Ultrafine nano-copper derived from dopamine polymerization & synchronous adsorption achieve electrochemical purification of nitrate to ammonia in complex water environments
Citation:
Xue Zhao, Mengshan Chen, Dan Wang, Haoran Zhang, Guangzhi Hu, Yingtang Zhou. Ultrafine nano-copper derived from dopamine polymerization & synchronous adsorption achieve electrochemical purification of nitrate to ammonia in complex water environments[J]. Chinese Chemical Letters,
2024, 35(8): 109327.
doi:
10.1016/j.cclet.2023.109327
Ultrafine nano-copper derived from dopamine polymerization & synchronous adsorption achieve electrochemical purification of nitrate to ammonia in complex water environments
English
Ultrafine nano-copper derived from dopamine polymerization & synchronous adsorption achieve electrochemical purification of nitrate to ammonia in complex water environments
Faculty of Chemistry and Chemical Engineering, Yunnan Normal University, Kunming 650092, China
b.
National Engineering Research Center for Marine Aquaculture, Marine Science and Technology College, Zhejiang Ocean University, Zhoushan 316004, China
c.
College of Chemistry Nankai University, Nankai University, Tianjing 300071, China
d.
Institute for Ecological Research and Pollution Control of Plateau Lakes, School of Ecology and Environmental Science, Yunnan University, Kunming 650504, China
Received Date:
04 September 2023 Accepted Date:
19 November 2023 Revised Date:
30 October 2023 Available Online:
15 August 2024
Abstract:
Electrochemical-nitrate-reduction-reaction (eNitRR) synthesis of ammonia is an effective way to treat nitrate wastewater and alleviate the pressure of the Haber-Bosch ammonia production industry. How to develop effective catalysts to electrochemically reduce nitrate to ammonia and purify sewage under complex environmental conditions is the focus of current research. Herein, the dopamine polymerization process and the [(C12H8N2)2Cu]2+ complex embedding process were run simultaneously in time and space, and ultrafine Cu nanoparticles (Cu/CN) were effectively loaded on nitrogen-doped carbon after heat treatment. Using Cu/CN as the catalyst, the ammonia yield rate and Faradaic efficiency of the electrochemical conversion of to NH3 are highly 8984.0 µg h−1 mgcat.−1 and 95.6%, respectively. Even in the face of complex water environments, such as neutral media, acidic media, coexisting ions, and actual nitrate wastewater, nitrate wastewater can be effectively purified to form high value-added ammonia. The strategy of simultaneous embedding increases the exposure rate of Cu sites, and the support of CN is also beneficial to reduce the energy barrier of *NO3 activation. This study rationally designed catalysts that are beneficial to eNitRR, and considered the situation faced by practical applications during the research stage, reducing the performance gap between laboratory exploration and industrial applications.
Energy and environmental issues are attacking the whole world, how to achieve clean production is crucial to effectively solve the energy/environmental crisis. As the most extensive industrial activity in the world, ammonia synthesis has continuously delivered raw materials for agricultural and industrial production to humans for more than 100 years, feeding more than seven billion people in the world and promoting the progress of civilization [1,2]. However, the energy-intensive Haber-Bosch ammonia synthesis process absorbs nearly 2% of the energy from fossil energy every year, and releases inestimable greenhouse gases (CO2) to the environment [3–6]. Today, the desire to find pathways to synthesize ammonia under environmental conditions is becoming more and more intense, and methods such as ammonia synthesis by microbial nitrogenase and photo/electrochemical synthesis of ammonia by nitrogen gas have been developed [7–10]. However, the slow kinetics of ammonia synthesis by biological nitrogenase and the difficulty of nitrogen photo/electrochemical ammonia synthesis have cast a veil on the vision of green ammonia synthesis, and there is still a long way to go to find alternative (or partial alternative) pathways about Haber-Bosch ammonia synthesis.
In recent years, it has been found that harmful nitrate ions (NO3−) can be converted into ammonia by electrochemical method, because the bond energy of the N=O bond is much lower than that of the N≡N triple bond, so it is easier to be protonated to form ammonia [11–13]. Nitrate is one of the environmental pollutants, which not only easily causes eutrophication of water body, but also poses a threat to human health [14]. The conversion of NO3− into high value-added ammonia by means of electrochemical reduction is not only beneficial to the purification of nitrate sewage, but also provides an effective way for green synthesis of ammonia [15–17]. For one thing, researchers have achieved initial breakthroughs in catalyst exploration. For example, density functional theory calculations have shown that most metallic materials can electrochemically reduce NO3− to ammonia [18–22], which creates a great space for the choice of catalysts. In the experiment, noble metal materials were first considered to be used to catalyze the electrochemical nitrate reduction reaction (eNitRR) to synthesize ammonia. For example, X. Li et al. constructed Pd nanocrystals on carbon-based materials by in-situ reduction strategy, which realized the electrochemical reduction of NO3−, and the ammonia yield rate reached 1730 µg h−1 cm−2 [23]. J. Li et al. developed a strained Ru noble metal nanocluster that achieves a higher ammonia yield rate and Faradaic efficiency (FE) when catalyzing eNitRR [24]. In addition, Ag-based and Ir-based materials also show fascinating activity in catalyzing eNitRR [25,26]. At the same time, non-noble metal-based materials also exhibit the ability to catalyze eNitRR to synthesize ammonia, especially Cu-based and Fe-based materials [27–33]. For example, X. Zhao et al. derived nano-Cu supported on BCN by constructing metal boron cluster organic polymer materials, and the ammonia yield rate reached 576.2 µmol h−1 mgcat.−1 when catalyzing the conversion of NO3− to NH3 [34]. In addition, materials such as Cu nanosheets and CuNi alloys also exhibit good activity in catalyzing eNitRR [28,35]. Most of the current research is still only on the screening of eNitRR catalysts with high activity and high selectivity. However, for the applicability of eNitRR synthesis, few studies have fully focused on issues related to practical applications, but this is the key to promoting eNitRR ammonia synthesis from experimental research to practical application.
Herein, the cross-linking and self-polymerization process of dopamine and the adsorption process of [(C12H8N2)2Cu]2+ complex were carried out simultaneously in time and cross-linked with each other in space, to achieve the purpose of synchronously intercalating 1,10-phenanthroline-Cu complex when dopamine was polymerized into polydopamine. Under the spatial confinement effect of polydopamine polymer, Cu species dispersed in the nitrogen-doped carbon support in the form of small particle size after heat treatment. Using Cu/CN as the catalyst, the ammonia yield rate and FE of eNitRR reach 8984.0 µg h−1 mgcat.−1 (−0.9 V vs. RHE) and 95.6% (−0.8 V vs. RHE), respectively, and possess electrochemical cycling stability and durability. The biocompatible Cu/CN can electrochemically convert nitrate into ammonia products in complex water bodies, and can reduce the NO3− concentration in actual sewage below the limit value set by WHO or US EPA. Through reasonable design, this research has produced advanced catalysts that are beneficial to eNitRR, and fully considered the possible situations of practical applications in the laboratory research stage, and comprehensively evaluated the application potential of the developed catalysts, which will help to realize the value-added vision (Fig. S1 in Supporting information) of low-value nitrate resources as soon as possible.
The Cu/CN catalyst was constructed by embedding Cu2+ in 1,10-phenanthroline while polydopamine polymerization. Specifically, 1.982 g (10 mmol) of 1,10-phenanthroline was dissolved in 80 mL of hot water (about 80 ℃) to form a homogeneous solution, and then added to 20 mL of a mixed solution containing 0.750 g (4 mmol) of copper nitrate under magnetic stirring (800 rpm). After standing for 2 h, a 1,10-phenanthroline copper ([(C12H8N2)2Cu]2+) complex solution was formed, which was then added to 200 mL Tris–HCl solution (0.417 mol/L, pH 8.5). Add 2.500 g of dopamine hydrochloride dissolved in 10 mL of water to the above mixed solution, stir magnetically at 800 rpm for 24 h, then collect the solid by centrifugation, and dry the solid in a vacuum oven at 80 ℃ for 2 h. Finally, the above solid was heat-treated to prepare Cu/CN according to the following procedure: N2 was used as the carrier gas, the sample was heated from room temperature to 600 ℃ at 5 ℃/min and kept for 2 h, and then cooled naturally under N2 atmosphere. Detailed experimental details, including the preparation of comparative materials, characterization methods and electrochemical experiments, were shown in Supporting information.
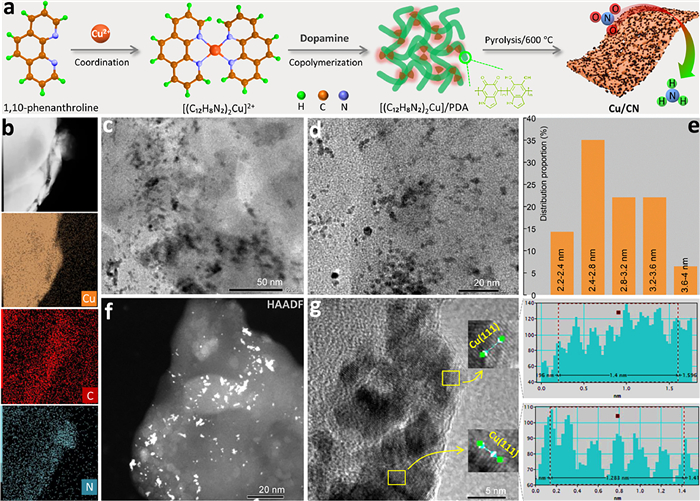
Copper-based materials have initially shown good activity as eNitRR catalysts, but it is still necessary to further improve their catalytic performance. To prevent the large-scale aggregation of copper species and thus reduce the utilization of copper sites, this study chose to simultaneously intercalate the complex ([(C12H8N2)2Cu]2+) formed by 1,10-phenanthroline and Cu2+ during the formation of polydopamine (Fig. 1a). On the one hand, after Cu2+ is complexed by 1,10-phenanthroline, it is not easy to aggregate and form large-particle Cu species during the heat treatment stage. In addition, the synchronous embedding of [(C12H8N2)2Cu]2+ complexes during the formation of polydopamine is beneficial to isolate Cu2+ species again and realize the double isolation effect. Therefore, after heat treatment of the formed composite, the Cu species are not easy to aggregate, and the particle size of the Cu species can be reduced to increase the utilization of Cu sites, thereby increasing the catalytic activity.
Figure 1
Figure 1.
(a) Schematic diagram of the preparation of Cu/CN material. (b) HAADF image and element mapping of Cu/CN. (c, d) TEM images of Cu/CN. (e) Particle size distribution of Cu nanoparticles in Cu/CN. HAADF image (f) and high resolution TEM image (g) of Cu/CN.
After the [(C12H8N2)2Cu]2+ complex was embedded in the polydopamine polymers, the Cu/CN material formed after heat treatment presents an irregular bulk structure with smooth surface (Fig. S2 in Supporting information). When there is no intercalated [(C12H8N2)2Cu]2+ complex in polydopamine, the CN material obtained by heat treatment shows a different morphology, and the overall morphology is regular granular (Fig. S3 in Supporting information). Therefore, it was shown that [(C12H8N2)2Cu]2+ complexes were effectively fused into polydopamine polymers and changed the overall morphology of polydopamine. Element mapping imaging shows that Cu, C, and N elements are uniformly distributed in the Cu/CN material (Fig. 1b), which verifies the effective doping of Cu species. In the high-resolution state, a large number of black particles with a particle size of 2–4 nm are distributed in the transmission electron microscope (TEM) images (Figs. 1c-e). When the TEM imaging mode was replaced by a high-angle annular dark field (HAADF) image mode, the particles distributed in the field of view appear as white bright spots (Fig. 1f). In general, the conventional TEM imaging mode collect signals of transmitted electrons, so atom with larger atomic weight have lower brightness in TEM images because they can block more electron energy. On the contrary, when the imaging mode is HAADF, the collected signal is the scattered electron beam signal, and the more scattered electron beamsignals, the brighter the contrast of the signal [36–38]. Since only one metal species was introduced in the preparation of Cu/CN materials, the nanoparticles in the TEM images should be Cu nanoparticles. Furthermore, the lattice spacing on these nanoparticles is around 0.208 nm (Fig. 1g and Fig. S4 in Supporting information), which is close to that of the (111) crystal plane of Cu with configuration face-centered cubic (fcc, JCPDS No. 04–0836, d(111) = 0.208). Therefore, Cu nanoparticles distributed in Cu/CN are Cu species in fcc configuration.
When [(C12H8N2)2Cu]2+ was not embedded into the polydopamine network, the particle size of Cu in the material (Cu/CN/NPDA) after direct pyrolysis of [(C12H8N2)2Cu]2+ was larger than that of Cu in Cu/CN (Fig. S5 in Supporting information). Therefore, synchronous embedding of [(C12H8N2)2Cu]2+ into the polydopamine network is beneficial to reduce the particle size of the Cu site. Even if it is difficult to change the conductivity (Fig. S6 in Supporting information), exposing more metal sites can increase the electrochemical surface area (ECSA) (Fig. S7 in Supporting information) of the catalyst.
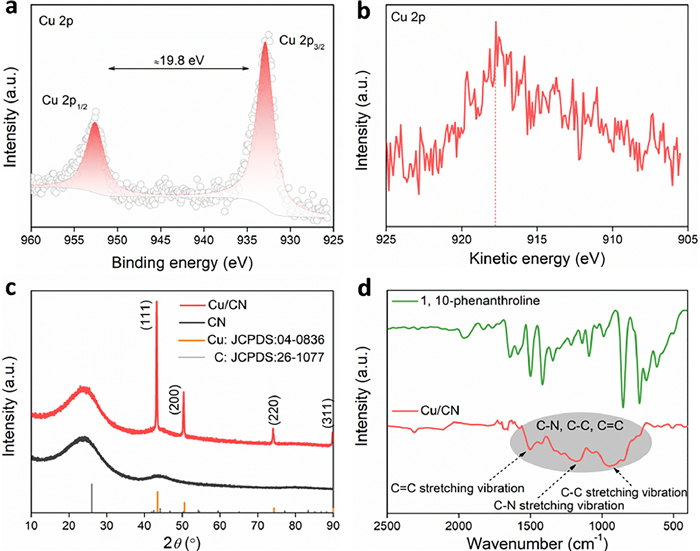
The X-ray photoelectron spectrum (XPS) signals of Cu/CN shows that there are only two signal peaks close to the normal distribution in the Cu binding energy region, and only two signal peaks can be fitted, so the Cu species in Cu/CN is a single component (Fig. 2a). Specifically, the binding energy gap between the signals of Cu 2p3/2 and Cu 2p1/2 is about 19.8 eV [34], while the signal peak of Cu 2p in the Auger spectrum (Fig. 2b) is only one (at 19.8 eV), so the valence of Cu in Cu/CN is zero [39]. In the X-ray diffraction (XRD) signal (Fig. 2c), only the signal of graphitic carbon appears in the CN material, while the obvious Cu signal peak appears in Cu/CN at the same time, which is consistent with the XRD signal of Cu with fcc configuration, which verifies the above inference. In addition, there is no characteristic FTIR signal of 1,10-phenanthroline in the Cu/CN material (Fig. 2d), and only some broad peak (contribution of C—C, C—N, C=N bonds [40]) appears in the range of 700–1500 cm−1, which is similar to the FTIR signal of inorganic graphite carbon nitrogen material, this state can improve the electron transport ability of the carrier. Specifically, organic matter will undergo dehydrogenation at high temperature to form reducing hydrogen ([H]) with reducing ability. After the H atom is removed from the organic matter, the carbon atoms are coupled to each other to form an inorganic carbon network, and the bonding state of the C atom is transformed from the sp3 configuration of the precursor to the sp2 configuration of the graphite carbon (Fig. S8 in Supporting information). This transformation will cause the infrared characteristic peak of the precursor to disappear, and the sp2 configuration can increase the electron transfer and conductivity of the carbon matrix. At the same time, [H] will couple with each other to form hydrogen and escape from the system, and some [H] act as reducing agents to reduce Cu2+ to Cu0. The introduced polydopamine also undergoes dehydrogenation reaction, which is coupled with the material after dehydrogenation of 1,10-phenanthroline, and finally forms a nitrogen-doped carbon matrix.
Figure 2
Figure 2.
XPS spectra (a) and Auger spectra (b) of Cu species in Cu/CN materials. (c) XRD spectra of Cu/CN and CN. (d) FTIR spectra of Cu/CN and 1,10-phenanthroline.
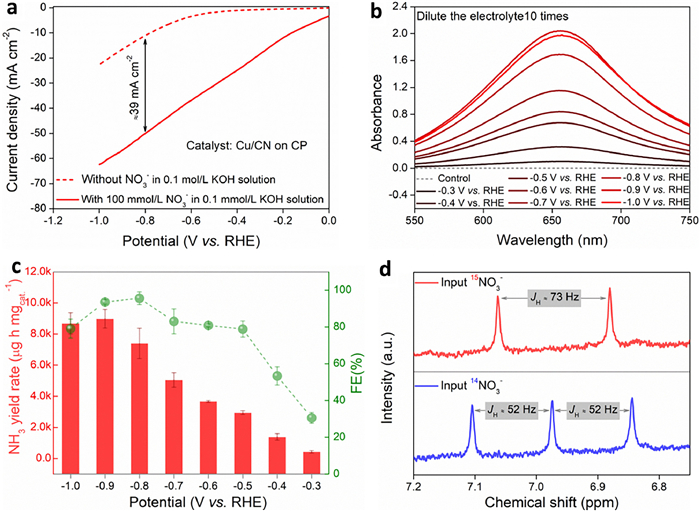
As a validation, the performance of Cu/CN as an eNitRR catalyst was evaluated in detail. When the electrolyte (0.1 mol/L KOH solution) without NO3−, the linear sweep voltammetry (LSV) curve shows that the current density is at a lower value, and the current signal at this time comes from the contribution of the hydrogen evolution reaction (HER) (Fig. 3a). When the electrolyte contained 100 mmol/L NO3−, the collected current signal was greatly improved, indicating that NO3− had reacted on the surface of the catalyst. The presence or absence of NO3− caused a significant difference in the current signal indicates that the Cu/CN material has the potential to catalyze the electrochemical reduction of NO3−. Further, −0.3, −0.4, −0.5, −0.6, −0.7, −0.8, −0.9, and −1.0 V vs. RHE were selected as the applied potentials for the chronoamperometry (CA) test according to the trend of HER activity increase in the LSV curve. After running CA for 1 h, the absorbance of the electrolyte after colorimetry first increased and then decreased slightly with the negative increase of the applied potential, and the absorbance reached the maximum when the applied potential was −0.9 V vs. RHE (Fig. 3b). The change trend of the ammonia yield rate obtained by the standard curve interpolation method is consistent with the change trend of the absorbance with the applied potential. The ammonia yield rate reaches 8984.0 µg h−1 mgcat.−1 and the FE reaches 93.6% when the applied potential is −0.9 V vs. RHE (Fig. 3c), which is superior to most reported catalysts (Fig. S9 and Table S1 in Supporting information). It is worth noting that although the rate of ammonia production was slightly lower at the applied potential of −0.8 V vs. RHE, the FE was higher than that of −0.9 V vs. RHE, up to 95.6%. As the reduction potential increases, the amount of charge (or electrons) through the electrode increases, which increases the rate of conversion of NO3− to NH3 per unit time and thus increases the rate of ammonia yield rate. However, the more negative reduction potential will aggravate the HER side reaction, resulting in a decrease in the proportion of the net increased charge for eNitRR, so the FE will decrease. When the applied reduction potential continues to increase, the HER side reaction is more intense and consumes more charge, resulting in a decrease in both the ammonia yield rate and FE of eNitRR. From the perspective of the utilization efficiency of electric energy, subsequent studies all used −0.8 V vs. RHE as the applied potential. It should be noted that when the applied potential is small (absolute value), both the ammonia yield rate and FE are at a lower value. Since the conversion of NO3− to NH3 undergoes an 8e− transfer process, it is difficult to completely protonate NO3− to NH3 at a low applied potential, so it is easy to accumulate by-products such as NO2−, resulting in a low value of the FE for the generation of NH3. As the negative direction of the applied potential increases, the amount of charge passing through the electrode per unit area and unit time increases, which can quickly and completely convert NO3− into NH3, so the ammonia yield rate and FE are both improved. When the applied potential increases negatively to a certain extent, the FE of synthetic ammonia will decrease because the intensity of HER side reactions will increase accordingly. 1HNMR is an effective method to monitor the source of NH3. Owing to the scalar interaction, the coupling between 1H and 15N in 15NH4+ leads to the 1H resonance splitting into equidistant double peaks, while the coupling between 1H and 14N in 14NH4+ leads to the 1H resonance splitting into equidistant triple peaks [41]. Here, when the NO3− in the electrolyte is all 14NO3−, only the isointense triplet peak of JH = 52 Hz appears in the 1H NMR signal of the electrolyte after running CA (Fig. 3d). However, when all the NO3− in the electrolyte is 15NO3−, only double peaks of equal intensity at JH = 73 Hz appear in the 1H NMR signal of the electrolyte after running CA. Therefore, the ammonia detected in this study all comes from the electrocatalytic reduction process of NO3− on the electrode surface [41], and there is no detectable external ammonia pollution.
Figure 3
Figure 3.
(a) The LSV curves collected with Cu/CN as the catalyst, with or without NO3− in the alkaline medium. (b) The ultraviolet-visible spectrum collected after the electrolyte was colorimetric. (c) Ammonia yield rate and FE at different applied potentials in alkaline media. (d) 1H NMR spectrum of the electrolyte after electrolysis for 1 h when NO3− is 14NO3− and 15NO3− respectively.
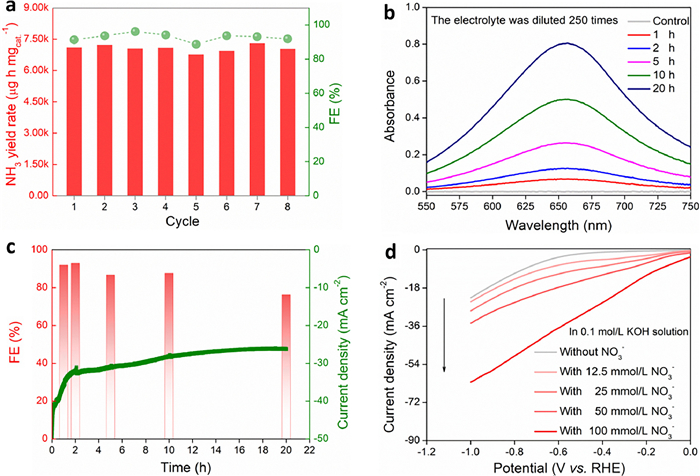
In addition, the electrochemical stability of Cu/CN as eNitRR catalyst was evaluated. On the one hand, after 8 cycles of CA test, the ammonia yield rate and FE of Cu/CN did not decrease significantly compared with the initial value, and only fluctuated slightly around the initial value (Fig. 4a). When running the CA test continuously for 20 h, the ammonia species in the electrolyte tends to gradually accumulate (reflected in the change of absorbance: Fig. 4b), and the FE can be close to 90% at 1 h, 2 h, 5 h, and 10 h (Fig. 4c). When the running time reaches 20 h, FE can maintain around 75%. It should be noted that eNitRR is heavily dependent on the initial concentration of NO3−, and it can also be seen from the LSV curves (Fig. 4d) that the current response is correlated with the concentration of NO3− [30,42]. With the prolongation of CA test time, the concentration of NO3− in the electrolyte decays continuously. At this time, the proportion of NO3− covered on the surface of the catalyst continued to decrease, while the proportion of H2O molecules or H+ species gradually increased, so the HER side reaction gradually strengthened, resulting in a downward trend in the FE of synthetic ammonia. After catalyzing eNitRR, the scanning electron microscope image (Fig. S10 in Supporting information) and XPS signal (Fig. S11 in Supporting information) of Cu/CN catalyst were collected. The morphology of Cu/CN material is similar to the initial state (irregular block structure), and the valence of Cu is still dominated by 0. Overall, Cu/CN has excellent cycle stability and long-term running durability when catalyzing the eNitRR for synthesis of ammonia, and is a catalyst with application-oriented capabilities.
Figure 4
Figure 4.
(a) Ammonia yield rate and FE (−0.9 V vs. RHE) of eNitRR when Cu/CN was reused. (b) Continuous electrolysis for 20 h (in alkaline medium), the UV–vis absorbance of the electrolyte (after colorimetry) in different time periods. (c) Continuous electrolysis for 20 h (in alkaline medium, −0.9 V vs. RHE), FE of eNitRR in different time periods. (d) Effect of initial concentration of NO3− on current response when Cu/CN was used as working electrode.
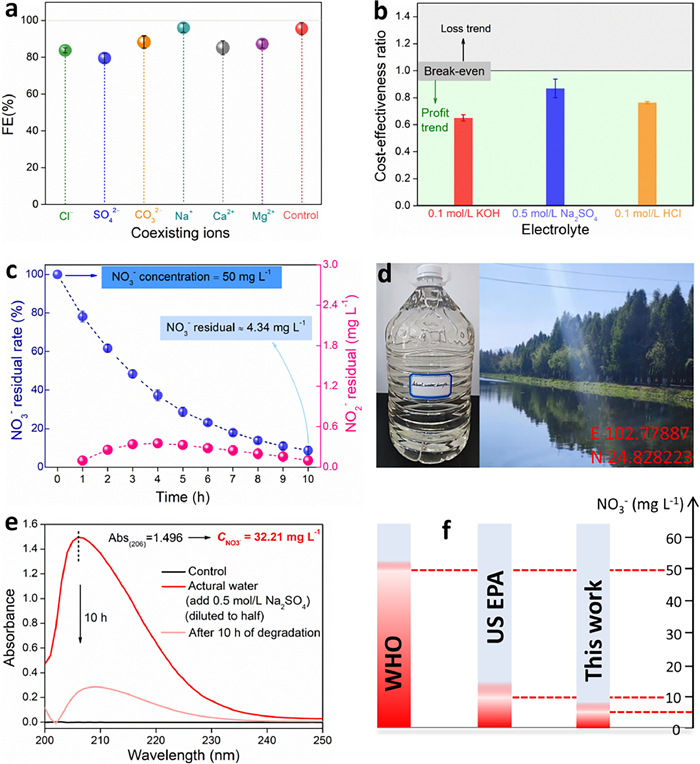
Considering the complexity of nitrate wastewater in the actual environment, some practical factors were introduced in this study. When the electrolyte was a neutral environment, the HER side reactions on the Cu/CN catalyst are not as strong as those in the alkaline environment (Fig. S12 in Supporting information). When containing NO3−, although the applied potential causing the current gain is more negative, it still shows that the eNitRR on the electrode surface occupies an absolute advantage. Although HER (more H+) is more likely to occur in acidic electrolyte, the current gain caused by HER on Cu/CN is only slightly increased compared with that in alkaline medium (Fig. S13 in Supporting information), and the eNitRR on the electrode surface also occupies an absolute advantage. After running the CA test, the ammonia yield rate and FE of eNitRR catalyzed by Cu/CN in neutral medium reached 1925.8 µg h−1 mgcat.−1 and 72.0% (at −0.8 V vs. RHE) (Fig. S14 in Supporting information), respectively, and the ammonia yield and FE in acidic medium also reached 5941.6 µg h−1 mgcat.−1 and 81.5% (at −0.8 V vs. RHE) (Fig. S15 in Supporting information), respectively. These results indicate that the performance of Cu/CN catalytic eNitRR will not change greatly due to the change of pH environment, and it has the characteristics of promoting the conversion of NO3− to NH3 in the whole pH range. Here, the influence of different coexisting ions was also considered. In the results, when the electrolyte contained 10 mmol/L Cl−, SO42−, CO32−, Na+, Ca2+, and Mg2+ the activity of Cu/CN catalytic eNitRR was not significantly inhibited, and the FE remained at a high level of more than 80% (Fig. 5a). Therefore, even if it is only at the stage of laboratory research, it has been shown that Cu/CN materials can serve as effective catalysts for the electrochemical conversion of NO3− to NH3 in complex environments. The results showed that the catalytic activity of Cu/CN for eNitRR was not significantly inhibited when the alkaline electrolyte contained 10 mmol/L Cl−, SO42−, CO32−, Na+, Ca2+, and Mg2+ coexisting ions, respectively, and the FE remained at a high level of more than 80% (Fig. 5a). Among them, Na+ has little effect on the selectivity of NO3− to NH3 on Cu/CN. Cl−, SO42−, CO32−, Ca2+, and Mg2+ ions slightly inhibited the catalytic performance of Cu/CN for eNitRR, but the degree of inhibition was less than 15%. It is worth noting that in neutral media, additional OH− ions are generated on the electrode surface when eNitRR was operated, which easily causes the introduced Ca2+ or Mg2+ to precipitate and cover some active sites of Cu/CN, thereby inhibiting eNitRR (Fig. S16 in Supporting information) [43].
Figure 5
Figure 5.
(a) Effects of coexisting ions on eNitRR driven by Cu/CN in alkaline media. (b) The cost-benefit ratio of ammonia synthesis by eNitRR. (c) Cu/CN as a catalyst to remove NO3− ions in simulated sewage by constant current electrolysis (50 mA, 70 mL electrolyte). (d) The actual sewage collection location and optical image. (e) Cu/CN as a catalyst to remove NO3− ions in actual sewage by constant current electrolysis (50 mA, 70 mL electrolyte). (f) With Cu/CN as the catalyst, the concentration of NO3− ions can be reduced to below the limits specified by WHO and US EPA under electric drive.
Usually, even in the face of severe pollution, the nitrate concentration in natural water will not reach the concentration of 100 mmol/L. However, there are often high concentrations of nitrate species in the sewage produced by some ammonia plants, nitric acid plants, printing and dyeing plants, electroplating plants or food processing plants. Therefore, for these high-concentration nitrate sewage, it can be converted into high value-added ammonia by means of electrochemical reduction, so as to achieve a state where the sewage can be allowed to be discharged. In the process of electrochemical converting nitrate into NO3−, the Cu/CN catalyst developed in this work, regardless of whether the electrolyte environment is alkaline, neutral or acidic, the benefits of the generated ammonia are higher than the cost input (electric energy), resulting in a cost-benefit ratio are lower than 1 (Fig. 5b). Therefore, while treating high-concentration nitrate sewage, it can also achieve a certain degree of profitability.
Considering that NO3− in natural water bodies is at a low level, and it is not operable to concentrate. Therefore, when dealing with excessive NO3− in natural water, this study focused on the residual NO3− concentration. By applying a current of 50 mA to the electrode, the NO3− concentration in the simulated nitrate sewage (50 mg/L NO3− in 0.5 mol/L Na2SO4 solution) decreased continuously with the extension of electrolysis time (Fig. 5c). When the electrolysis time reached 10 h, the residual concentration of NO3− was as low as 4.34 mg/L, and the removal rate was as high as 92.32%, while the production of NO2− was close to zero. This performance outperforms most of the results recently reported (Fig. S17 in Supporting information). When dealing with natural water from river (Laoyu river, Fig. 5d) polluted by NO3−, the concentration of NO3− can be reduced from 32.21 mg/L to 5.14 mg/L within 10 h under constant current electrolysis (Fig. 5e), which is lower than the thresholds set by the World Health Organization (WHO) and the US Environmental Protection Agency (US EPA) (Fig. 5f). It should be noted that the produced ammonia is easier to remove by membrane interception than NO3−. Therefore, from the perspective of purifying nitrate sewage, a reasonable amplification device can be designed and Cu/CN can be used as a catalyst to continuously eliminate excessive NO3− in natural water. In addition, Cu in Cu/CN is zero-valent metal copper, which will not dissolve in aqueous solution, and the CN support is a kind of substance similar to natural biochar. Therefore, Cu/CN has good biocompatibility (Fig. S18 in Supporting information) and will not pose a threat to aquatic animals and plants.
It should be noted that the generated ammonia is easier to be removed than NO3−, and only a reasonable ammonia removal device needs to be set. For example, high-concentration ammonia can be recovered directly using reclaimed resins or extracting methods (for industrial high-concentration nitrate wastewater) (Fig. S19 in Supporting information) [44–46]. The low concentration of ammonia solution can directly flow into the farmland as "liquid fertilizer" after removing the supporting electrolyte (or ionic ammonia) through the flow electrode capacitive deionization device. Clean water can be obtained after the water flow containing low concentration of NH3 undergoes regenerated resins or extracting device again. The vision of electrochemically eliminating nitrates and enriching ammonia requires direct use of clean energy as a driving force to reduce electricity costs.
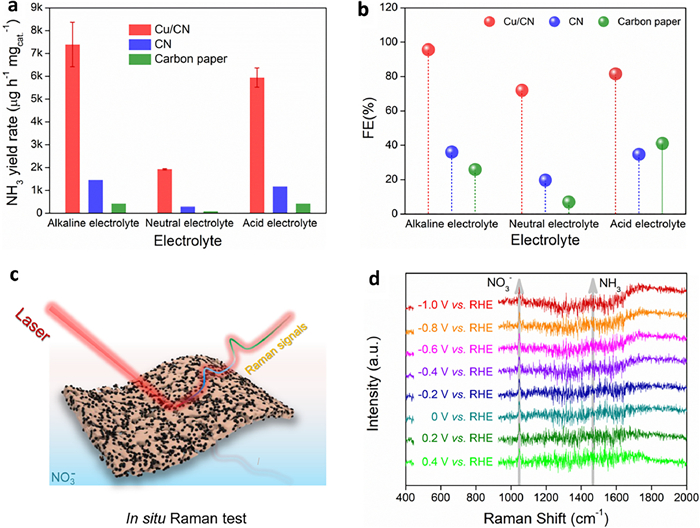
When "CN on carbon paper" and "Carbon paper" were used as the working electrode, respectively, the ammonia yield rate of eNitRR was much lower than that of 'Cu/CN on carbon paper' as the catalyst (Fig. 6a). In addition, when "Carbon paper" was used as the working electrode, the ammonia yield rate of eNitRR is only about 1/3 of that of "CN on carbon paper" as the catalyst. Therefore, the catalytic activity of Cu/CN comes from the Cu species rather than the CN matrix, and the carbon paper as the catalyst carrier has poor catalytic activity, which is not the source of the catalytic activity. In addition, the electrochemical conversion of NO3− to NH3 on "CN on carbon paper" and "Carbon paper" no satisfactory selectivity, and the FE is lower than 40% in alkaline, neutral and acidic media (Fig. 6b).
Figure 6
Figure 6.
The ammonia yield rate (a) and FE (b) of eNitRR when "Cu/CN on carbon paper", "CN on carbon paper", and carbon paper were used as catalysts, respectively. (c) Schematic diagram of testing in situ Raman spectroscopy. (d) Raman spectra collected on the catalyst surface when Cu/CN catalyzed eNitRR.
Using Cu/CN as the working electrode, the Raman signal on the catalyst surface was collected in real time on situ device (Fig. 6c) during the operation of eNitRR. When running eNitRR, the surface of the catalyst can adsorb NO3− (1045 cm−1) [47], and as the applied potential increases negatively, the signal (1470 cm−1) of NH3 gradually appears, indicating that the NH3 in the solution was derived from the surface of the catalyst (Fig. 6d). In addition, it is difficult to find obvious signals represented by NO2− and NO in the in situ Raman spectrum, indicating that NO3− was converted to NH3 with high selectivity, and the short residence time of intermediates makes it difficult to form stable by-products. In the experiment, the FE of NO2− is as low as 3.29% (Fig. S20 in Supporting information), and the ultra-low selectivity leads to obvious in-situ Raman signals that are difficult to collect. In addition, during the conversion of NO3− to NH3, NO2− as an intermediate will be further reduced, and it is difficult to accumulate enough NO2− to present a clear Raman signal.
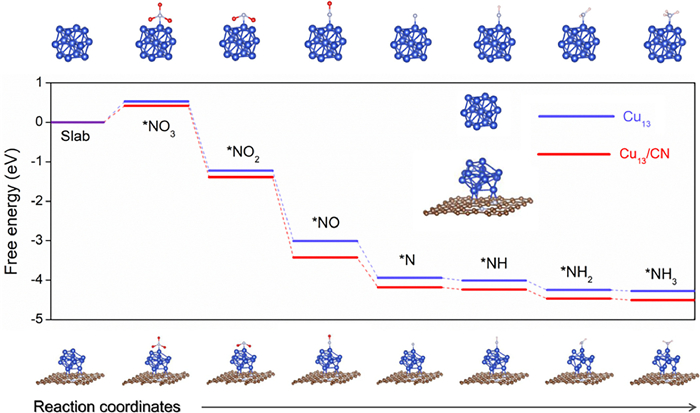
Based on the preparation strategy of Cu/CN, TEM images, XPS and FTIR results, we anchored Cu atom clusters on CN support as a possible model to describe Cu/CN material. Among them, a cluster (Cu13) containing 13 Cu atoms was constructed to replace Cu nanoparticles (Fig. S21 in Supporting information), and graphitic nitrogen was used as the N doping type in the graphitic carbon network (graphitic nitrogen is dominant in Cu/CN, Fig. S22 in Supporting information). Considering that Cu nanoparticles are in contact with the Cu surface in more than one direction in CN, and the CN carrier will affect the performance of the Cu site, a Cu13 cluster with moderate Cu atoms was constructed on the CN matrix (Fig. S23 in Supporting information). In the XPS of Cu/CN material, the fine spectrum of N element shows that the N in Cu/CN is mainly graphite nitrogen (Fig. S24 in Supporting information), with a ratio of more than 80%. Therefore, in the density functional theory (DFT) calculation model, N is doped into the C network in the form of graphite N. During the heat treatment of [(C12H8N2)2Cu]2+, a part of Cu aggregates to form 2–4 nm nanoparticles, where a small number of Cu atoms are still coordinated (or bonded) with C or N. When constructing the Cu13/CN model, the Cu atoms near the CN side were selected to bond with the CN support. Considering that N2H4 was not detected in the electrolyte (the absorbance was consistent with the blank control) (Fig. S25 in Supporting information), and the FE of NO3− to NH3 was as high as 95.6%, a general conversion path was used here: NO3− → *NO3 → *NO2− → *NO → *N→ *NH → *NH2 → *NH3. DFT calculations show (Fig. 7) that the step of adsorbing *NO3− and forming *NO3 is the rate determining step (RDS), which corresponds to the characteristic that the reaction is related to the initial concentration of NO3−. Regardless of whether Cu13 clusters contain carriers or not, the process of further protonation of *NO3− to *NH3 is a process of energy accumulation, so NO3− can be effectively converted into NH3. When Cu13 clusters are supported on the support CN, the energy barrier required for RDS is reduced by about 0.11 eV, which promotes the initiation process of eNitRR and thus promotes the conversion of NO3−. Therefore, the introduction of [(C12H8N2)2Cu]2+ complexes in the polydopamine polymerization stage can not only increase the exposure of Cu species, but also help reduce the energy barrier of NO3− activation, thus promoting the smooth progress of eNitRR. In addition, by introducing polydopamine as the adsorption carrier of [(C12H8N2)2Cu]2+, the particle size of Cu nanoparticles was effectively reduced, and the performance of electrochemical reduction of NO3− to NH3 was improved (Fig. S26 in Supporting information).
Figure 7
Figure 7.
DFT calculations. Pathways for the conversion of NO3− to NH3 on model Cu13 and Cu13/CN, respectively, including the Gibbs free energy change of each transition state. Where, the blue ball is Cu atom, the light blue ball is N atom, the red ball is O atom, the brown ball is C atom, and the pink ball is H atom.
In this study, the dopamine polymerization process and the [(C12H8N2)2Cu]2+ complex embedding process were run simultaneously in time and space, and ultrafine Cu nanoparticles (Cu/CN) were effectively loaded on nitrogen-doped carbon after heat treatment. Using Cu/CN as the catalyst, the ammonia yield rate and Faradaic efficiency of the electrochemical conversion of NO3− to NH3 are highly 8984.0 µg h−1 mgcat.−1 (−0.9 V vs. RHE) and 95.6% (−0.8 V vs. RHE), respectively. Even in the face of complex water environments, such as neutral media, acidic media, coexisting ions, and actual nitrate wastewater, the electrochemical reduction pathway using Cu/CN as a catalyst can effectively purify nitrate sewage and form high value-added ammonia, and this process has the characteristics of profitability. The strategy of simultaneous embedding increases the exposure rate of Cu sites, and the support of CN is also beneficial to reduce the energy barrier of *NO3 activation. This study rationally designed catalysts that are beneficial to eNitRR, and considered the situation faced by practical applications during the research stage, reducing the performance gap between laboratory exploration and industrial applications.
Declaration of competing interest
The authors declare no competing financial interest.
Acknowledgments
This work was financially supported by the Zhejiang Province Key Research and Development Project (No. 2023C01191), the Construction of the Scientific Research Platform of Yunnan Normal University (No. 01100205020503202), the "Union University Innovation Team" of Yunnan Normal University (No. 01100205020503209), and the "Spring City Plan: the High-level Talent Promotion and Training Project of Kunming (No. 2022SCP005)". The authors are also grateful for the support of the "shiyanjia lab" (www.shiyanjia.com) platform in materials characterization.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109327.
[1]
C. Smith, A.K. Hill, L. Torrente-Murciano, Energy Environ. Sci. 13 (2020) 331–344. doi: 10.1039/C9EE02873K
[2]
Y. Liu, E. Huixiang Ang, X. Zhong, et al., J. Colloid Interface Sci. 652 (2023) 418–428. doi: 10.1016/j.jcis.2023.08.106
[3]
B.H.R. Suryanto, H.L. Du, D. Wang, et al., Nat. Catal. 2 (2019) 290–296. doi: 10.1038/s41929-019-0252-4
[4]
F.Y. Chen, Z.Y. Wu, S. Gupta, et al., Nat. Nanotechnol. 17 (2022) 759–767. doi: 10.1038/s41565-022-01121-4
[5]
Q.S. Hong, T.Y. Li, S.S. Zheng, et al., Chin. J. Struct. Chem. 40 (2021) 519–526.
[6]
X. Zhong, E. Yuan, F. Yang, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2306673120. doi: 10.1073/pnas.2306673120
[7]
K.M. Lancaster, M. Roemelt, P. Ettenhuber, et al., Science 334 (2011) 974–977. doi: 10.1126/science.1206445
[8]
C.C. Lee, W. Kang, A.J. Jasniewski, et al., Nat. Catal. 5 (2022) 443–454. doi: 10.1038/s41929-022-00782-7
[9]
Z. Yan, M. Ji, J. Xia, et al., Adv. Energy Mater. 10 (2020) 1902020. doi: 10.1002/aenm.201902020
[10]
X. Zhao, G. Hu, G.F. Chen, et al., Adv. Mater. 33 (2021) 2007650. doi: 10.1002/adma.202007650
[11]
H. Xu, Y. Ma, J. Chen, et al., Chem. Soc. Rev. 51 (2022) 2710–2758. doi: 10.1039/D1CS00857A
[12]
Y. Wang, C. Wang, M. Li, et al., Chem. Soc. Rev. 50 (2021) 6720–6733. doi: 10.1039/D1CS00116G
W. Gao, K. Xie, J. Xie, et al., Adv. Mater. 35 (2023) 2202952. doi: 10.1002/adma.202202952
Figure 1
(a) Schematic diagram of the preparation of Cu/CN material. (b) HAADF image and element mapping of Cu/CN. (c, d) TEM images of Cu/CN. (e) Particle size distribution of Cu nanoparticles in Cu/CN. HAADF image (f) and high resolution TEM image (g) of Cu/CN.
Figure 2
XPS spectra (a) and Auger spectra (b) of Cu species in Cu/CN materials. (c) XRD spectra of Cu/CN and CN. (d) FTIR spectra of Cu/CN and 1,10-phenanthroline.
Figure 3
(a) The LSV curves collected with Cu/CN as the catalyst, with or without NO3− in the alkaline medium. (b) The ultraviolet-visible spectrum collected after the electrolyte was colorimetric. (c) Ammonia yield rate and FE at different applied potentials in alkaline media. (d) 1H NMR spectrum of the electrolyte after electrolysis for 1 h when NO3− is 14NO3− and 15NO3− respectively.
Figure 4
(a) Ammonia yield rate and FE (−0.9 V vs. RHE) of eNitRR when Cu/CN was reused. (b) Continuous electrolysis for 20 h (in alkaline medium), the UV–vis absorbance of the electrolyte (after colorimetry) in different time periods. (c) Continuous electrolysis for 20 h (in alkaline medium, −0.9 V vs. RHE), FE of eNitRR in different time periods. (d) Effect of initial concentration of NO3− on current response when Cu/CN was used as working electrode.
Figure 5
(a) Effects of coexisting ions on eNitRR driven by Cu/CN in alkaline media. (b) The cost-benefit ratio of ammonia synthesis by eNitRR. (c) Cu/CN as a catalyst to remove NO3− ions in simulated sewage by constant current electrolysis (50 mA, 70 mL electrolyte). (d) The actual sewage collection location and optical image. (e) Cu/CN as a catalyst to remove NO3− ions in actual sewage by constant current electrolysis (50 mA, 70 mL electrolyte). (f) With Cu/CN as the catalyst, the concentration of NO3− ions can be reduced to below the limits specified by WHO and US EPA under electric drive.
Figure 6
The ammonia yield rate (a) and FE (b) of eNitRR when "Cu/CN on carbon paper", "CN on carbon paper", and carbon paper were used as catalysts, respectively. (c) Schematic diagram of testing in situ Raman spectroscopy. (d) Raman spectra collected on the catalyst surface when Cu/CN catalyzed eNitRR.
Figure 7
DFT calculations. Pathways for the conversion of NO3− to NH3 on model Cu13 and Cu13/CN, respectively, including the Gibbs free energy change of each transition state. Where, the blue ball is Cu atom, the light blue ball is N atom, the red ball is O atom, the brown ball is C atom, and the pink ball is H atom.

 DownLoad:
DownLoad:

 下载:
下载:








 下载:
下载:
