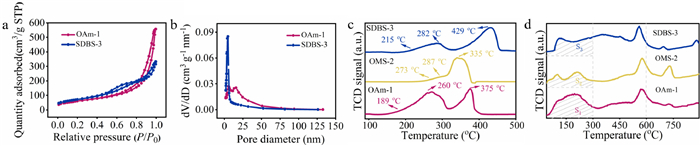
Figure 1.
(a) N2 adsorption/desorption isotherms, (b) pore analysis of OAm-1 and SDBS-3, (c) H2-TPR and (d) O2-TPD.
Mediated electron transfer process in α-MnO2 catalyzed Fenton-like reaction for oxytetracycline degradation
Yiqian Jiang , Zihan Yang , Xiuru Bi , Nan Yao , Peiqing Zhao , Xu Meng

Antibiotic pollution, especially oxytetracycline, tetracycline and chlortetracycline pollution, has become a serious environmental issue. It is reported that the intensive use and inappropriate disposal of antibiotics have contaminated over 60% rivers globally [1,2]. Moreover, the long-term existence of antibiotic pollution will damage people's health and even cause antimicrobial resistance-related deaths [3,4]. Therefore, developing cost-effective technologies to remove these contaminants is imperative. Advanced oxidation processes (AOPs) are regarded as the promising technologies for the degradation and mineralization of refractory antibiotic pollutants [5–7]. AOPs that rely on the in-situ generation of various reactive oxygen species (ROS) from oxidants can degrade organic pollutants into harmless mineralized products [8–16]. Due to the non-selectivity of these ROSs, such as •OH, SO4•− and •O2−, it is difficult to utilize AOPs for the selective degradation of pollutants in a complex aquatic environment. The existence of dissolved organic matter, halides and some certain anions in the environmental matrix also leads to an invalid consumption of oxidants [17]. The shortages of free radicals increase the cost of AOPs and limit their application.
Recently, nonradical oxidation processes, including electron transfer processes, singlet oxygenation and activated high-valent metal, were found to be useful rotes for the degradation of organic pollutants in water [18]. Among these processes, electron transfer processes among catalysts, oxidants and pollutants exhibit oxidation selectivity, high utilization efficiency of oxidants, and tolerance of background interference and pH values in the degradation [19–22]. For example, Wang et al. recently reported an electron transfer process over a Fe-Co dual-atom catalyst via the regulation of peroxymonosulfate (PMS) adsorption mode on the catalyst surface [23]. Li et al. synthesized a mullite oxide with rich surface labile oxygen that conducted the bisphenol A degradation with electron transfer via PMS activation [24]. Although many endeavors have been devoted to the nonradical oxidation for the removal of antibiotic pollutants, most of them focused on the activation of persulfates. Comparatively, the knowledge about activation of H2O2 via a mediated electron transfer mechanism for the degradation of organic contaminants is rather limited.
Manganese oxide catalysts have been extensively exploited in environmental catalysis, such as volatile organic compounds and wastewater treatment, because they are conductive, cheap and environmentally friendly, and have excellent adsorption and oxidation ability to organics [25–28]. α-MnO2-based materials usually can proceed fast electron transfer processes, which makes them good catalysts for a number of chemical transformations [29,30]. Therefore, it is important to fabricate novel α-MnO2 materials with outstanding redox and electron transfer abilities for promoting chemical oxidation reaction. Previously, our study focused on the fabrication and modification of cryptomelane-type α-MnO2 (OMS-2), and we found that OMS-2-based catalysts could activate various oxidants for the synthesis of organic compounds and the degradation of pollutants in water [31–34]. Recently, Chen et al. found that Mn(Ⅲ) of MnO2 catalyst was critical to proceed the degradation of acetaminophen via an electron transfer pathway in PMS-based AOPs [35]. Zhang et al. reported that the oxygen vacancies of MnO2 was responsible for the PMS activation via an electron transfer mechanism [36]. MnO2 materials were used as catalysts in the degradation of biorefractory organics via the activation of H2O2 and further formation of ROSs [37,38]. However, limited attention was paid to the electron transfer involved nonradical processes in H2O2 activation. More importantly, the textural properties, adsorption capacity, redox ability and vacancy concentration of α-MnO2-based catalysts need to be modified and enhanced for catalytic application.
In this study, we used a self-assembly method with the assistance of a surfactant to prepare α-MnO2 nanoribbons (OAm-1) from potassium permanganate (KMnO4) and ethylene glycol. The textural properties, redox ability and conductivity of OAm-1 were studied by various characterization techniques. Compared to OMS-2, OAm-1 had promoted textural properties and enhanced electron transfer ability, although a large amount of n-hexane and ethanol was needed to wash off the surfactant used in the synthesis process. The adsorption and catalytic properties of OAm-1 were investigated in the degradation of oxytetracycline (OTC) via the activation of H2O2, and OAm-1 showed a synergistic effect of adsorption and catalysis for the oxidative removal of oxytetracycline. For the mechanism study, free radical quenching experiments and EPR analysis were conducted to identify the ROSs and nonradical pathway. XPS, FT-IR analysis and more control experiments were performed to illustrate the critical role of Mn(Ⅲ) in OAm-1 and the behavior of adsorption between OTC and OAm-1. Linear sweep voltammetry (LSV) and open-circuit potential (OCP) analysis were used to monitor the electron transfer process during the degradation. Chronoamperometry (CP) analysis was also applied to further elucidate the change of electric current in the OAm-1/H2O2 system. This work explains the effect of Mn(Ⅲ) of the catalyst for the removal of OTC, and gets insight into the mediated electron transfer mechanism between MnO2 and H2O2 through systematic electrochemical methods.
The crystal structures of OAm-1, OMS-2 and SDBS-3 that was synthesized using sodium dodecylbenzenesulphonate as the surfactant with KMnO4 (for details, see Supporting information) were characterized by X-ray diffraction (XRD). In Fig. S1 (Supporting information), seven characteristic peaks (2θ = 12.8°, 18.1°, 28.8°, 37.5°, 41.9°, 49.8°, 60.2°) were detected in OAm-1, OMS-2 and SDBS-3, which indicates they had the pure α-MnO2 (JCPDS, PDF 44–0141) crystal structures consisting of MnO6 octahedra [39]. A peak shift at 65.1° of OAm-1 was observed, which suggests the surfactant used in the preparation caused lattice distortion to some extent and more defects might be generated. Next, FESEM images exhibit that the materials synthesized with surfactants had a similar nest morphology (Fig. S2 in Supporting information), which is different from the morphology of OMS-2. The MnO2 nests were composed by nanoribbons with a diameter of 10–15 nm and a length of several micrometers. And, the density of nanoribbons of SDBS-3 was much higher than OAm-1. With the help of high-resolution TEM, we could see the distinct interface defect existed on the surface of nanoribbons possibly because of the addition of sulfuric acid during the catalyst preparation (Fig. S3 in Supporting information). With the corresponding selected-area electron diffraction (SAED) pattern in hand, the well-identified periodic lattice fringes of 3.1 Å and 4.9 Å were corresponded to the interplanar distance of (310) facet in OAm-1 and (200) facet in SDBS-3, respectively.
N2 adsorption/desorption isotherms were applied to explore the textural properties of the samples (Figs. 1a and b). OAm-1 and SDBS-3 both manifested type-Ⅳ N2 adsorption/desorption isotherms with a H3 hysteresis loop at the pressure (P/P0) range of 0.6–1.0 and 0.5–1.0, reflecting the existence of mesoporous due to the phenomenon of capillary condensation [40]. The pore diameter distribution curves revealed the different porous structures of OAm-1 and SDBS-3 (Fig. 1b). OAm-1 had mesopores with a larger pore size from 5 nm to 20 nm, while SDBS-3 had mesopores with a very small pore size around 3–5 nm and narrow pore diameter distribution. For OMS-2, it had mesopores and macropores (Fig. S4 in Supporting information). Generally, the mesoporous catalysts with smaller and uniform pore size may lead to a long retention time of the in-situ generated complex from the substrate in the void space and accelerate the catalytic performance [41]. Table S1 (Supporting information) lists the textural parameters of the MnO2 samples. Impressively, OAm-1 and SDBS-3 both had a larger specific surface area (221.17 m2/g and 259.85 m2/g, respectively) and total pore volume (0.87 cm3/g and 0.53 cm3/g, respectively) than those of OMS-2 (67.11 m2/g and 0.38 cm3/g). The nest architecture with large surface area and rich porous structure of OAm-1 might benefit the catalytic performance because it affords more active sites and large contact areas for the substrates in the adsorption and degradation.
H2-temperature programmed reduction (H2-TPR) were performed to evaluate the reducibility of the samples and distinguish their surface adsorbed oxygen species (Fig. 1c). All samples experienced the reduction reaction from MnO2 to Mn2O3/Mn3O4 and MnO with the increase of reduction temperature [42]. Compared with OMS-2, OAm-1 and SDBS-3 started to show reduction peaks at around 189 ℃ and 215 ℃, respectively. These reduction peaks mean the consumption of surface labile oxygen species on the materials [43]. OMS-2 showed the reduction peaks at around 273 ℃. Thus, α-MnO2 prepared with the surfactants had better reducibility and more abundant surface labile oxygen species. Between 250 ℃ and 300 ℃, three materials all showed the peaks from the reduction of MnO6-vacancy and MnO6-K+ at the similar temperature region (260 ℃, 287 ℃ and 282 ℃). The reduction peaks from 300 ℃ to 450 ℃ were assigned to the reduction of the lattice oxygen. Compared with OMS-2, the lattice oxygen of OAm-1 and SDBS-3 were harder to be reduced. Next, O2 temperature programmed desorption (O2-TPD) was carried out to investigate the oxygen species in the MnO2 samples. In general, O2-TPD spectra is explained by three sequential stages of oxygen species: the release of surface oxygen molecules/active surface oxygen (low temperatures, < 300 ℃), subsurface lattice oxygen (medium temperatures, 300–600 ℃) and bulk lattice oxygen (high temperatures, > 600 ℃) [44]. The most important observation from Fig. 1d is OAm-1 showed the biggest desorption area at the low temperature region. The amount of surface adsorbed oxygen species from the lower desorption temperature follows the order: OAm-1 > SDBS-3 > OMS-2. Since oxygen is usually adsorbed at the oxygen vacancies of a material, it is reasonable to deduce that OAm-1 owned the largest number of oxygen vacancies and exhibited strong oxygen adsorption and activation ability. Moreover, the Mn-O bond in OAm-1 was looser because the oxygen species released at a very low temperature. These results indicate that the surfactant oleylamine better the physicochemical properties of OAm-1 and led to some positive effects on its catalyst activity in the further degradation reaction.
To investigate the surface chemical properties of the samples, X-ray photoelectron spectroscopy (XPS) analysis was conducted (Figs. S5 and S6 in Supporting information). The XPS survey spectra show the main elements (Mn, O, K) of the as-prepared MnO2 (Fig. S6a). And the XPS spectra of Mn 2p3/2 of all samples were divided into three peaks corresponding to the Mn3+ (642.6 eV), Mn4+ (643.9 eV) and Mn2+ (641.5 eV), respectively [33]. The ratio of different valence Mn was calculated by their peak areas and the results were listed in Table S1. The ratios of low valence Mn (Mn2+ and Mn3+) to Mn4+ in the samples follow the order: OAm-1 (2.92) > SDBS-3 (2.56) > OMS-2 (1.81). Accordingly, the average oxidation state (AOS) of Mn in all samples followed the same order. These results also indicate that OAm-1 with the largest amount of low valence Mn exhibited the highest concentration of oxygen vacancies [45,46]. In the O 1s spectra of the samples, three kinds of surface oxygen species could be distinguished (Fig. S5b). The peaks at 530.0 eV, 531.5 eV and 533.1 eV corresponded to lattice oxygen (Olatt), surface adsorbed oxygen (Osurf) and surface adsorbed water (OadsH), respectively [47]. OAm-1 owned the much higher ration of Oads/Olatt and the most abundant Oads that is tended to participate into the oxidation reaction. In Fig. S7 (Supporting information), the signal intensity in the ESR spectra at g = 2.005 attributed to the unpaired electrons at the oxygen vacancy sites also indicates the abundant presence of oxygen vacancies in OAm-1 [48], which is consistent with the results of O2-TPD and XPS analysis. Thus, OAm-1 might be highly active in oxidation reaction.
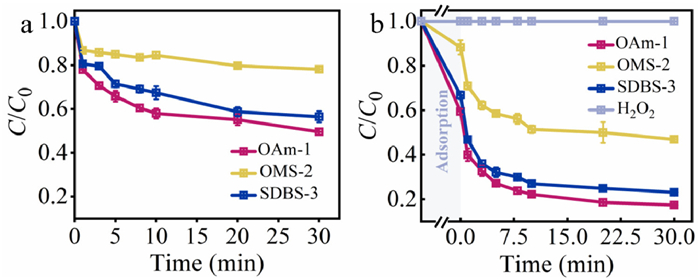
In order to study the catalytic ability of OAm-1, OMS-2 and SDBS-3, OTC was used as the target pollutant to degrade via the activation of H2O2. In Fig. 2a, the adsorption of these catalysts was discussed firstly. The adsorption rate of OTC followed the order: OAm-1 (50%) > SDBS-3 (44%) > OMS-2 (22%). It is worth noting that no characteristic peaks of generated organic intermediates were found by UV–vis spectrophotometer among the reaction of OTC and OAm-1 (Fig. S8 in Supporting information), and this result excluded the oxidation role of OAm-1. FT-IR spectra of OAm-1 in different conditions suggested that OTC might adsorb on the catalyst via chemosorption because blue- and red-shift of adsorbed OTC peaks was observed (Fig. S9 in Supporting information). Fig. S10 (Supporting information) revealed that the adsorption over OAm-1 was better consistent with the pseudo-second-order model and chemosorption played a major role in the rate-limiting step in the adsorption of OTC [49–51]. It is believed that the better adsorption performance of OAm-1 resulted from its large specific surface area, mesoporous structure and abundant oxygen vacancies. Subsequently, the degradation of OTC was studied in the Fenton-like reaction (Fig. 2b). First of all, H2O2 could not oxidize OTC without any catalysts. The OTC degradation efficiency was obviously improved when the as-synthesized α-MnO2 materials were introduced in the reaction. OAm-1, OMS-2 and SDBS-3 led to degradation rates of 82%, 53% and 76%, respectively. And, the degradation rate constant (kobs) of 0.115 min−1, 0.072 min−1 and 0.099 min−1 was obtained from OAm-1, OMS-2 and SDBS-3, respectively. Considering the adsorption ability of the as-synthesized α-MnO2 materials, the removal of OTC was attributed to a synergistic effect between adsorption and degradation. The observation showed that OAm-1 is the optimal catalyst among the as-obtained α-MnO2 catalysts.
Next, the reaction parameters, such as the dosage of H2O2 and catalyst, OTC concentration and pH value, were examined (Fig. S11 in Supporting information). The H2O2 dosage was studied under the standard conditions (Fig. S11a). The degradation efficiency rose gradually with the increase of H2O2 dosage from 5 mmol/L to 200 mmol/L. When the H2O2 dosage was added in 400 mmol/L, the degradation rate did not significantly enhance. It tends to believe that the excessive H2O2 was catalyzed by OAm-1 through an invalid decomposition pathway [52]. In order to enhance the utilization efficiency of H2O2, the addition of H2O2 in portions was applied for the subsequent experiments and the degradation rate reached 82%. According to Fig. S11b, the degradation rate of OTC grew rapidly from 75% to 88% with the increase of catalyst dosage from 1.15 mmol/L to 5.75 mmol/L, and the value of kobs complied with the same tendency. Based on this, the optimum OAm-1 dosage was 3.45 mmol/L. In Fig. S11c, with the increase of OTC concentration, the degradation rate decreased with a falloff in the kobs value. In addition, the influence of the solution pH value and co-existed ions on the degradation were systematically studied. OAm-1 exhibited superior tolerance in acid (pH 3) and basic (pH 10) conditions, showing the similar OTC degradation performance (Fig. S11d). When inorganic anions (Cl−, SO42−, NO3− and HPO42−) and natural organic matters (humic acid) existed in the degradation system, the degradation efficiency was not influenced too much (Fig. S11e). Interestingly, a significant promotion effect of HCO3− for the OAm-1/H2O2 system was observed, which may be about bicarbonate activated H2O2 system to produce more active species for the highly efficiency [53]. Lastly, the degradation kinetics suggested that the OAm-1/H2O2 system gave the lowest apparent activation energy (Ea = 20.604 kJ/mol) in the degradation compared with the OMS-2/H2O2 system (Ea = 32.569 kJ/mol) and the SDBS-3/H2O2 system (Ea = 21.096 kJ/mol) (Fig. S11f). The lowest apparent activation energy of OAm-1 further indicates its catalytic superiority in the degradation process via the activation of H2O2.
To unveil the degradation mechanism, the ROSs were identified by using quenching experiments. The three free radical quenching reagents, including methanol (CH3OH, a sacrificial for •OH radical), sodium azide (NaN3, a sacrificial for 1O2 radical), chloroform (CHCl3, a sacrificial for •O2− radical) were employed. The degradation was almost not affected by the scavenging reagents and only CH3OH showed a slight inhibition (Fig. 3a) [54–56]. However, the signal of •OH and •O2− was detected using EPR measurement with 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO) and 2,2,6,6-tetramethyl-4-piperidinol (TEMP) as the spin-trapping reagents (Fig. 3b). No 1O2 signal was detected. So, it means that free radicals were produced in the catalytic system [57], but they did not directly contribute to the degradation of OTC. These results implied that radical and 1O2-dominated processes were not involved in the degradation reaction.
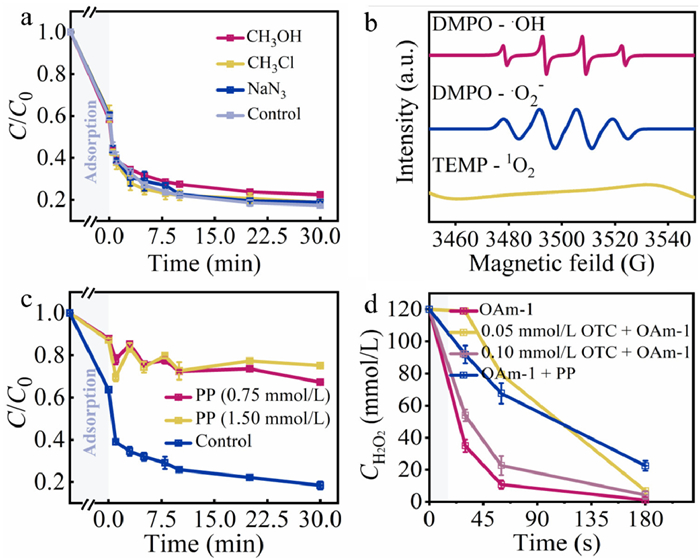
For studying the degradation mechanism, the impact of Mn in the system was investigated. It is known that sodium pyrophosphate (PP) can be applied as a probe molecule for scavenging Mn(Ⅲ) [58–60]. When PP was added in the degradation reaction, the removal rate of OTC decreased significantly (Fig. 3c). Thus, Mn(Ⅲ) of OAm-2 might be the active site during the degradation. Besides, H2O2 decomposition was investigated under the different conditions (Fig. 3d). Normally, MnO2 is inclined to decompose H2O2 through a two electrons transfer [37]. As expected, almost 100% of H2O2 was decomposed by OAm-1, and PP could decrease the conversion of H2O2 because it scavenged Mn(Ⅲ) of the catalyst. Interestingly, we found that the amount of pollutant could affect the decomposition of H2O2 over the catalyst. The addition of OTC (0.05 mmol/L) unexpectedly inhibited H2O2 decomposition rate with OAm-1 although H2O2 was almost decomposed at 180 s. Increasing the addition of OTC from 0.05 mmol/L to 0.10 mmol/L, the conversion rate of H2O2 surprisedly increased. It is concluded that H2O2 decomposition might be related to a complex generated via the adsorption of OTC by OAm-1.
Subsequently, we focused on the electron transfer process in the degradation process through a series of electrochemical methods. First, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were applied to explore the electrochemical performance of the as-synthesize α-MnO2 materials. The CV within a potential window from 0 V to 1 V in 0.5 mol/L Na2SO4 electrolyte was conducted to evaluate the α-MnO2 electrode charge propagation properties (Fig. S12a in Supporting information). The CV curves of OMS-2 and SDBS-3 showed a very narrow area and small current, indicating a minimal specific capacitance contribution [61]. OAm-1 exhibited good charge propagation and irregular rectangular shape at the same scan rates, indicating its excellent electrochemical performance. On the other hand, the interfacial conductivity of the three materials was discussed by means of EIS because more conductive catalysts can lead to faster electron transfer (Fig. S12b in Supporting information). OAm-1 presented the lowest Rct (15.48 Ω) that was smaller than 81.89 Ω of SDBS-3 and 241.1 Ω of OMS-2. The smallest Rct indicates that OAm-1, which has the lowest charge transfer, can facilitate electron transfer in the degradation process [62]. These results prove that the good reducibility and abundant oxygen vacancies of OAm-1 made it more conducive to interfacial charge transfer in the degradation of pollutants [63]. Next, linear sweep voltammetry (LSV) was used to study the electron transfer process in the OAm-1/H2O2 system. As shown in Fig. 4a, when a bare glassy carbon electrode (GCE) was used as the working electrode, no apparent current response was observed in the electrolyte containing both OTC and H2O2. Interestingly, the current density of OAm-1 electrode suddenly increased with the addition of OTC. And a slight red shift occurred in Raman spectra also confirm the consequence of electron transfer during the reaction (Fig. S13 in Supporting information) [64]. Moreover, it should be mentioned again that new peaks appeared in the FT-IR spectra when OAm-1 adsorbed OTC (for detailed discussion, see Fig. S9 in Supporting information). These results suggest that the electron transfer process existed between OAm-1 and OTC and a complex was formed via the chemosorption of OTC on OAm-1 [65].
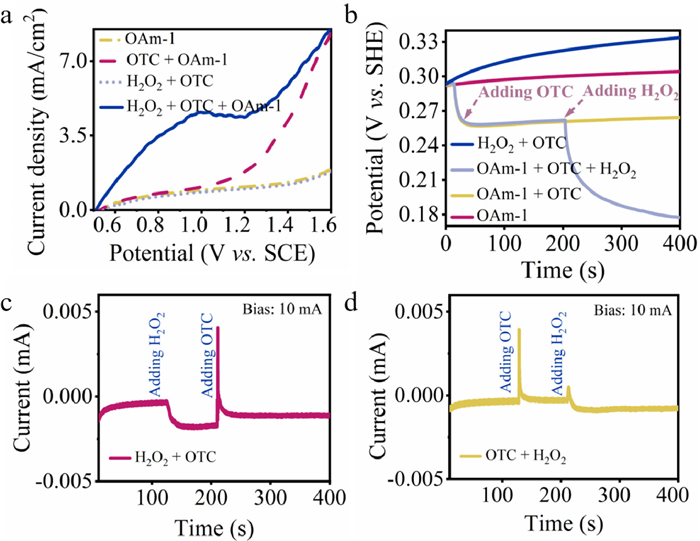
The fast electron transfer in OAm-1/H2O2 system was next revealed by open circuit potential (OCP) test (Fig. 4b). The OCP of bare GCE raised slightly once H2O2 and OTC were added into the solution, which means H2O2 cannot directly degrade OTC without any catalysts. The OCP of OAm-1-coated GCE was stable and below 0.30 V (vs. SHE) after being immersed into the Na2SO4 electrolyte. With OTC in the solution, the OCP decreased immediately and subsequently approached to an equilibrium value at 0.26 V. The observation indicates the complex were formed on the catalyst surface via chemosorption of OTC over OAm-1. If H2O2 was subsequently added, a significant negative offset for the electrode was observed again with an electron transfer.
Lastly, chronoamperometry (CP) was applied to reveal the direction of electron transfer from OTC (electron supplier) to H2O2 (electron withdrawer) via the changes of current signal in the presence of OAm-1-coated GCE. If H2O2 was added firstly, a negative change in the current was observed compared with the current of addition of oxidant before. Then, the addition of OTC initiated a significant positive current. These results imply that the electron transfer took place from OTC to OAm-1 (Fig. 4c). If OTC was added firstly, we detected a slight time-dependent positive change compared with current which no OTC added before. Then H2O2 was added, another relative negative current change was observed (Fig. 4d). Based on these results, it can be inferred that the electron transfer process firstly occurred from OTC to OAm-1 for the formation of complex, and then H2O2 preferred to be an electron withdrawer from the complex in the second process of electron transfer. Thus, the OAm-1 mediated electron transfer process is existed in the degradation process.
According to the XPS spectra of the fresh catalyst and used one (Fig. S14a in Supporting information), the peaks of Mn2+, Mn3+ and Mn4+ in used catalyst were almost maintained at the same position. The proportion of Mn4+ did not change significantly. However, the percentage of Mn3+ decreased from 58.2% to 42.7% and the percentage of Mn2+ increased from 16.3% to 28.9%. The AOS of the used OAm-1 also decreased from 3.66 to 3.43 (Fig. S15 in Supporting information). Therefore, the redox reaction of Mn2+/Mn3+ (and Mn3+/Mn4+) happened during the degradation, which plays a crucial role in the electron transfer process between OTC and H2O2 via the chemosorption. Meanwhile, the increased intensity of Olatt and the decreased proportion of Osurf in used catalyst were observed (Fig. S14b in Supporting information), which demonstrates that immense amount of Osurf participated into the electron transfer process during the degradation.
Based on the above discussion and previous reports [66,67], a catalyst-mediated electron transfer mechanism was proposed in Scheme 1. Firstly, OTC orientally adsorbed onto the Mn(Ⅲ) species of OAm-1 through the chemosorption to form a complex. After that, the electron transferred from OTC to Mn(Ⅲ) and OTC was oxidized at the same time. Subsequently, the second electron transfer process occurred from the electron donor Mn(Ⅱ) to H2O2 that acted as the electron withdrawer. Finally, H2O2 decomposed to H2O and O2. Meanwhile, a small amount of H2O2 was activated by the catalyst to yield hydroxyl free radicals for the degradation. And, in-situ generated O2 accepted electron to yield superoxide radicals.

The recycling experiments of OAm-1 were conducted, which shows that its adsorption ability to OTC decreased after five cycles and the degradation rate still reached around 70% (Fig. S16a in Supporting information). OAm-1 had a good structural stability because the morphology of OAm-1 kept unchanged after the degradation (Fig. S18 in Supporting information). The metal leaching of the catalyst was analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-AES), which shows that 2.289 mg/L of Mn and 1.270 mg/L of K were detected in the filtrate (Table S2 in Supporting information). A Mn2+ salt was used as a homogeneous catalyst instead of OAm-1 in the Fenton-like reaction and only a 20% degradation rate was obtained (Fig. S17 in Supporting information). The result rules out the involvement of the leaching metal in the degradation. On the other hand, the OAm-1/H2O2 system demonstrated excellent degradation activity for chlortetracycline and tetracycline, and the degradation rate of examined antibiotics was significantly accelerated with the addition of H2O2 (Fig. S16b in Supporting information). Especially for TC, an excellent degradation rate of 88% over OAm-1 was realized.
In summary, we successfully prepared α-MnO2 nanoribbons (OAm-1) with the help of oleylamine as a surfactant. As-synthesized material had a large specific surface area of 221 m2/g, abundant mesoporous structures and a large proportion of Mn(Ⅲ). More characterization confirmed that it had outstanding reducibility and conductivity. OAm-1 could be used as a catalyst in the degradation of OTC via Fenton-like oxidation. And, 82% of OTC could be degraded in the OAm-1/H2O2 system. According the free radical quenching experiments, EPR, FT-IR and XPS analysis, the degradation proceeded via a mediated electron transfer pathway. Mn(Ⅲ) was found to be the adsorption sites for OTC and related to the complex formation. More importantly, comprehensive electrochemical characterization and experiments demonstrated that the first electron transfer process occurred between the catalyst and the pollutant to form the complex, and the complex induced the second electron transfer process for the H2O2 decomposition. This study provides a quite novel perspective for understanding a mediated electron transfer in Fenton-like oxidation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Youth Innovation Promotion Association, CAS (No. 2018456), Major Program of Lanzhou Institute of Chemical Physics, CAS (No. ZYFZFX-10), and State Key Laboratory Program of the Lanzhou Institute of Chemical Physics, CAS (No. CHGZ-202211). Also, the authors would like to thank Qian Zhang from Shiyanjia Lab (
Supplementary material associated with this article can be found, in the online version, at doi:
X. Liu, S. Lu, W. Guo, B. Xi, W. Wang, Sci. Total Environ. 627 (2018) 1195–1208. doi: 10.1016/j.scitotenv.2018.01.271
S. Li, Y. Liu, Y. Wu, et al., Natl. Sci. Open 1 (2022) 20220029. doi: 10.1360/nso/20220029
Y. Li, F. Kong, S. Li, et al., J. Hazard. Mater. 456 (2023) 131706. doi: 10.1016/j.jhazmat.2023.131706
A. Frei, A.D. Verderosa, A.G. Elliott, J. Zuegg, M.A.T. Blaskovich, Nat. Rev. Chem. 7 (2023) 202–224. doi: 10.1038/s41570-023-00463-4
J. Wang, R. Zhuan, Sci. Total Environ. 701 (2020) 135023. doi: 10.1016/j.scitotenv.2019.135023
C.H. Han, H.D. Park, S.B. Kim, et al., Water Res. 172 (2020) 115514. doi: 10.1016/j.watres.2020.115514
Y. Liu, Y. Zhao, J. Wang, J. Hazard. Mater. 404 (2021) 124191. doi: 10.1016/j.jhazmat.2020.124191
J. Wang, S. Wang, Chem. Eng. J. 334 (2018) 1502–1517. doi: 10.1016/j.cej.2017.11.059
Y. Pan, Y. Zhang, M. Zhou, J. Cai, Y. Tian, Water Res. 153 (2019) 144–159. doi: 10.1016/j.watres.2018.12.063
L. Zhang, J. Chen, Y. Zhang, et al., Water Res. 216 (2022) 118322. doi: 10.1016/j.watres.2022.118322
Z. Yang, C. Shan, B. Pan, J.J. Pignatello, Environ. Sci. Technol. 55 (2021) 8299–8308. doi: 10.1021/acs.est.1c00230
Y. Zhao, S. Chen, H. Qie, et al., Water Res. 236 (2023) 119957. doi: 10.1016/j.watres.2023.119957
A.A. Isari, M. Mehregan, S. Mehregan, et al., J. Hazard. Mater. 390 (2020) 122050. doi: 10.1016/j.jhazmat.2020.122050
C. Tang, M. Cheng, C. Lai, et al., J. Environ. Chem. Eng. 11 (2023) 110395. doi: 10.1016/j.jece.2023.110395
H. Peng, W. Xiong, Z. Yang, et al., Chem. Eng. J. 457 (2023) 141317. doi: 10.1016/j.cej.2023.141317
C. Guo, M. Cheng, G. Zhang, et al., Environ. Sci. : Nano 10 (2023) 1528–1552. doi: 10.1039/D3EN00007A
J. Dou, Y. Tang, Z. Lu, et al., Environ. Sci. Technol. 57 (2023) 5703–5713. doi: 10.1021/acs.est.3c00022
W. Ren, C. Cheng, P. Shao, et al., Environ. Sci. Technol. 56 (2022) 78–97. doi: 10.1021/acs.est.1c05374
X. Zhou, Q. Zhao, J. Wang, Z. Chen, Z. Chen, Chem. Eng. J. 410 (2021) 128312. doi: 10.1016/j.cej.2020.128312
M. Kohantorabi, G. Moussavi, S. Giannakis, Chem. Eng. J. 411 (2021) 127957. doi: 10.1016/j.cej.2020.127957
Y. Yang, W. Ren, K. Hu, et al., Chem Catal. 2 (2022) 1858–1869. doi: 10.1016/j.checat.2022.05.021
X. Duan, H. Sun, Z. Shao, S. Wang, Appl. Catal. B: Environ. 224 (2018) 973–982. doi: 10.1016/j.apcatb.2017.11.051
F. Wang, Y. Gao, H. Fu, et al., Appl. Catal. B: Environ. 339 (2023) 123178. doi: 10.1016/j.apcatb.2023.123178
C. Yin, Q. Xia, J. Zhou, et al., Sep. Purif. Technol. 280 (2022) 119924. doi: 10.1016/j.seppur.2021.119924
R. Yang, Y. Fan, R. Ye, et al., Adv. Mater. 33 (2021) 2004862. doi: 10.1002/adma.202004862
X. Bi, T. Tang, X. Meng, et al., Catal. Sci. Technol. 10 (2020) 360–371. doi: 10.1039/C9CY01968E
W. Yang, Z. a. Su, Z. Xu, et al., Appl. Catal. B: Environ. 260 (2020) 118150. doi: 10.1016/j.apcatb.2019.118150
M. Zhong, M. Li, Z. Fan, et al., Chin. Chem. Lett. 34 (2023) 107189. doi: 10.1016/j.cclet.2022.01.082
Q. Shi, S. Pu, X. Yang, et al., Chin. Chem. Lett. 33 (2022) 2129–2133. doi: 10.1016/j.cclet.2021.07.063
X. Zeng, C. Shan, M. Sun, D. Ding, S. Rong, Chin. Chem. Lett. 33 (2022) 4771–4775. doi: 10.1016/j.cclet.2021.12.085
L. Zhang, X. Bi, M. Gou, et al., Sep. Purif. Technol. 263 (2021) 118397. doi: 10.1016/j.seppur.2021.118397
X. Bi, Y. Huang, X. Liu, et al., Sep. Purif. Technol. 275 (2021) 119141. doi: 10.1016/j.seppur.2021.119141
N. Yao, H. Zhao, X. Liu, et al., Sep. Purif. Technol. 302 (2022) 122047. doi: 10.1016/j.seppur.2022.122047
X. Wang, X. Bi, N. Yao, et al., Chem. Eng. J. 442 (2022) 136180. doi: 10.1016/j.cej.2022.136180
S. Shen, X. Zhou, Q. Zhao, et al., J. Hazard. Mater. 439 (2022) 129613. doi: 10.1016/j.jhazmat.2022.129613
Y. Deng, P. Gao, L. Wang, et al., J. Environ. Chem. Eng. 10 (2022) 107481. doi: 10.1016/j.jece.2022.107481
A.L.T. Pham, F.M. Doyle, D.L. Sedlak, Water Res. 46 (2012) 6454–6462. doi: 10.1016/j.watres.2012.09.020
C. Wang, J. Fu, Y. Zhang, et al., Chem. Commun. 54 (2018) 7330–7333. doi: 10.1039/C8CC02958J
S. Rong, P. Zhang, F. Liu, Y. Yang, ACS Catal. 8 (2018) 3435–3446. doi: 10.1021/acscatal.8b00456
K.S.W. Sing, D.H. Everett, R.A.W. Haul, et al., Pure Appl. Chem. 57 (1985) 603–619. doi: 10.1351/pac198557040603
D. Chen, G. Zhang, M. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 6377–6381. doi: 10.1002/anie.202013667
P. Vidinha, S. Barreiros, J.M.S. Cabral, et al., J. Phys. Chem. C 112 (2008) 2008–2015.
X. Meng, J. Zhang, B. Chen, Z. Jing, P. Zhao, Catal. Sci. Technol. 6 (2016) 890–896. doi: 10.1039/C5CY01433F
Y. Yang, J. Huang, S. Wang, et al., Appl. Catal. B: Environ. 142 (143) (2013) 568–578.
V.R. Galakhov, M. Demeter, S. Bartkowski, et al., Phys. Rev. B 65 (2002) 113102. doi: 10.1103/PhysRevB.65.113102
X. Liu, K. Zhou, L. Wang, B. Wang, Y. Li, J. Am. Chem. Soc. 131 (2009) 3140–3141. doi: 10.1021/ja808433d
Y. Wang, H. Arandiyan, H.A. Tahini, et al., Nat. Commun. 8 (2017) 15553. doi: 10.1038/ncomms15553
J. Jung, M.J. Lee, Water Res. 36 (2002) 3359–3363. doi: 10.1016/S0043-1354(02)00038-6
J. Ai, W. Zhang, G. Liao, F. Chen, D. Wang, Water Res. 150 (2019) 473–487. doi: 10.1016/j.watres.2018.11.085
J. Wang, X. Guo, J. Hazard. Mater. 390 (2020) 122156. doi: 10.1016/j.jhazmat.2020.122156
J. Wang, X. Guo, Chemosphere 258 (2020) 127279. doi: 10.1016/j.chemosphere.2020.127279
H. Zhou, Y.F. Shen, J.Y. Wang, et al., J. Catal. 176 (1998) 321–328. doi: 10.1006/jcat.1998.2061
H. Pan, Y. Gao, N. Li, et al., Chem. Eng. J. 408 (2021) 127332. doi: 10.1016/j.cej.2020.127332
M. Golshan, B. Kakavandi, M. Ahmadi, M. Azizi, J. Hazard. Mater. 359 (2018) 325–337. doi: 10.1016/j.jhazmat.2018.06.069
N. Motohashi, Y. Saito, Chem. Pharm. Bull. 41 (1993) 1842–1845. doi: 10.1248/cpb.41.1842
A.L. Teel, R.J. Watts, J. Hazard. Mater. 94 (2002) 179–189. doi: 10.1016/S0304-3894(02)00068-7
J. Wang, S. Wang, Chem. Eng. J. 401 (2020) 126158. doi: 10.1016/j.cej.2020.126158
J.K. Klewicki, J.J. Morgan, Geochim. Cosmochim. Acta 63 (1999) 3017–3024. doi: 10.1016/S0016-7037(99)00229-X
E. Hu, Y. Zhang, S. Wu, et al., Water Res. 111 (2017) 234–243. doi: 10.1016/j.watres.2017.01.013
B. Sun, X. Guan, J. Fang, P.G. Tratnyek, Environ. Sci. Technol. 49 (2015) 12414–12421. doi: 10.1021/acs.est.5b03111
J. Huang, S. Zhong, Y. Dai, C.C. Liu, H. Zhang, Environ. Sci. Technol. 52 (2018) 11309–11318. doi: 10.1021/acs.est.8b03383
Y.F. Yuan, J.X. Lin, D. Zhang, et al., Electrochim. Acta 227 (2017) 303–309. doi: 10.1016/j.electacta.2017.01.002
A. Zhang, R. Gao, L. Hu, et al., Chem. Eng. J. 417 (2021) 129186. doi: 10.1016/j.cej.2021.129186
L. Wu, Z. Sun, Y. Zhen, et al., Environ. Sci. Technol. 55 (2021) 15400–15411. doi: 10.1021/acs.est.1c04600
H. Wang, W. Guo, B. Liu, et al., Water Res. 160 (2019) 405–414. doi: 10.1016/j.watres.2019.05.059
Q. Zhang, M. Yu, N. Wang, et al., ACS Appl. Nano Mater. 5 (2022) 9117–9128. doi: 10.1021/acsanm.2c01454
Z. Ma, X. Wei, S. Xing, J. Li, Catal. Commun. 67 (2015) 68–71. doi: 10.1016/j.catcom.2015.04.010

Figure 1 (a) N2 adsorption/desorption isotherms, (b) pore analysis of OAm-1 and SDBS-3, (c) H2-TPR and (d) O2-TPD.

Figure 2 (a) Adsorption and (b) degradation performance on OTC removal in different reaction systems. Reaction conditions: [OTC] = 0.05 mmol/L, [catalyst] = 3.45 mmol/L, [H2O2] = 200 mmol/L, room temperature.

Figure 3 (a) Inhibition effect of radical scavengers on OTC degradation in OAm-1/H2O2 system. (b) EPR spectra of OAm-1/H2O2 system with DMPO and TEMP as the trapping reagents. (c) The effect of PP dosage on OTC degradation in OAm-1/H2O2 system. (d) The effect of the variation of H2O2 concentrations on H2O2 conversion with or without PP. Reaction conditions: [OTC] = 0.05 mmol/L, [OAm-1] = 3.45 mmol/L, [H2O2] = 200 mmol/L, room temperature.

Figure 4 (a) LSV and (b) OCP curves in the different systems by the bare electrode (bare GCE) and OAm-1 electrode. (c, d) I-t curve of OAm-1/H2O2 system in the presence of OTC. Reaction conditions: [OTC] = 0.05 mmol/L, [OAm-1] = 3.45 mmol/L, [H2O2] = 120 mmol/L, room temperature.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: