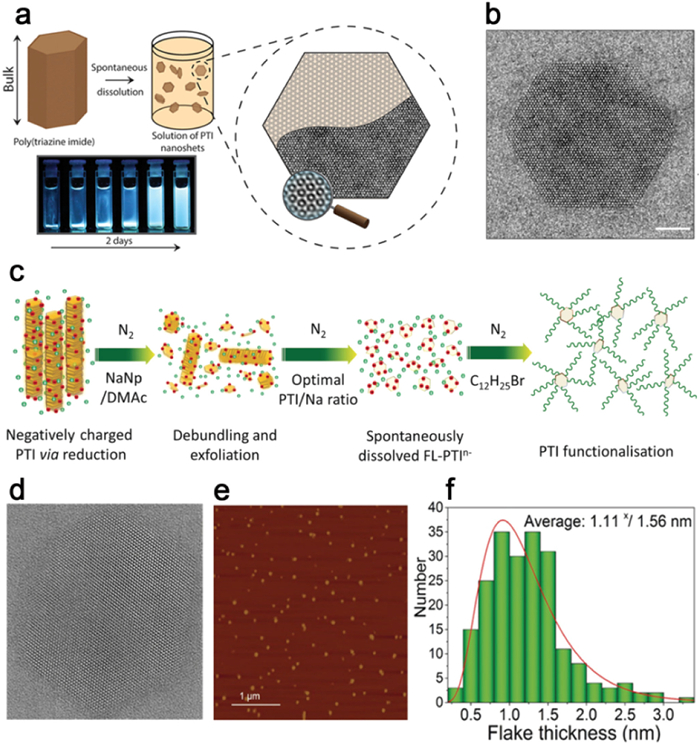
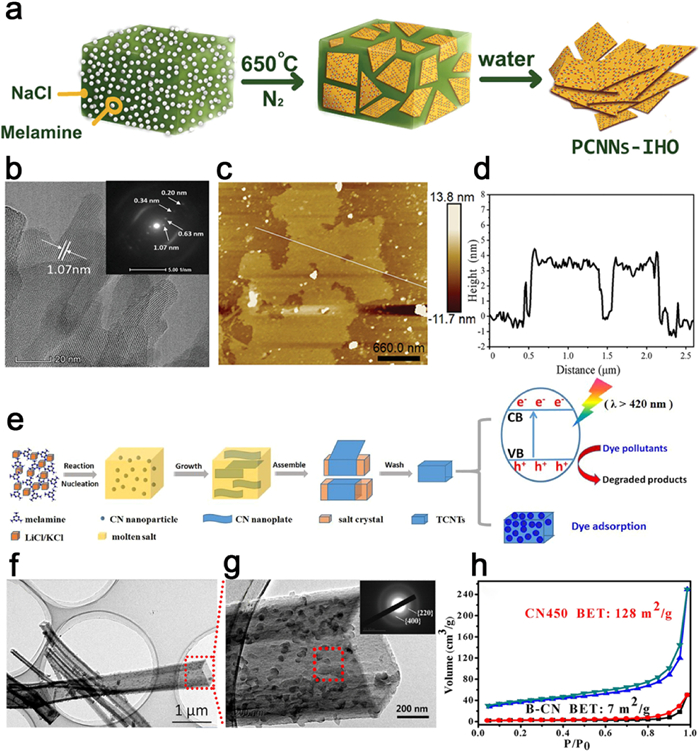
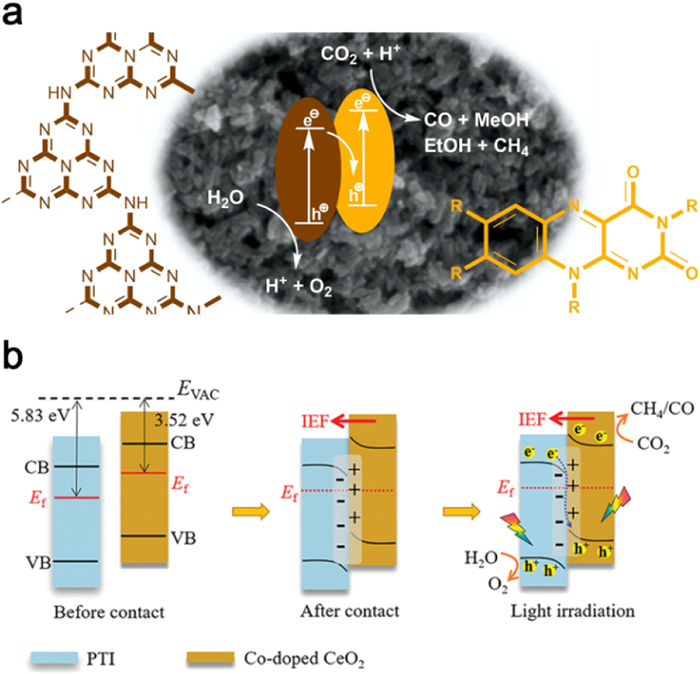
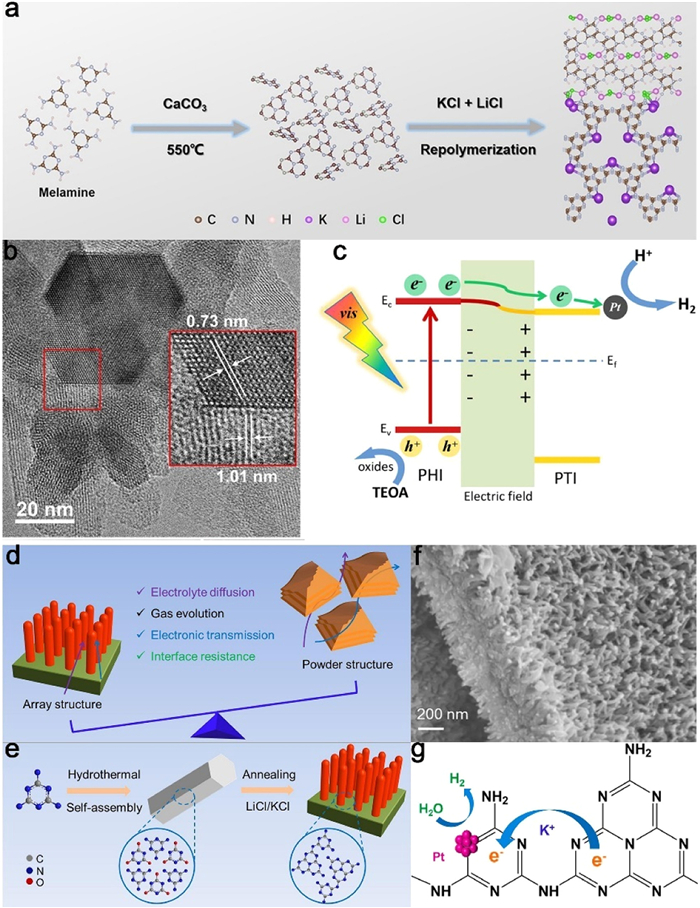
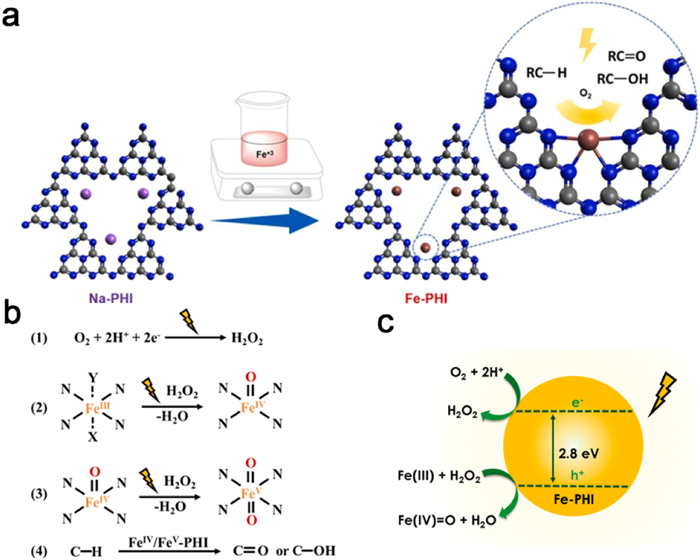
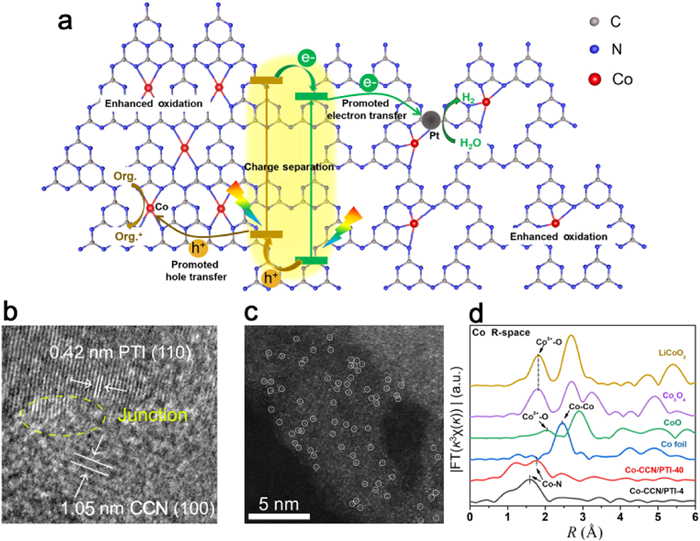
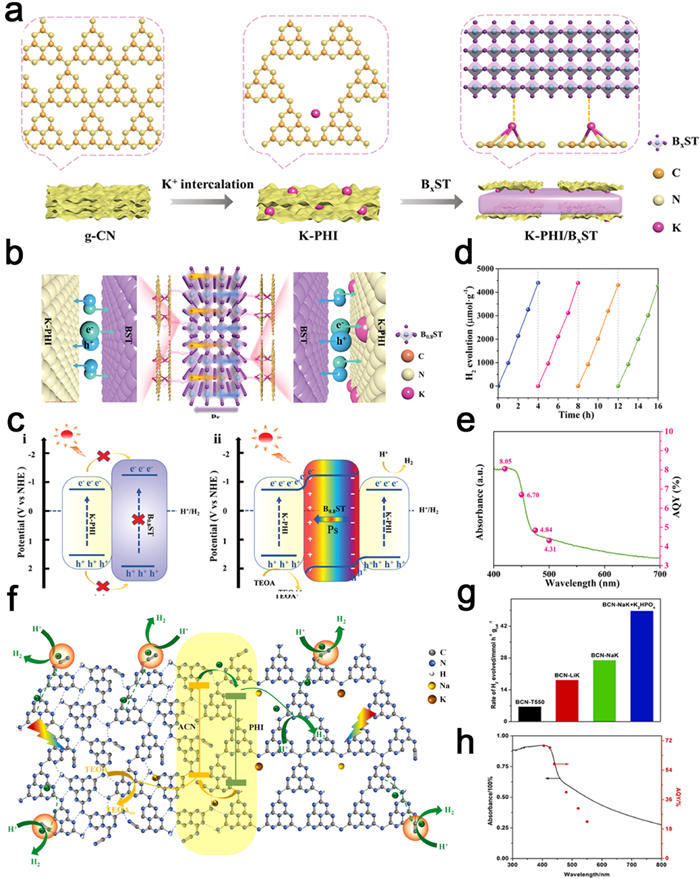
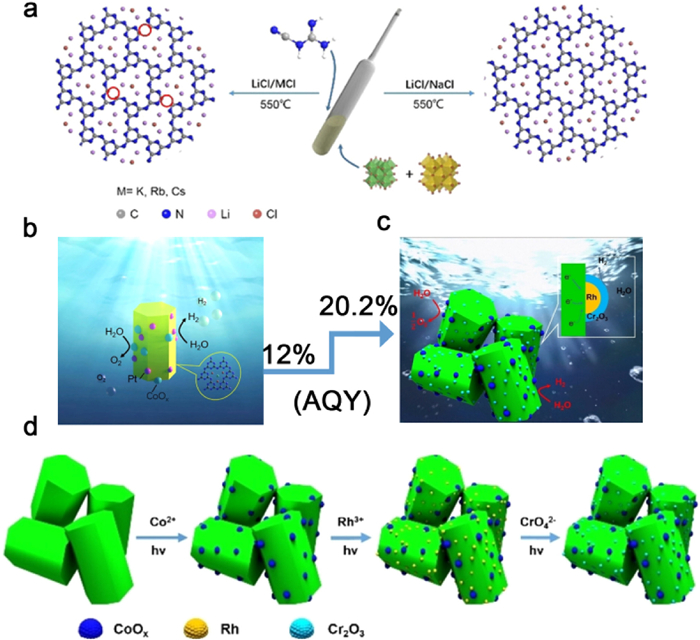
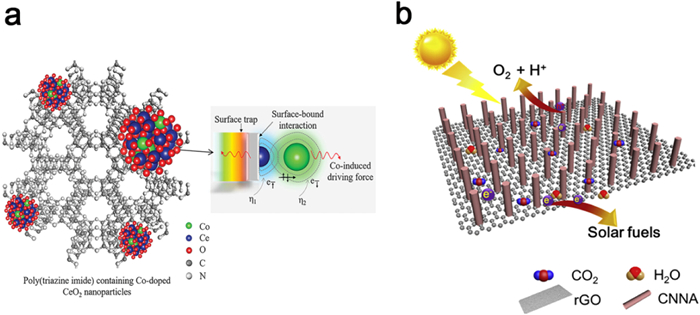
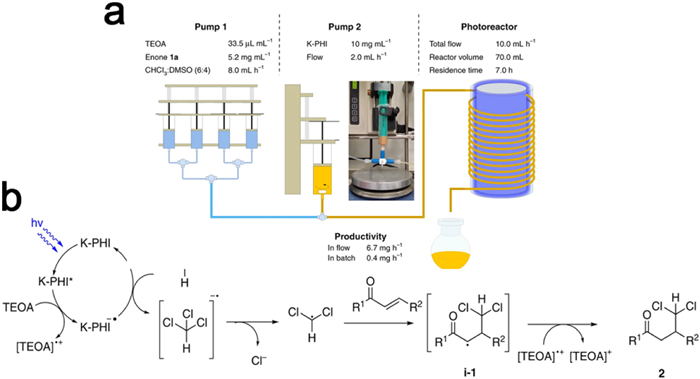
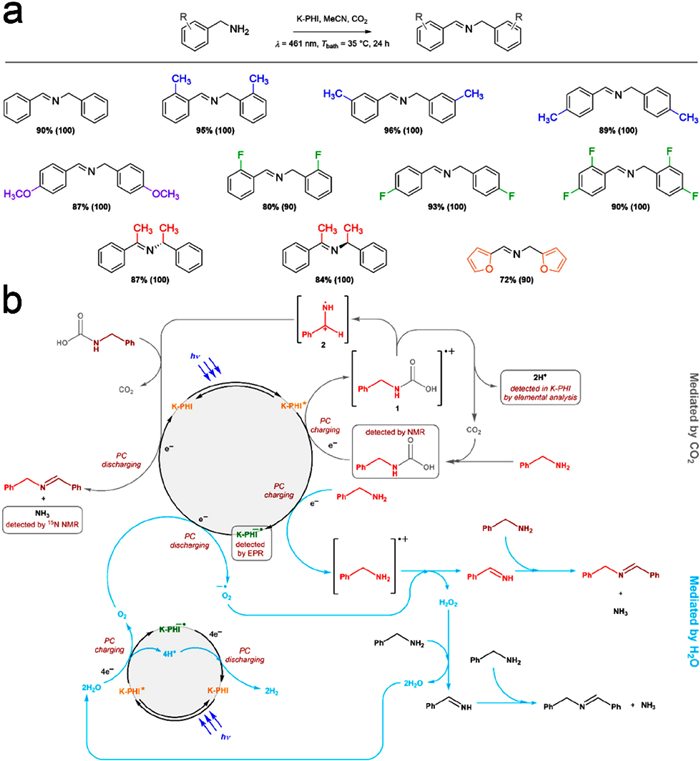
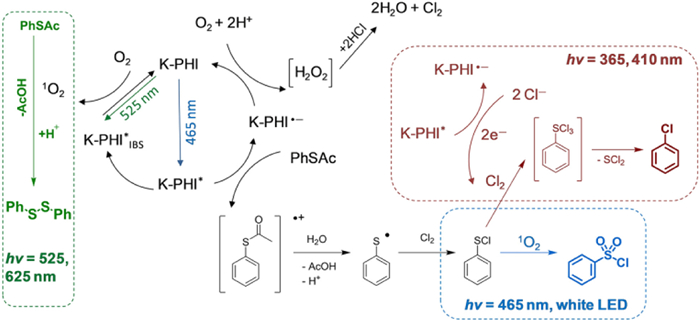
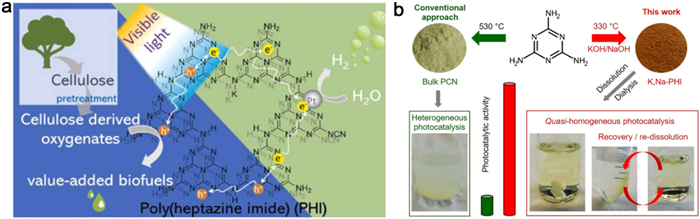
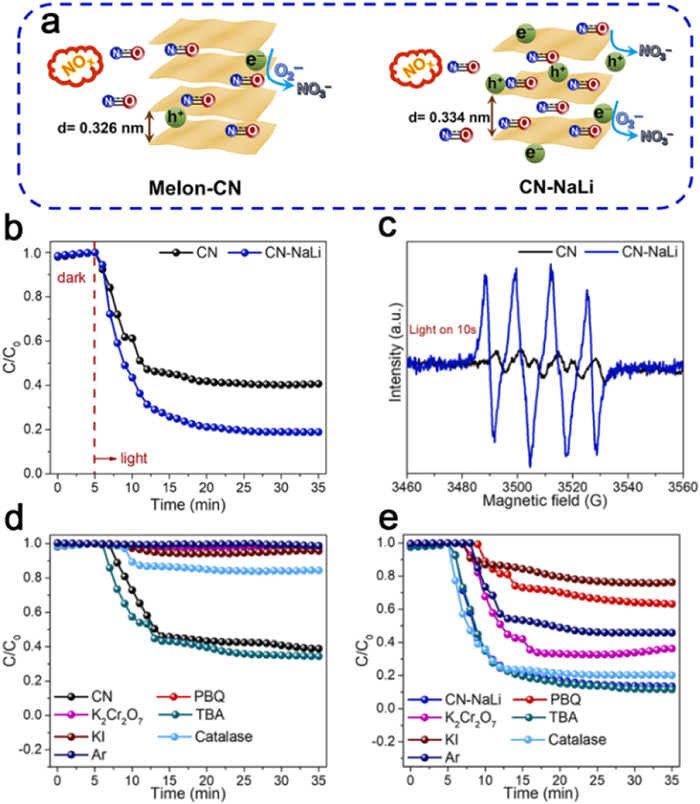
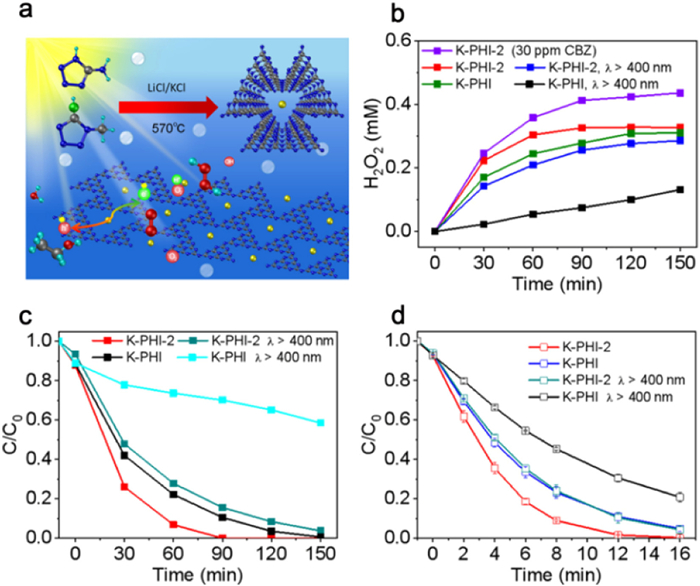
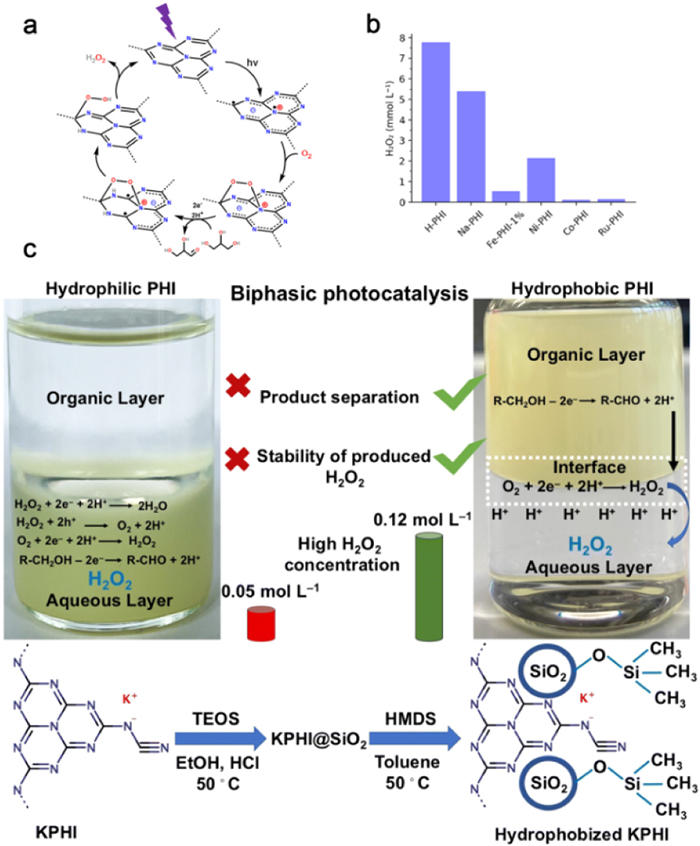
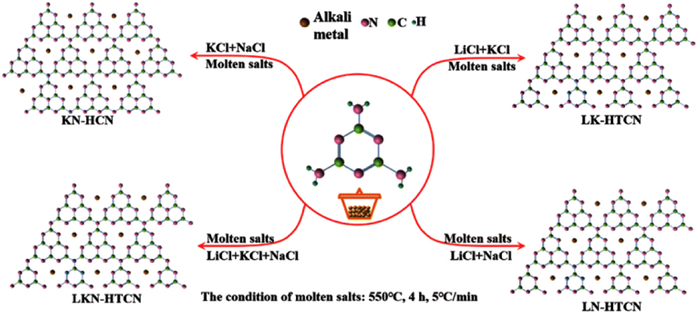

 下载:
下载:





























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 27
- 文章访问数: 2832
- HTML全文浏览量: 241
 下载:
下载:
