Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Jiangsu Provincial Key Lab for the Chemistry and Utilization of Agro-Forest Biomass, Jiangsu Key Lab of Biomass-Based Green Fuels and Chemicals, International Innovation Center for Forest Chemicals and Materials, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China
b.
Department of Radiology and Imaging Sciences, Emory University, Atlanta, GA 30322, United States
Received Date:
17 August 2023 Accepted Date:
27 November 2023 Revised Date:
14 November 2023 Available Online:
15 August 2024
Abstract:
As a powerful noninvasive imaging technology, positron emission tomography (PET) has been playing an important role in disease theranostics and drug discovery. The successful application of PET relies on not only the biological properties of PET tracers but also the availability of facile and efficient radiochemical reactions to enable practical production and widespread use of PET tracers. Most recently, photochemistry is emerging as a novel, mild and efficient approach to generating PET agents. In this review, we focus on the recent advances in newly developed photocatalytic radiochemical reactions, innovation on automated photochemical radiosynthesis modules, as well as implementation in late-stage radiolabeling and radiopharmaceutical synthesis for PET imaging. We believe that this review will inspire the development of more promising radiolabeling protocols for the preparation of clinically useful PET agents.
As an ideal non-invasive imaging technology, positron emission tomography (PET), has been getting widespread attention in disease diagnosis and treatment assessment in recent years [1-11]. In contrast to traditional imaging technologies, such as X-ray, ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI), which offer structural and anatomical information, PET could provide in vivo real-time functional information via the interaction between the biological system and a "radiotracer" installed with a positron-emitting radionuclide. With only trace doses of radiotracers (< 10–100 µg/subject), PET allows to non-invasively monitor the biological processes with negligible pharmacological effects as well as to support and mitigate the risk for the clinical translation of a candidate therapeutic drug via a better understanding of its target engagement, pharmacokinetics, and mode of action. Moreover, PET also indicates the state of disease by monitoring the changes in the expression level of a specific biological target between the disease and healthy state and further enables examining the treatment efficacy of a specific drug in a longitudinal investigation. The incremental role of PET in disease theranostics and drug discovery has stimulated great efforts in developing novel PET tracers that exhibit satisfactory biological properties by academia and industry. Notably, apart from the biological properties of PET tracers, the key to the advance and successful clinical translation of PET also hinges on the availability of facile and efficient radiochemical reactions to enable practical production and widespread use of PET tracers. Such radiochemical reactions are greatly impacted by the properties of positron-emitting radionuclides including their half-lives and chemical reactivities. For translational molecular PET imaging, carbon-11 (11C) and fluorine-18 (18F) are among the most commonly used positron-emitting radionuclides. A number of publications have been reported to capture the basic principles of PET and provide an overview of classical PET tracer development which requires installation of an appropriate functional group into the radiolabeling precursor.
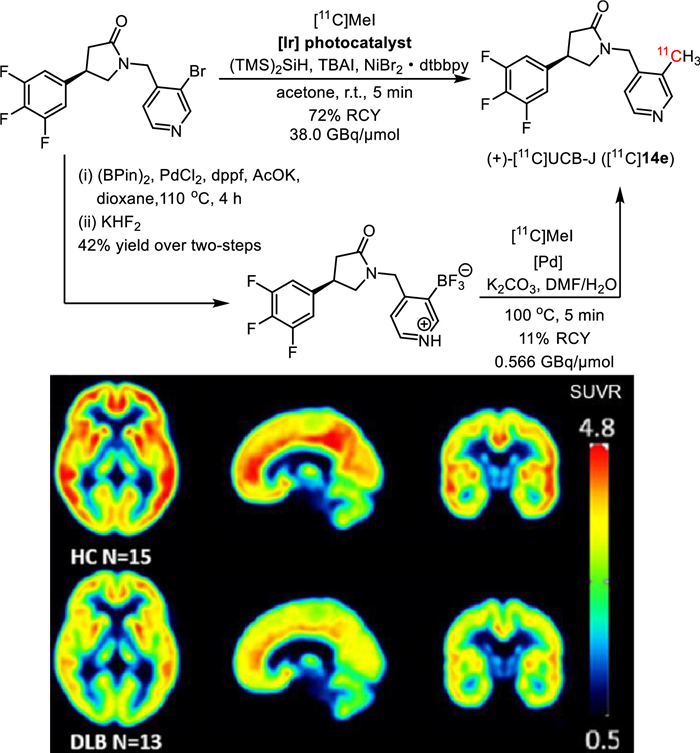
In the last decade, photochemistry and particularly photocatalysis have emerged as a powerful synthetic transformation approach to offering a plethora of new opportunities to the communities of synthetic organic chemistry, pharmacy, and life sciences [12-22]. Of particular note, photocatalysis has been embraced by the radiochemistry community to enable successful radiolabeling of divergent previously elusive precursors (aryl halides, phenol ethers, carboxylic acids, and carboxylic esters) and even native molecules via direct C−H functionalization, since the first attempt by Britton and co-workers in 2017 [23]. More importantly, photocatalysis has enabled late-stage radiolabeling of plenty of complex pharmaceuticals and natural products to facilitate clinical application in PET imaging benefiting its selective and mild catalytic pattern with broad functional group tolerance. For instance, (+)-[11C]UCB-J ([11C]14e) (Fig. 1) is a clinically investigational PET tracer for synaptic density imaging in neurodegenerative diseases, such as Parkinson's disease (PD), Alzheimer's disease (AD) and cognitive impairment [24-27]. For instance, in PD with dementia with Lewy bodies (DLB), the uptake of (+)-[11C]UCB-J is significantly lower than that in healthy controls (HC), which is consistent with the synaptic loss in PD with DLB (Fig. 1) [24]. The conventional approach to synthesizing (+)-[11C]UCB-J involves a palladium-catalyzed [11C]methylation of the trifluoroborate radiolabeling precursor, which was prepared in 42% overall yield from the corresponding aryl bromide [28]. Under high temperature (100 ℃), the desired (+)-[11C]UCB-J was isolated in 11% decay-corrected radiochemical yield (RCY, all the RCY included in this review was decay corrected, unless otherwise specified) with moderate molar activity (0.566 GBq/µmol). By contrast, under photocatalysis conditions, (+)-[11C]UCB-J was obtained from aryl bromide at room temperature in significantly improved isolated RCY (72%) and molar activity (38.0 GBq/µmol), which entirely meet the requisition for clinical PET imaging (Fig. 1) [29].
Figure 1
Figure 1.
Preparation of (+)-[11C]UCB-J and PET imaging in healthy controls (HC) and Parkinson's disease with dementia with Lewy bodies (DLB). Reproduced with permission from [24]. Copyright 2023. The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
With our continuous interest in radiochemistry [8,9], PET imaging [21,30-35], and photocatalytic C−H functionalization [36], in this review, we aim to provide the most comprehensive and up-to-date summary covering the recent advances in newly developed photocatalytic radiochemical reactions. Of particular note, the innovation of automated photochemical radiosynthesis modules as well as implementation in late-stage functionalization and radiopharmaceutical synthesis for PET imaging were also highlighted. We believe that this review will gain much attention from researchers with organic synthetic chemistry, radiochemistry, and radiopharmaceutical discovery background, stimulate more future efforts towards this area, and facilitate the successful implication of these novel reactions in the production of clinically useful radiopharmaceuticals.
2.
Photocatalytic 18F-radiolabeling reactions
Among the various existing positron-emitting radionuclides, fluorine-18 represents the most widely used PET radionuclide befitting from its unique physicochemical properties over other radionuclides for PET [37]. For instance, the clear positron emission (97%) profile and low positron range (< 1 mm) of fluorine-18 enable to generate images with high spatial resolution. The relatively long half-life (t1/2 = 110 min) allows for shipment over fairly large distances and off-site use in satellite facilities without a cyclotron. Traditional strategies including electrophilic and nucleophilic 18F-fluorination have been widely used to prepare 18F-labeled PET tracers. While such application has been limited to a certain extent either due to the low selectivity or by the harsh reaction conditions [38,39], photocatalysis provides an alternatively effective and rapid strategy to achieve radiofluorination including aliphatic radiofluorination, aromatic radiofluorination, and aromatic radiodifluoromethylation in mild conditions, which is introduced as follows.
2.1
Photocatalytic aliphatic 18F-fluorination
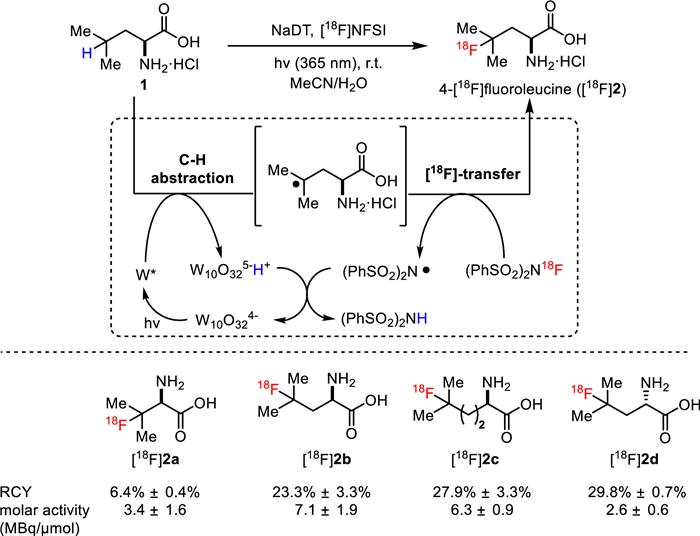
Britton and co-workers have been devoted to developing effective photocatalytic systems for the (radio)fluorination of aliphatic amino acids and peptides [40]. In 2017, they disclosed a mild and photocatalytic direct C−H 18F-fluorination reaction to selectively radiolabel branched aliphatic amino acids at the tertiary C−H bond sites [23]. As displayed in Fig. 2, with electrophilic [18F]-N-fluorobenzenesulfonimide ([18F]NFSI) as the radiofluorinating reagent, a mixture of H2O and MeCN as the co-solvent system, sodium decatungstate (Na4W10O32, NaDT) as the photocatalyst under UV light (365 nm) irradiation, several 18F-labelled amino acids [18F]2a-2d were obtained in moderate to good RCY ranging from 6.4% to 29.8% despite with low molar activity (2.6–7.1 MBq/µmol) due to the use of electrophilic [18F]NFSI. This reaction displayed sufficient synthesis time (50 min) compared to the half-life decay of fluorine-18 (110 min), which suggested the potential of this radiolabeling method for the preparation of 18F-labeled branched amino acids. More importantly, this approach obviates the conventional implementation of prosthetic groups as well as the complicated synthesis of precursors [41]. This reaction was proposed to be initiated by a hydrogen atom transfer (HAT) from amino acids to the photoactivated decatungstate catalyst [42,43], generating a tertiary carbon radical. Subsequent 18F-fluorine atom transfer proceeded in the presence of [18F]NFSI to arrive at the desired 18F-labeled amino acids. Notably, 18F-labeled and native amino acid mixture could be purified from other impurities with strong cation exchange resin, and directly used without further purification with high performance liquid chromatography (HPLC), since the low plasma concentrations ranging from 100 µmol/L to 150 µmol/L of native amino acids was unlikely to interfere with the pharmacokinetic effect of the 18F-labeled amino acids.
Figure 2
Figure 2.
Photocatalytic [18F]fluorination of the tertiary C(sp3)−H bonds in branched aliphatic amino acids.
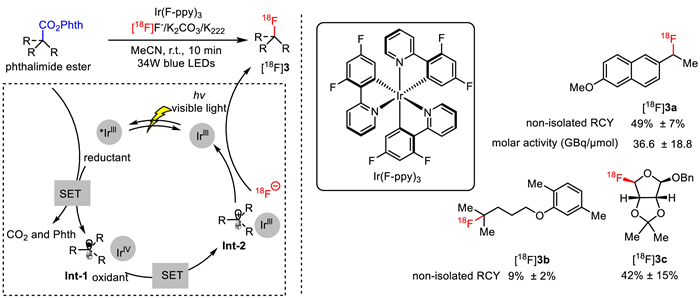
Notwithstanding the great superiority of [18F]NFSI in photocatalytic electrophilic 18F-fluorination of amino acids, its complicated synthesis from [18F]F2, equipment corrosion caused by the requisite use of F2 as a carrier gas, and the relatively low molar activity of produced radioligands strongly limit its widespread use for PET. In this scenario, a reliable alternative approach to effect aliphatic 18F-fluorination resides in direct nucleophilic fluorination with [18F]fluoride, which has been commonly utilized to produce 18F-labeled radioligands with high molar activity and RCY. Towards this end, Doyle and co-workers developed a redox-neutral approach for photocatalytic decarboxylative radiofluorination of N-hydroxyphthalimide esters with nucleophilic [18F]KF (Fig. 3) [44]. With Ir(F-ppy)3 as photocatalyst under blue light-emitting diode (LED) irradiation, various radioligands containing a secondary or tertiary C(sp3)−18F bond were generated very fast at room temperature with excellent molar activity and good non-isolated RCYs, which remains a formidable challenge by conventionally direct nucleophilic radiofluorination of alkyl sulfonates. Notably, even for primary or activated secondary substrates, the harsh reaction conditions (high temperature and high basicity) requisite for sufficient substitution with conventional approaches frequently generate inseparable olefin by-products via β-hydrogen elimination, which is not suitable for late-stage radiofluorination of biomolecules [1,9]. For the reaction mechanism, it was proposed that single-electron transfer (SET) first occurred between the excited Ir(F-ppy)3 and the phthalimide ester to generate phthalimide ester radical anion, which then underwent fragmentation and concomitant CO2 extrusion to produce a carbon-centered radical Int-1 (Fig. 3). This radical intermediate then proceeded another SET process with the oxidized photocatalyst IrIV complex, giving rise to a carbocation Int-2 and turning over the photocatalyst. Finally, the desired 18F-labeled radioligand was obtained by trapping the carbocation with the [18F]fluoride source.
Figure 3
Figure 3.
Photocatalytic [18F]fluorination of N-hydroxyphthalimide esters.
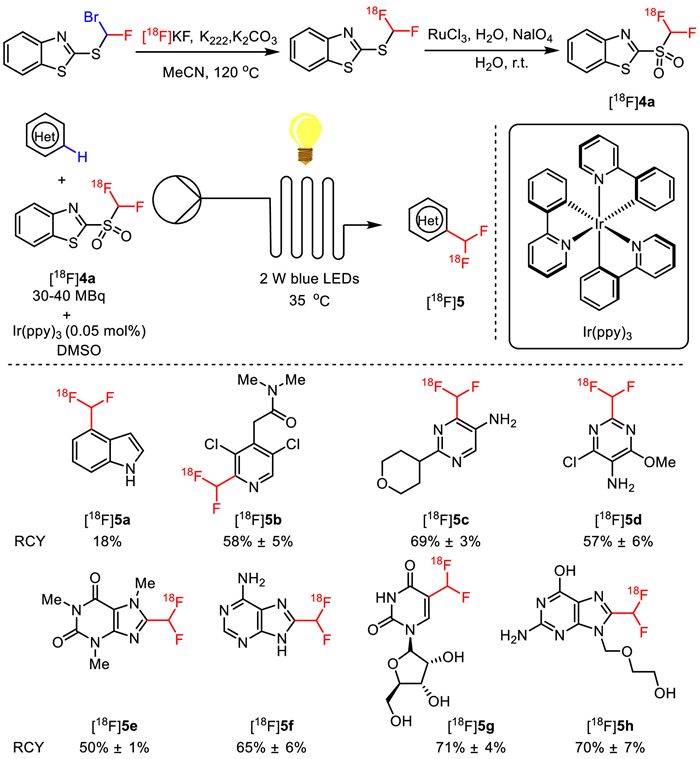
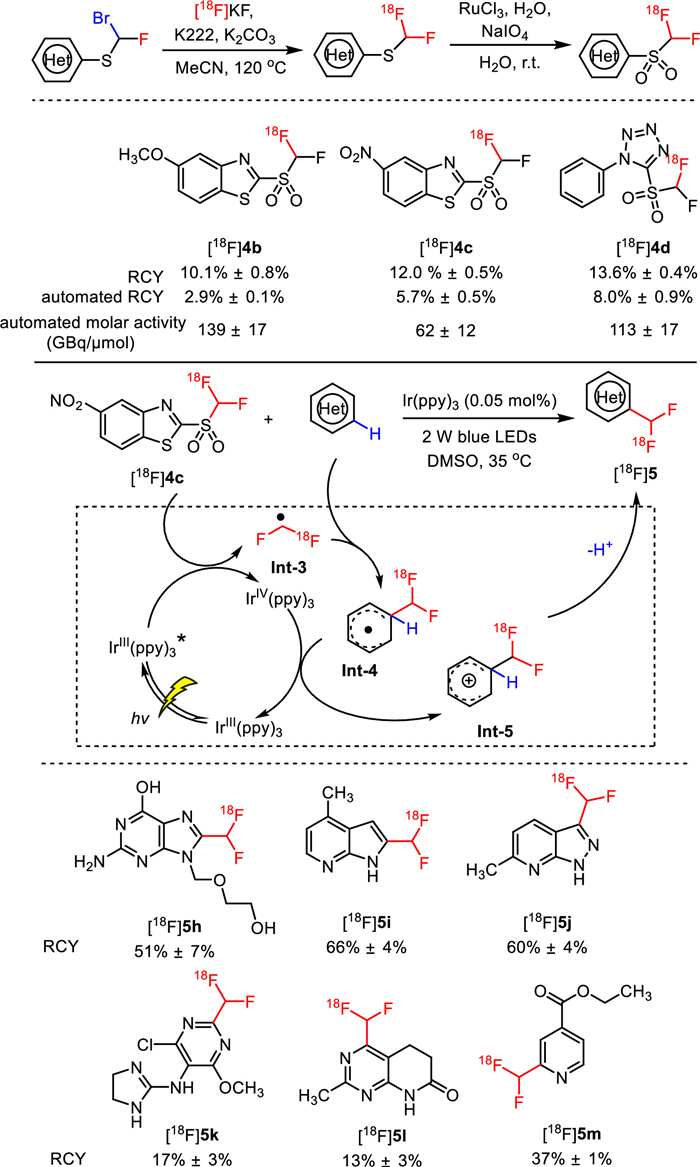
As a continuation of aliphatic 18F-fluorination, Genicot and co-workers developed a new 18F-difluoromethylation reagent based on benzothiazole sulfone (2-[18F]((difluoromethyl)-sulfonyl)benzo[d]thiazole) ([18F]4a) via a two-step nucleophilic radiofluorination with [18F]fluoride and RuCl3 catalyzed oxidation of sulfide to sulfone (Fig. 4). Furthermore, they applied this new CHF18F reagent in photocatalyzed 18F-difluoromethylation of N-heteroaromatics using flow chemistry to accelerate the reaction rate by maintaining a better solution irradiation. With Ir(ppy)3 as the photocatalyst under irradiation of 2 W blue LEDs at 35 ℃, a wide range of N-heteroaromatics including indoles, pyridines, pyridazines, pyrimidines, 1H benzoimidazoles, 1H-pyrrolo[2,3-b]pyridine and 1H-pyrazolo[3,4-b]pyridine were successfully 18F-difluoromethylated in 2 min via direct aromatic C−H activation. As a result, various specific [18F]difluoromethylated N-heterocycles [18F]5 including compounds with scaffolds of medicinal interest, such as xanthine scaffolds ([18F]5e, RCY = 50% ± 1%), nucleic bases scaffolds ([18F]5f, RCY = 65% ± 6%), nucleosides scaffolds ([18F]5g, RCY = 71% ± 4%), and even drug scaffolds ([18F]5h, RCY = 70% ± 7%), were achieved in moderate to good nonisolated RCY. It should be mentioned that the total radiosynthesis time of this strategy was 90 min, which did not exceed the half-life decay of fluorine-18 atom (110 min) [45].
Figure 4
Figure 4.
Synthesis of reagent [18F]4a and its application in [18F]difluoromethylation of N-heteroaromatics.
In 2020, Genicot and co-workers further evaluated the influence of different 18F-difluoromethylation reagents on the photocatalytic 18F-difluoromethylation reaction (Fig. 5) [46]. In this work, six structure-related [18F]difluoromethyl heteroaryl-sulfones were prepared, and fully automated two-step production for three of them were realized with favorable isolated RCYs (2.9% ± 0.1% for [18F]4b, 5.7% ± 0.5% for [18F]4c, and 8.0% ± 0.9% for [18F]4d) and improved molar activity (139 ± 17 GBq/µmol for [18F]4b, 62 ± 12 GBq/µmol for [18F]4c, and 113 ± 17 GBq/µmol for [18F]4d). These three new 18F-difluoromethylation reagents all enabled to radiolabel a variety of N-heteroaromatics despite with slightly reduced nonisolated RCYs (13%−66%) compared with that using [18F]4a. The underlying mechanism of this reaction was proposed to involve radical intermediates as revealed in Fig. 5. The [18F]CF2H radical Int-3 was first generated by the oxidative quenching between [18F]difluoromethylating reagents with the excited state of Ir(ppy)3 under light irradiation. Then [18F]heteroaryl-CF2H radical species Int-4 was produced via the addition of radical Int-3 onto N-heteroaromatic rings, which was further oxidized by the intermediate Ir(IV)(ppy)3 to afford cationic intermediate Int-5, concomitantly turning over the Ir(ppy)3 photocatalyst. Finally, deprotonation of intermediate Int-5 arrived at the desired [18F]heteroaryl-CF2H derivatives.
Figure 5
Figure 5.
Influence of different 18F-difluoromethylation reagents on the photocatalytic [18F]difluoromethylation reaction of N-heteroaromatics.
The development of powerful approaches for aromatic 18F-fluorination has gained widespread attention from researchers with (radio)pharmaceutical background given the incremental significance of C(sp2)–F bonds in small-molecule therapeutics and probes [47,48]. Conventional approaches for aromatic 18F-fluorination with [18F]fluoride include aromatic nucleophilic substitution (SNAr) of electron-deficient arenes, N-arylsydnones, triarylsulfonium salts, diaryliodonium salts, and spirocyclic iodonium salts (SCIDY), copper-promoted coupling of aryl boronic acids, esters, and arylstannanes, [18F]fluorodemetalation of arene-Pd(IV) or arene-Ni(II) complexes, and [18F]deoxyfluorination of phenols via uranium intermediates [8,9,37]. Notwithstanding the great advances in aromatic 18F-fluorination, these approaches typically suffer from high temperature and requisite prefunctionalization of the aromatic subunits, which is very challenging for a number of molecules of medicinal interest. Moreover, the involvement of stoichiometric or superstoichiometric metal reagents would complicate the quality control (QC) for clinical translation, since additional analysis on the levels of residual metals is necessary. Given these challenges and the prevalence of aromatic C–H bonds [47,48], the direct conversion of aromatic C–H into C(sp2)–18F bonds represents an ideal approach to aromatic 18F-fluorination.
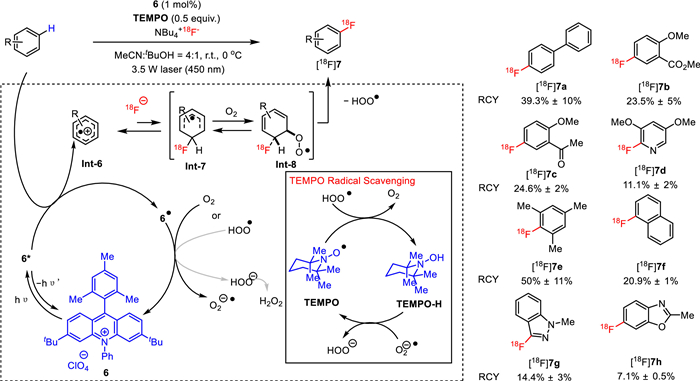
Towards this end, in 2019, Li, Nicewicz and co-workers disclosed a very mild photocatalytic 18F-fluorination of arene C–H bonds with [18F]fluoride [49], which surmounted the problems regarding to functionality incompatibility and poor molar activity when electrophilic [18F]F2, [18F]NFSI, or [18F]Selectfluor was applied to enable C–H 18F-fluorination of aromatics. As shown in Fig. 6, with an acridinium based compound 6 as a photocatalyst, TEMPO (2,2,6,6-tetramethyl-1-piperidine 1-oxyl) as a redox co-mediator, 18F-tetrabutylammonium fluoride (NBu4+18F−, [18F]F−NBu4+, [18F]TBAF) as a radiofluorinating agent, tetrabutylammonium bisulfate as a phase transfer agent, and oxygen air as oxidant, a wide range of 18F-labeled aromatics [18F]7 were produced from the corresponding aromatic substrates under the irradiation of a 3.5 W laser (450 nm) in a solvent of MeCN and t-BuOH (4:1) for 30 min. A moderate to good RCY ranging from 4.9% to 50% was achieved by this method. The total radiosynthesis time of this reaction, including purification, was about 50–60 min, which supported the practical translation of this photocatalytic 18F-labeling approach into human use. A probable reaction mechanism was also proposed. As depicted in Fig. 6, a radical cation intermediate Int-6 was first generated via a single electron transfer between arene and activated photocatalyst 6. Subsequent combination of Int-6 with fluorine-18 proceeded to give the radical species Int-7, which was then trapped by oxygen to afford Int-8. Finally, elimination of a hydroperoxyl radical (HOO•) from Int-8 occurred to arrive at the desired [18F]fluorinated products.
Figure 6
Figure 6.
Photocatalytic direct C(sp2)-H radiofluorination of arenes.
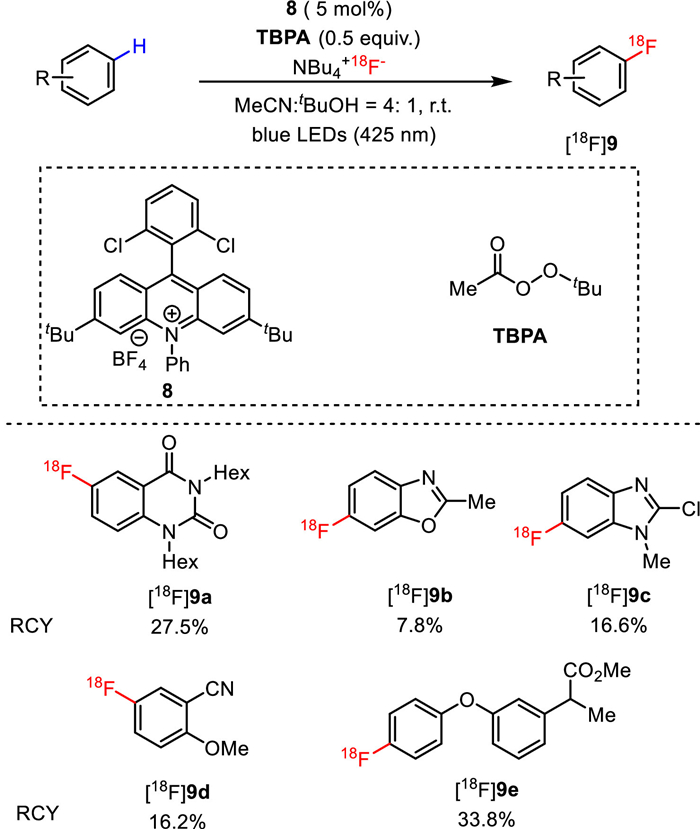
Later in 2020, Li, Nicewicz and co-workers improved this approach by replacing the previously used laser with a readily available low-energe blue LED light source and involving tert‑butyl peroxyacetate (TBPA) as the terminal oxidant (Fig. 7) [50]. A microtubing reactor was utilized to increase the light influx by enhancing the surface area that was exposed to the LED light. The feasibility to omit the azeotropic drying process for C−H radiofluorination was also demonstrated, which remarkably simplified the radiolabeling workflow. As a result, direct 18F-fluorination of various C−H bonds in a wide range of electron-rich aromatics and heteroaromatics were obtained with moderate to good RCYs under the optimized conditions. For instance, in the presence of [18F]F−NBu4+, TBPA, photocatalyst 8 and a solvent mixture of MeCN and t-BuOH (4:1) under blue LED illumination, [18F]9a, [18F]9b, [18F]9c, [18F]9d, and [18F]9e were produced in 27.5%, 7.8%, 16.6%, 16.2% and 33.8% RCYs, respectively.
Figure 7
Figure 7.
Photocatalytic C(sp2)-H radiofluorination of arenes with peroxide as terminal oxidant.
Although SNAr has been routinely applied for radiofluorination of aromatic molecules, this strategy is generally limited to electron-deficient arenes, which is ascribed to the kinetic barriers during the processes of C–F bond formation. In 2020, Li, Nicewicz and co-workers successfully developed a polarity-reversed photocatalyzed deoxyfluorination reaction to achieve radiofluorination of electron-rich arenes via cation-radical-accelerated SNAr [51]. As displayed in Fig. 8, 18F-fluorination of a broad array of arylethers was site-selectively achieved at the more electron-rich aromatic moiety over the less-oxidizable arene nucleofuge by empolying [18F]TBAF as a fluorine source, tetrabutylammonium bicarbonate (TBAHCO3) as a phase transfer agent, acridinium 6 as the photocatalyst in the solvent of 1,2-dichloroethane (DCE): t-BuOH: MeCN (3:4:1) under laser light irradiation (450 nm) for 30 min. Additionally, deoxyradiofluorination was also feasible at the meta position relative to the electron-rich substituent ([18F]10d), suggesting that the substates for this reaction are not limited to those with ortho or para electron-donating groups ([18F]10a-c). With this approach, 18F-fluorinated oestrogen derivatives were obtained in moderate to good RCYs ([18F]10f), providing potential alternatives to radioligand [18F]FES, which serves to visualize and quantify progesterone and oestrogen receptors in primary and metastatic breast cancers [52,53]. The practability of this approach was further highlighted by the highly efficient synthesis of [18F]10g in excellent RCY, which can be readily deprotected to afford [18F]FU ([18F]10g-10l), an important PET tracer for colon cancer [54], with excellent molar activity.
Figure 8
Figure 8.
Photocatalytic radiofluorination of electron-rich arenes.
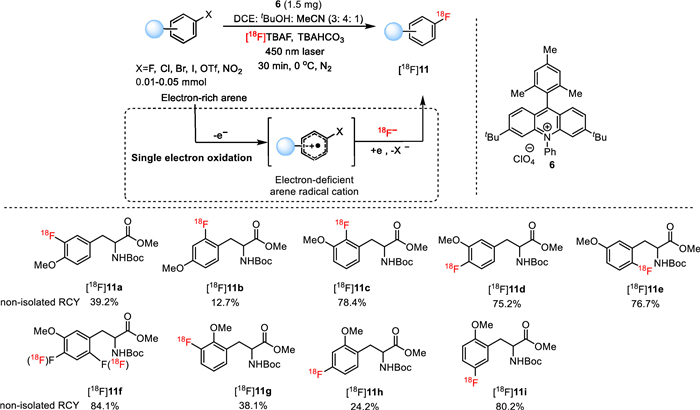
In 2022, Li, Nicewicz and co-workers elegantly utilized photocatalysi to effect the construction of aryl C(sp2)−18F bonds via direct halide/18F exchange in electron-rich arenes under mild conditions [55,56]. As exhibited in Fig. 9, aryl halides were readily converted into their 18F-labelled congers with high non-isolated RCY in the presence of photocatalyst 6, [18F]TBAF, TBAHCO3 under laser irradiation (450 nm) for 30 min in a co-solvent of DCE: t-BuOH: MeCN. The application of this approach was demonstrated by the successful preparation of ten 18F-labelled O-methyl tyrosine analogues [18F]11a-11i in good to excellent non-isolated RCYs (12.7%−84.1%). The underlying mechanism of this reaction was proposed as shown in Fig. 9. In brief, via single electron oxidation, the electron-rich arene was initially turned into electron-deficient cation radical. The further capture of this cation radical by 18F− at the halide-bearing carbon and follow-up reduction proceeded to arrive at the expected radioflurinated arene. Such direct halide/18F exchange approach provides an facile access to preparing PET agents by avoiding synthesizing complicated precursors.
Figure 9
Figure 9.
Photocatalyzed radiofluorination of aryl halides via direct halide/18F exchange.
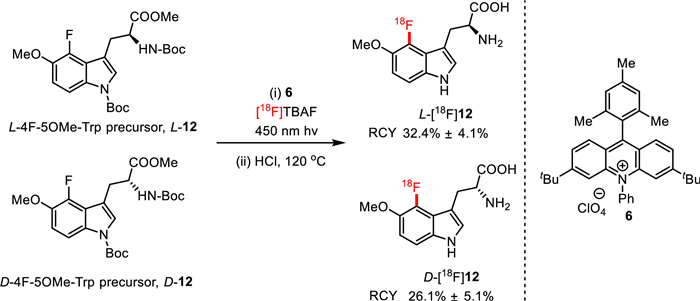
The synthesis of tryptophan-based PET ligands, which have great potential to monitor tryptophan metabolism in various diseases, has been hampered by complicated synthetic procedures. Most recently, Li, Moschos and co-workers provided a novel and facile method to prepare [18F]F-5-OMe-tryptophans via photoredox aromatic radiofluorinantion under mild reaction conditions, by which enantiopure precursors and PET tracers can be directly prepared without racemization [56]. As displayed in Fig. 10, using [18F]TBAF as the radiolabeling reagent, and acridinium 6 as photocatalyst, [18F]F-5-OMe-tryptophans L-[18F]12 and D-[18F]12 were easily generated from the corresponding precursor in the solution of DCE/t-BuOH/MeCN under light (450 nm) irradiation. These tryptophan-based PET ligands were obtained in high radiochemical yields of 2.6% ± 0.5% and 32.4% ± 4.1%, respectively.
Figure 10
Figure 10.
Radiolabeling of tryptophan-based PET agents.
Although fluorine-18 exhibits favorable physicochemical properties for PET, researchers have to be aware of a fact that most bioactive molecules do not contain a fluorine atom. Therefore, fluorine-18 is typically introduced via replacement of a bioisosteric functionality such as a hydrogen atom or hydroxyl group without the generation of significant steric effects [1-3]. Notwithstanding, introducing strongly electronegative fluorine atoms may result in substantial alteration of the pharmacodynamic and pharmacokinetic properties of the target molecules [47]. Considering that carbon atoms are ubiquitous in all virtually bioactive molecules, 11C-radiolabeling is usually regarded as a beneficially alternative approach to overcome the problems caused by bioisosteric replacement with fluorine-18, despite the relatively higher positron range and shorter half-life (t1/2 = 20.3 min) of carbon-11 compared with fluorine-18 [9,44,57,58]. Additionally, the replacement of one carbon atom with its isotope has minimal effect on the intrinsic biological properties of target molecules [59]. Indeed, the past five years have witnessed great progress in photo-mediated 11C-labeling reactions. Several previously elusive precursors have been successfully radiolabeled with carbon-11, which significantly expanding the chemical space of substrates amenable to PET. In this scenario, the recent advances in photocatalytic radiocarboxylation, radiocyanation and radiomethylation are herein summarized.
3.1
Photocatalytic 11C-carboxylation
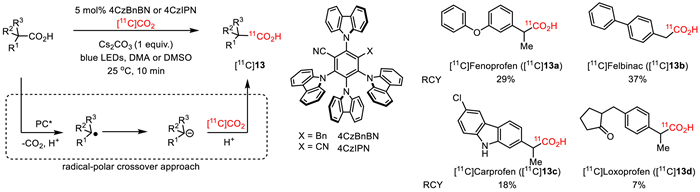
Given the prevalence of carboxylic acids in various pharmaceuticals and natural products, the direct CO2 group exchange offers an opportunity for simple and cost-effective radiolabeling of carboxylic acids. Towards this end, Lundgren, Rotstein and co-workers disclosed an organo-photoredox catalytic direct exchange of CO2 groups of carboxylic acids without the need of high temperatures or prior carboxylate activation [60]. As depicted in Fig. 11, using 4CzBnBN as a photocatalyst, Cs2CO3 as a base, DMA (dimethylacetamide) as a solvent, a broad range of aliphatic carboxylic acids readily underwent fast carbon isotope exchange with [11C]CO2 under mild conditions. This reaction involves a radical-polar crossover process contributed by the low-barrier C—CO2 bond cleavage initiated by carboxylate single-electron oxidation and the following effective recombinantion of carbanion intermediates with [11C]CO2 [61]. A wide range of substrates, including pharmaceutical carboxylic acids and precursors, were tolerated by this reaction. For example, in the presence of 4CzBnBN and Cs2CO3 in DMA, 11C-labeled drug molecules [11C]Fenoprofen ([11C]13a), [11C]Felbinac ([11C]13b), [11C]Carprofen ([11C]13c), and [11C]Loxoprofen ([11C]13d), were successfully prepared by the radiolabeling of corresponding precursors with [11C]CO2 in moderate to good RCY (7%−37%), following 10 min of LED irradiation. [11C]Fenoprofen could be achieved in a lower RCY (9.5%) with a molar activity reaching 0.029 GBq/µmol, when the reaction time was increased to 20 min. It was worth mentioning that this approach is appliable to substrates that were neither tolerated by the transition-metal-catalyzed degradation−reconstruction nor by the thermally induced reversible decarboxylation.
Figure 11
Figure 11.
Photocatalytic carbon isotope exchange of carboxylic acids with [11C]CO2.
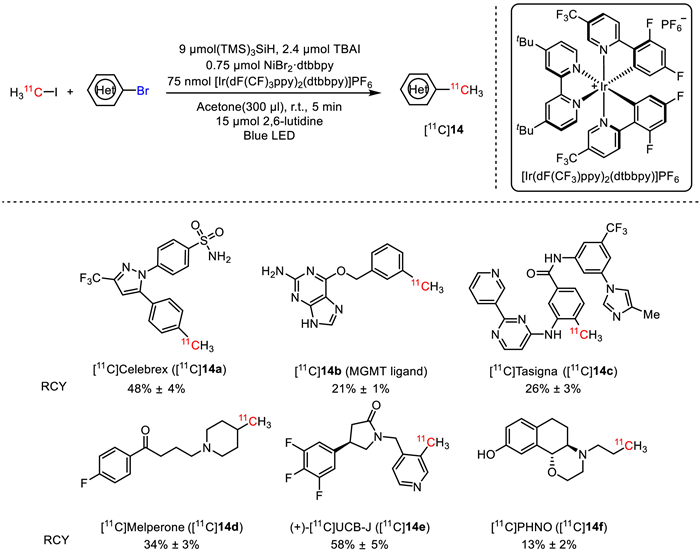
Photochemistry was also employed to effect rapid aryl and alkyl radiomethylation in mild conditions. In 2020, MacMillan's group reported a metallophotoredox-catalytic reaction for late-stage 11C-methylation of pharmaceutical precursors containing aryl and alkyl bromides [29]. As depicted in Fig. 12, the radiolabeling was conducted by using [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (dF(CF3)ppy: 2-(2,4-difluorophenyl)−5-(trifluoromethyl)pyridine) as photocatalyst, NiBr2·dtbbpy (dtbbpy: 4,4′-di‑tert‑butyl‑2,2′-bipyridine) as metal catalyst, (TMS)3SiH (TMS: trimethylsilyl) as reductant, tetrabutylammonium iodide (TBAI) as addictive, 2,6-lutidine as base, and acetone as solvent. With blue light irradiation at room temperature for 5 min, a number of 11C-labelled complex pharmaceuticals and PET radioligands such as [11C]Celebrex ([11C]14a), [11C]14b (a MGMT (O6-methylguanine DNA methyltransferase) ligand), [11C]Tasigna ([11C]14c), and [11C]Melperone ([11C]14d) were effectively prepared in high RCYs ranging from 13% to 58%, indicating the broad applicability of this method. The feasibility of this method was further highlighted by the facile one-step radiosynthesis of the clinically used PET tracers, giving rise to (+)-[11C]UCB-J ([11C]14e) and [11C]PHNO ([11C]14f) in 58% and 13%, respectively.
Figure 12
Figure 12.
Photocatalytic radiomethylation of aryl bromides.
The development of facile approaches to 11C-cyanation of aromatic molecules has attracted great attention due to the ubiquity of aromatic nitriles in pharmaceuticals, natural products, pigments, agrochemicals and herbicides [62-64]. Conventional approaches to 11C-aryl nitriles typically suffer from use of transition metals, high temperature, tedious precursor synthesis, and complicated operation [64]. To address these limitations, several mild and effective photocatalytic 11C-cyanation reactions of aromatic scaffolds have been successfully developed [59,65].
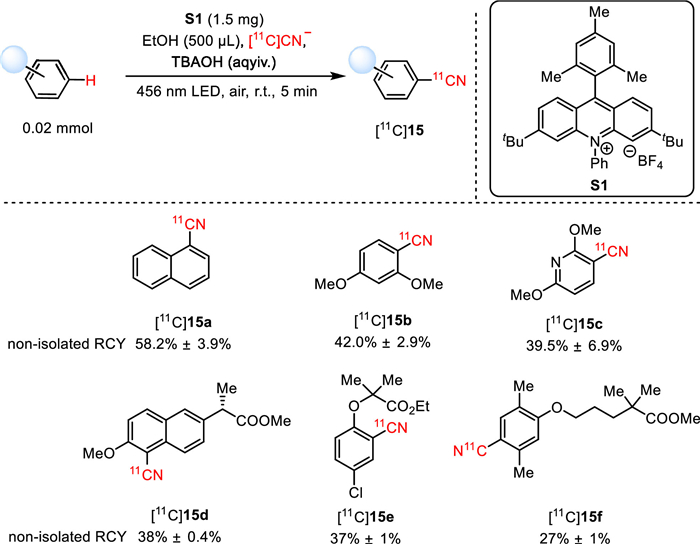
In 2022, Li, Nicewicz, and co-workers innovatively disclosed a direct C(sp2)-H [11C]cyanation method, which incorporates [11C]cyanide into electron-rich arenes via organo-photoredox catalysis (Fig. 13) [59]. By employing [11C]CN− as the radiolabeling source, ethanol as the solvent, tetrabutylammonium hydroxide aqueous solution (TBAOH) as the trapping solution/base, and LED light (465 nm) as the light source, a broad array of [11C]aryl nitriles were obtained in favorable non-isolated RCYs ranging from 16.5% ± 1.8% to 76.0% ± 0.7% with excellent molar activity up to 3.55 Ci/µmol. In particular, late-stage C−H radiocyanation of drugs and bioactive substrates, such as [11C]15d-15f, was successfully effected in mild conditions, affording the 11C-cyanated derivatives in moderate to good non-isolated RCYs (38% ± 0.4% for [11C]15d, 37% ± 1% for [11C]15e, and 27% ± 1% for [11C]15f). Notably, this reaction is not sensitive to moisture and can be performed in the air environment, rendering operationally simple and scalable setup and facilitating feasible production of 11C-cyanated PET tracers without preparing challenging precursors.
Figure 13
Figure 13.
Photocatalytic [11C]cyanation of arenes via direct C−H functionalization.
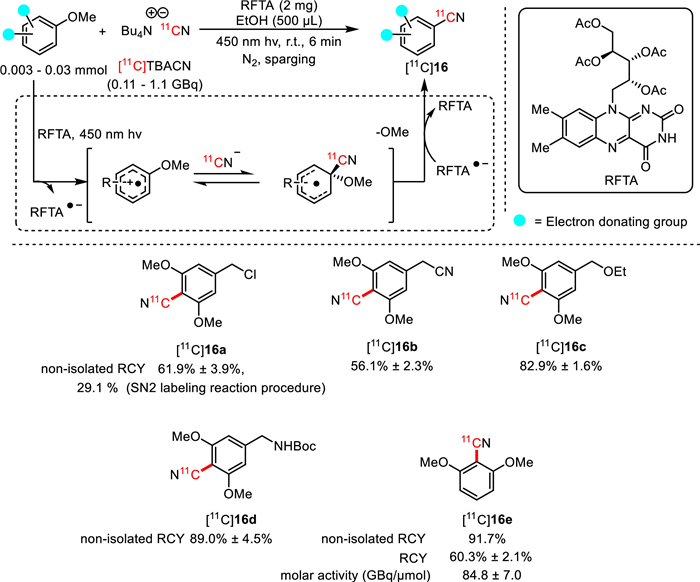
Further in 2023, Li, Nicewicz, and co-workers took advantage of the cation-radical-accelerated SNAr to accomplish 11C-cyanation of electron-rich methoxyarenes via metal-free photocatalysis [65,66]. 11C-cyanation of a wide range of electron-rich methoxyarenes, including super electron-rich substrates, were achieved in excellent non-isolated RCYs (Fig. 14). A particularly compelling characteristic of this photocatalytic approach resided in its different chemo-selectivity from a typical SN2 radiolabeling method. With benzylic chloride as substrate, while the thermal SN2 radiolabeling conditions provided benzylic [11C]nitrile ([11C]16a) in 29.1% non-isolated RCY, the photocatalytic [11C]cyanation conditions provided [11C]16a in 61.9% non-isolated RCY without any benzylic [11C]nitrile product. Additionally, the relatively low precursor loading (0.003–0.03 mmol) also simplified the process for radioligand purification. Notably, excellent molar activity could be achieved to fulfill the demand for translational use in humans (84.8 ± 7.0 GBq/mmol for [11C]16e). It was hypothesized that the cation radical intermediates generated from the corresponding aryl ether substrates might play an important role in this reaction by accelerating the nucleophilic aromatic substitution involving TBA+[11C]CN−.
Figure 14
Figure 14.
Photocatalytic [11C]cyanation of methoxyarenes.
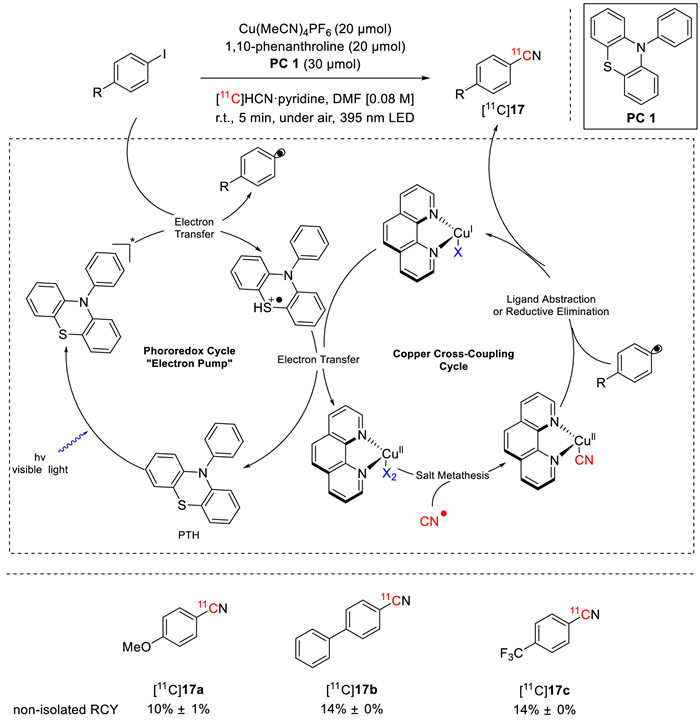
Also in 2023, Scott's group developed a novel and efficient copper-mediated photo-catalytic system for the radiocyanation of aryl iodides in remarkably mild reaction conditions [67]. As shown in Fig. 15, in the presence of Cu(MeCN)4PF6, 1,10-phenanthroline, 10-phenylphenothiazine (PC 1), and [11C]HCN·pyridine in DMF, irradiated with 395 nm LEDs, [11C]cyanation of aryl iodides were feasible at room temperature under air in 5 min, providing expected products [11C]17a-17c in good non-isolated RCYs (10% ± 1% for [11C]17a, 14% ± 0% for both [11C]17b and [11C]17c). The underlying mechanism has been proposed (Fig. 15). Aryl radicals were first generated from aryl iodides via copper-mediated visible-light photoredox catalysis with PC-1. The aryl radicals were then captured by a Cu-11CN species to achieve aryl-11CN coupling, which finally arrived at the expected product.
Figure 15
Figure 15.
Photocatalytic [11C]cyanation of aryl iodides.
The successful translation of a PET radiopharmaceutical with favorable biological properties into clinical use hinges not only on the availability of an efficient radiochemical approach for its benchtop synthesis, but also the feasibility of this benchtop approach for translation into automated or remotely-controlled radiopharmaceutical production under GMP (Good Manufacturing Practice)-required quality assurance levels. To qualify a novel radiolabelling approach as translational radiochemistry [8] for human use, apart from its RCY and molar activity (> 37 GBq/µmol), the translational feasibility for automated or remotely-controlled radiopharmaceutical production in a hot cell also needs to be evaluated. For photocatalytic radiochemistry, its translatability and feasibility of remotely-controlled or automated operation in part relies on the challenging equipment innovation to combine a photoreaction apparatus with the traditional automated radiosynthetic equipment and further movement towards marketization.
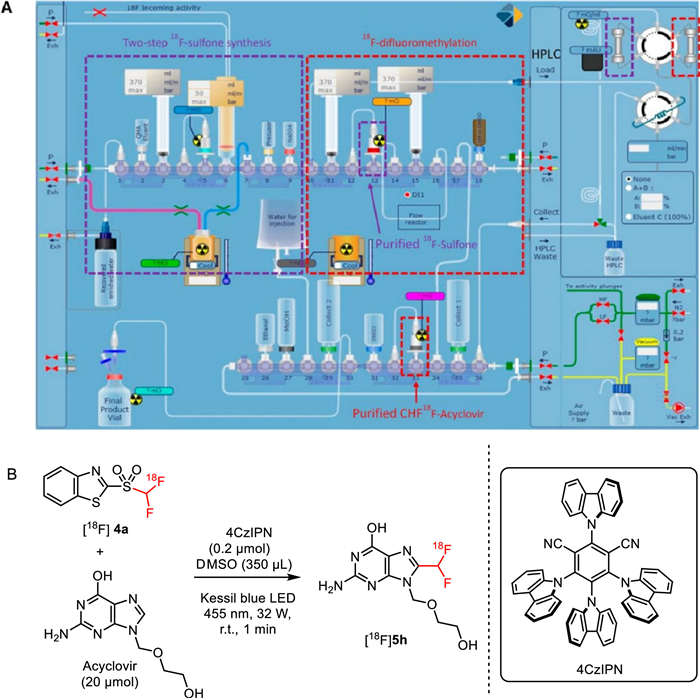
In this scenatio, in 2020, Genicot and co-workers realized the full automation for their previously developed three-step [18F]difluoromethylation of N-heteroarenes including two-step radiosynthesis of reagent and flow photoredox [18F]difluoromethylation using antiherpetic drug acyclovir as a model substrate on a "AllinOne" (AIO) module from Trasis, Belgium (Fig. 16A) [23]. In this automatic system, 2-[18F]((difluoromethyl)-sulfonyl)benzo[d]thiazole was prepared by a one-pot two step reaction, which was similar as the method they reported previously [45] except that the second oxidation step was modified to a liquid phase rather than a solid phase. In the automated photoredox reaction, 4CzlPN was used as a photocatalyst instead of their previously used Ir(ppy)3 to achieve higher RCY. Notably, the automated flow photoredox radiochemistry was the most challenging process, which had never been implemented before this work. To beat off this challenge, Genicot and co-workers designed a perfluoroalkoxyalkane (PFA) tubing reactor via three-dimensional (3D) spiral print, which was perfectly adjusted to a Kessil blue LED. As a consequence, the desired [18F]difluoromethylated acyclovir ([18F]5h) (Fig. 16B) was obtained via 95 min' three-step automation with 1.4% ± 0.1% RCY and a molar activity of 35 GBq/µmol [68].
Figure 16
Figure 16.
Automated three-step synthesis of 18F-difluoromethylatated arenes: (A) AllinOne layout; (B) Application in the sysnthesis of [18F]difluoromethylated acyclovir ([18F]5h). Reproduced with permission [68]. Copyright 2020, American Chemical Society.
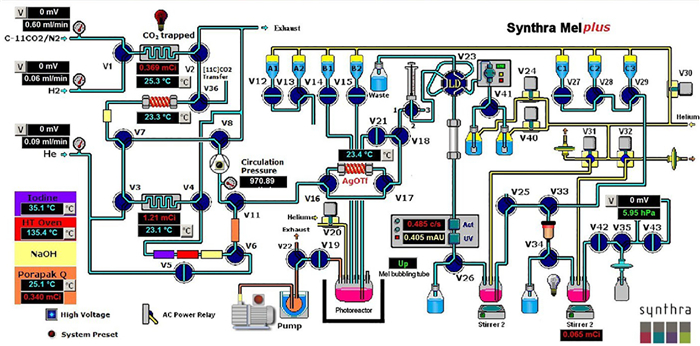
To ensure the reproducibility and enable clinical translation of photocatalyzed [11C]methylation of aryl bromides, MacMillan and co-workers collaborated with the Siemens Molecular Imaging Biomarker Research (North Wales, Pennsylvania) to setup a pair of robotic manipulator arms, which allowed for remotely controlled radiosynthesis of (+)-[11C]UCB-J, a PET tracer used to monitor synaptic density in neurodegenerative disorders [29]. Furthermore, MacMillan and co-workers adapted their previously developed integrated photoreactor [69] with a Synthra MeIplus module, which allowed for a fully automated production of 11C-labeled nonsteroidal anti-inflammatory drug Celebrex ([11C]Celebrex, [11C]14a) within GMP criterion (Fig. 17) [29]. Under identical conditions, the fully automated radiosynthesis of [11C]14a was achieved in 35% isolated RCY (decay-corrected, 43.2 mCi) and good molar activity (2.237 Ci/µmol) within 29 min from the corresponding Celebrex-Br precursor. Notably, quality control (QC) indicated that the residue iridium and nickel contents with [11C]14a accorded with the international recommendations of elemental impurities for clinical radiopharmaceuticals [70].
Figure 17
Figure 17.
Schematic of the Synthra/photoreactor setup. Reproduced with permission [29]. Copyright 2020, The Author(s), under exclusive licence to Springer Nature Limited.
5.
Late-stage functionalization and radiopharmaceutical synthesis for PET imaging
The radiolabeling of bioactive natural products and drug candidates is far from trivial, which frequently requires reconsidering retrosynthetic analysis [71]. With traditional approaches, a complicated or multi-step de novo synthesis is often unavoidable, suffering from step-, resource-, and cost-inefficiency. By contrast, late-stage functionalization represents a more direct, alternative strategy to access radiolabeled natural products or drug candidates [12,72]. As Ritter and co-workers defined, "late-stage functionalization is a desired chemo-selective transformation on a complex molecule to provide at least one analog in sufficient quantity and purity for a given purpose without the necessity for installation of a functional group that exclusively serves the purpose to enable said transformation" [73]. In this regard, photocatalysis involving radical processes has witnessed promising utilization in late-stage functionalization with divergent chemoselectivity from traditional radiolabeling approaches, offering an orthogonal strategy to radiolabeling complex scaffolds and drug molecules. Given that photocatalysis could generate a broad array of radical precursors in a selective and mild pattern, it provides a compelling approach for late-stage radiolabeling different functional groups or even C—H bonds in an atom-economic and chemo-selective pattern.
In 2018, Britton and co-workers successfully effect the site-selective, late-stage C−H 18F-fluorination of peptides using their previously developed photoredox radiochemical approach (Fig. 18) [23]. Under mild reaction conditions, a plethora of unprotected peptides containing a leucine residue were successfully radiolabeled with fluorine-18 at branched leucine position with good RCYs [74]. Likewise, low molar activity ranging from 1.3 MBq/µmol to 5.3 MBq/µmol was obtained for the resultant 18F-labeled peptides by this photo-catalyzed radiolabeling. Particularly, a NAAG (N-acetyl-L-aspartyl-L-glutamate) peptidase inhibitor ZJ-43 was successfully 18F-fluorinated to produce radioligand [18F]ZJ-43 ([18F]18a) in 14% ± 2% RCY, which acted as a prostate cancer imaging agent. Moreover, an analogue of [18F]ZJ-43 ([18F]18b), and [18F]FAfLGEA-NH2 ([18F]18c) targeting cancer-specific receptor EGFRvIII were also successfully prepared with 24% ± 4% RCY and 8% ± 1% RCY, respectively. They demonstrated that both photoactivated NaDT and [18F]NFSI played important roles in the site-selective 18F-fluorinantion at the isopropyl position in leucine residues in unprotected and unaltered peptides [74].
Figure 18
Figure 18.
Late-stage radiofluorination of leucine-containing peptides.
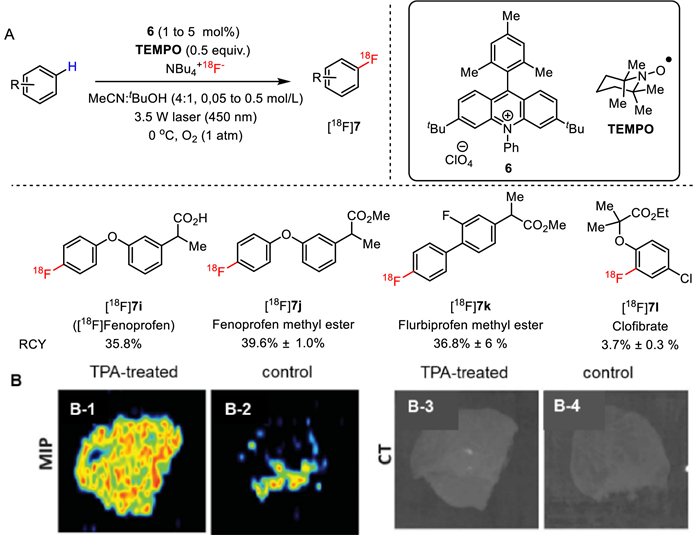
In 2019, Li, Nicewicz co-workers disclosed their success in late-stage radiolabeling of readily available bioactive molecules or their derivatives via a direct photocatalytic arene C−H radiofluorination approach without originally required arene prefunctionalization [49]. Radiolabeling of various nonsteroidal anti-inflammatory drugs (NSAIDs), which belong to an allimportant class of cyclooxygenase (COX) inhibitors to alleviate inflammation and pain, were investigated with this approach, thus giving rise to PET radioligands such as [18F]7i-7l in 3.7%−39.6% RCY (Fig. 19A). Notably, existing 18F-labeled COX radioligands typically introduce fluorine-18 as part of fluorine-containing alkyl chain appended to a phenol, which are inclined to metabolic decomposition and may thus be less effective. The feasibility of translating the NSAID fenoprofen into a clinically relevant PET agent by this approach were also evaluated, resulting in [18F]fenoprofen ([18F]7i) in 35.8% RCY via a one-pot two-step sequence including C−H [18F]fluorination and hydrolysis. Preliminary ex vivo PET imaging studies of [18F]fenoprofen ([18F]7i) exhibited remarkably elevated radioactive levels in the 12-o-tetradecanoylphorbol-13-acetate (TPA)-treated mouse ear (ear inflammation model, B-1) compared with that of control (B-2) (Fig. 19B). These results indicated that [18F]fenoprofen ([18F]7i) may be a promising PET ligand to visualize inflammation in vivo, despite that further biological investigation are still needed.
Figure 19
Figure 19.
Late-stage C(sp2)−H radiofluorination of bioactive molecules (A) and the representative maximum intensity projection (MIP) PET and CT imaging of [18F]7i in TPA-treated and control mouse ears (B). Reproduced with permission [49]. Copyright 2019, The American Association for the Advancement of Science.
Utilizing their developed photocatalytic [11C]cyanation approaches, Li, Nicewicz, and co-workers successfully realized late-stage radiolabeling of a wide range of drug candidates and natural products with a pyrogallol core. As shown in Fig. 20A, with riboflavin tetraacetate (RFTA) as photocatalyst, late-stage [11C]cyanation of podophyllptoxin, colchicine, benzyl, guanidine derivative, and troxipide readily proceeded to afford the expected radioligands [11C]16f-16i in 20.9%−62.2% non-isolated RCYs. A preliminary PET imaging study of [11C]16i was performed in rodents. As depicted in Fig. 20B, high radioactivity levels were observed in the liver, kidneys, and intestines after intravenous injection of [11C]16i, which is consistent with in vivo distribution pattern of colchicine.
Figure 20
Figure 20.
Late-stage [11C]cyanation of bioactive molecules (A) and representative small-animal PET images of [11C]16i (B). Reproduced with permission [66]. Copyright 2022, Elsevier Inc.
The development of small-molecule PET tracers has long been impeded by the stringent constraints endowed by challenges with the short-lived carbon-11 and fluorine-18, the complicated or tedious precursor synthesis for radiolabeling, as well as the incompatibility of most existing radiolabeling approaches with complex scaffolds. Recent breakthroughs in photocatalytic radiochemistry have undoubtedly broadened the practicality of divergent precursors, which are less effective or even inert in traditional radiolabeling methods, such as aryl halides, phenol ethers, carboxylic acids, carboxylic esters, and even various C(sp3)-H and C(sp2)-H bonds. More importantly, photocatalytic radiochemistry enables late-stage radiolabeling of a variety of complex scaffolds in a selective and mild pattern with broad functional group tolerance.
It is important, however, to be aware that although photocatalytic radiochemistry provides plenty of opportunities to radiolabel simple substrates with minimal functionalization or complex bioactive molecules via late-stage functionalization, it typically requires oxygen-free conditions to prevent the destroying of photocatalysts and guarantee the success of radiolabeling, thus complicating the operations to prepare the reaction solution. Additionally, most emerging photocatalytic radiolabeling reactions are still handled manually, which can't be applied to most clinical production. This is partially attributed to the lack of commercially available automated radiosynthesis module, despite that several groups have combined photocatalytic device with existing module or setup robotic manipulator arms to enable automated or remotely-controlled photocatalytic radiochemistry. Therefore, one important direction deserving more efforts in the future resides in the development of highly efficient automated photochemical radiosynthesis modules as well as their further promotion and popularization onto the market. It should also be noted that the use of iridium photocatalysts in some cases may induces issues with regard to purifications and quality controls for further clinical usage. Consequently, it's highly desirable to develop organic photocatalysts as surrogates for iridium complexes to enable radiosynthesis of clinically useful PET ligands.
Declaration of competing interest
The authors declare no competing financial interest.
Acknowledgment
We gratefully acknowledge the financial support from the Basic Research Program of Jiangsu Province (No. BK20220408).
E.W. Webb, K. Cheng, J.S. Wright, et al., J. Am. Chem. Soc. 145 (2023) 6921–6926. doi: 10.1021/jacs.3c00422
[68]
L. Trump, A. Lemos, J. Jacq, et al., Org. Process Res. Dev. 24 (2020) 734–744. doi: 10.1021/acs.oprd.9b00442
[69]
C.C. Le, M.K. Wismer, Z.C. Shi, et al., ACS Cent. Sci. 3 (2017) 647–653. doi: 10.1021/acscentsci.7b00159
[70]
Quality Guidelines, International council for harmonisation of technical requirements for pharmaceuticals for human use (ICH). URL: https://www.ich.org/page/quality-guidelines.
Z. Yuan, M.B. Nodwell, H. Yang, et al., Angew. Chem. Int. Ed. 57 (2018) 12733–12736. doi: 10.1002/anie.201806966
Figure 1
Preparation of (+)-[11C]UCB-J and PET imaging in healthy controls (HC) and Parkinson's disease with dementia with Lewy bodies (DLB). Reproduced with permission from [24]. Copyright 2023. The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Figure 16
Automated three-step synthesis of 18F-difluoromethylatated arenes: (A) AllinOne layout; (B) Application in the sysnthesis of [18F]difluoromethylated acyclovir ([18F]5h). Reproduced with permission [68]. Copyright 2020, American Chemical Society.
Figure 17
Schematic of the Synthra/photoreactor setup. Reproduced with permission [29]. Copyright 2020, The Author(s), under exclusive licence to Springer Nature Limited.
Figure 19
Late-stage C(sp2)−H radiofluorination of bioactive molecules (A) and the representative maximum intensity projection (MIP) PET and CT imaging of [18F]7i in TPA-treated and control mouse ears (B). Reproduced with permission [49]. Copyright 2019, The American Association for the Advancement of Science.
Figure 20
Late-stage [11C]cyanation of bioactive molecules (A) and representative small-animal PET images of [11C]16i (B). Reproduced with permission [66]. Copyright 2022, Elsevier Inc.

 DownLoad:
DownLoad:




 下载:
下载:





















 下载:
下载:
