Received Date:
19 May 2023 Accepted Date:
23 October 2023 Revised Date:
14 October 2023 Available Online:
15 August 2024
Abstract:
Idiopathic pulmonary fibrosis (IPF) is a chronic and fatal lung disease characterized by pulmonary inflammation, oxidative stress, and excessive extracellular matrix (ECM) deposition. Current anti-fibrotic drugs for IPF treatment in the clinic lack selectivity and demonstrate unsatisfactory efficacy, highlighting the urgent necessity for a novel therapeutic strategy. Taraxasterol (TA), which has biological activities against lung injury induced by various factors, is a potential anti-IPF drug due to its anti-inflammatory, antioxidant and lung-protective effects. However, the protective effect of TA on IPF has not been confirmed, and its clinical application is limited due to its poor aqueous solubility. In this study, we demonstrated that TA could inhibit epithelial-mesenchymal transition (EMT) and migration of A549 cells by inhibiting the transforming growth factor-β1 (TGF-β1)/Smad signaling pathway. To improve the aqueous solubility and pulmonary administration performance of TA, we prepared TA loaded methoxy poly(ethylene glycol)-poly(d, l-lactide) (mPEG-PLA)/d-α-tocopheryl polyethylene glycol succinate (TPGS) mixed polymeric micelles (TA-PM). Then a MicroSprayerⓇ Aerosolizer was used to deliver TA-PM once every two days for three weeks to evaluate their therapeutic effects on bleomycin (BLM)-induced IPF mice. Our results demonstrated that inhaled TA-PM significantly inhibited BLM-induced inflammation, oxidative stress and fibrosis in lung tissue. Furthermore, TA-PM exhibited high pulmonary deposition and retention by pulmonary administration, along with a favorable safety profile. Overall, this study emphasizes the potential of inhaled TA-PM as a promising treatment for IPF, providing a new opportunity for their clinical application.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and irreversible interstitial pneumonia with the pathological characteristics of fibrotic tissue in the lung parenchyma and greatly diminished lung function [1]. These pathological changes of lung tissue bring about a poor prognosis with a median survival of only 2–4 years [2]. Many factors, such as virus infection, chemotherapy, and genetic susceptibility, are able to cause IPF [2]. Clinical reports have shown that IPF is a meaningful clinical feature and sequela of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection [3]. In the progression of IPF, injured type Ⅱ alveolar epithelial cells (AEC Ⅱ) are abnormally activated and release various cytokines to facilitate the proliferation and differentiation of fibroblasts, resulting in myofibroblastic foci formation, excessive extracellular matrix (ECM) deposition, and abnormal lung structural remodeling [4–6]. Furthermore, it has been considered a key event in IPF that AEC Ⅱ differentiate into myofibroblasts through the process of epithelial-mesenchymal transition (EMT) [7]. Currently, only two drugs, pirfenidone (PFD) and nintedanib, have been approved by the U.S. Food and Drug Administration (FDA) for IPF treatment because they have slowed disease progression compared to placebo [8]. Unfortunately, these medications have shown poor selectivity, undesired gastrointestinal side effects, and limited effectiveness, hence remaining far from achieving IPF revision [9]. Therefore, there is a critical need to develop a novel drug and an exceptional delivery system for IPF treatment.
In traditional Chinese medicine, dandelion is widely used to alleviate upper respiratory tract infections, bronchitis, and pneumonia [10]. Taraxasterol (TA), one of the main bioactive components of dandelion, is a pentacyclic triterpenoid compound with anti-inflammatory, antioxidant and antitumor effects [11]. TA has shown good biological activities in respiratory diseases such as acute lung injury, acute respiratory distress syndrome (ARDS) and asthma [12–14]. It has been reported to ameliorate lung injury by inhibiting the release of inflammatory cytokines and inflammatory mediators [12,15,16]. Moreover, TA could significantly increase the activity of antioxidants such as superoxide dismutase (SOD) and catalase to treat sepsis-induced ARDS [13]. However, whether TA has a protective effect on IPF remains unclear, and its low oral bioavailability and poor aqueous solubility, as a phytosterol, often restrict its application in the pharmaceutical field [17].
Inhalation has been regarded as a non-invasive and appealing method to deliver active ingredients to pulmonary lesions, improving efficacy while diminishing systemic side effects [18–21]. However, the respiratory epithelium is covered by a mucus layer, which poses a challenge for inhalation therapy as drugs have to penetrate the mucus barrier in order to reach the lower cells rapidly [22–24]. As an excellent penetration enhancer, d-α-tocopheryl polyethylene glycol succinate (TPGS) can reduce the mucus-nanoparticle interaction and improve the mucus-penetrating property of the nanoparticles [25,26]. Its amphiphilic structure also makes it useful as a solubilizer and emulsifier in nanomedicines [27]. Additionally, TPGS can act as an absorption enhancer owing to the presence of vitamin E in its structure, which can induce cellular uptake through membrane receptors [28,29]. However, the relatively high critical micelle concentration (CMC) of TPGS (approximately 0.2 g/L) leads to its internal instability [30]. Therefore, it is usually combined with other amphiphilic materials to increase the stability of the system [31,32]. Methoxy poly(ethylene glycol)-poly(d, l-lactide) (mPEG-PLA), as an FDA approved excipient, has been popularly used as a safe drug nanocarrier due to its good biocompatibility, excellent self-assembly ability, and high drug loading efficiency [33–35]. Therefore, it can be hypothesized that the mixed micelles prepared from TPGS and mPEG-PLA can facilitate drug permeation and ensure stable dilution performance.
In this study, we found that TA could inhibit EMT in A549 cells, demonstrating its potential anti-IPF effect in vivo. To improve the water-solubility of TA and augment its accumulation in the lungs, the TA loaded mPEG-PLA/TPGS mixed polymeric micelles (TA-PM) were developed for IPF treatment in vivo. All animal studies were conducted in accordance with the guidelines of the Experimental Animal Ethics Committee of Ocean University of China (No. OUC-AE-2023134). As expected, TA-PM exhibited superior pulmonary deposition and retention by pulmonary administration. Furthermore, they showed the ability to inhibit bleomycin (BLM)-induced IPF through anti-inflammatory and antioxidant effects. Therefore, our mPEG-PLA/TPGS mixed polymeric micelles-based delivery system for pulmonary drug delivery may provide a promising paradigm and pave the way for overcoming the obstacles of IPF treatment.
The viability of A549 cells, which have important characteristics of AEC Ⅱ [36,37], was evaluated using the cell counting kit-8 assay. No cytotoxicity of TA (Fig. S1A in Supporting information) was observed at concentrations of 2.5, 5, and 10 µg/mL within 24 h of administration (Fig. S1B in Supporting information), so the subsequent cell experiments were performed at these concentrations.
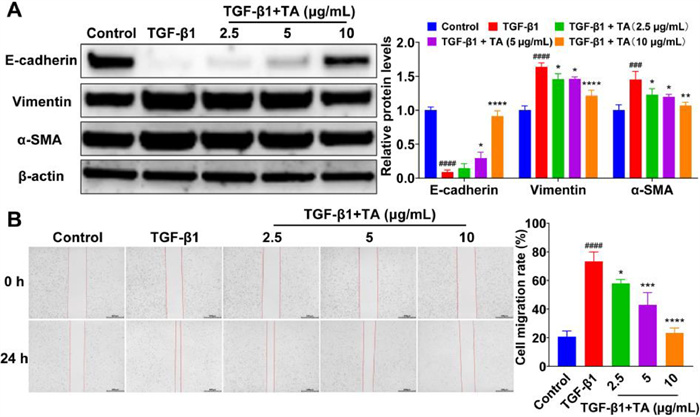
Transforming growth factor-β1 (TGF-β1) is considered a crucial fibrogenic cytokine that stimulates AEC Ⅱ to undergo EMT in IPF [38]. To evaluate the effect of TA on the EMT process, TGF-β1 was used to trigger EMT in A549 cells. As shown in Fig. 1A, TGF-β1 treatment significantly upregulated the mesenchymal markers vimentin and α-smooth muscle actin (α-SMA) while significantly downregulating the epithelial marker E-cadherin. Instead, TA treatment significantly downregulated vimentin and α-SMA, and significantly upregulated E-cadherin in a concentration-dependent manner when compared to those in TGF-β1-only-treated cells, indicating that TA could inhibit TGF-β1-induced EMT (Fig. 1A). The migratory and invasive abilities of epithelial cells were potentiated when the adhesion between cells was diminished during EMT [39,40]. To investigate the effect of TA on A549 cell migration under TGF-β1 stimulation, we performed wound healing assay. As shown in Fig. 1B, TGF-β1 treatment significantly increased A549 cell migration ability while TA treatment suppressed TGF-β1-induced cell migration, which might result from the antagonizing effect of TA on E-cadherin expression (Fig. 1A). The inhibitory effect of TA on the EMT urged us to further explore its mechanism. It has been reported that TGF-β1 primarily activates EMT through activation of the TGF-β1/Smad signaling pathway, leading to the development of IPF [41]. In this study, TA treatment significantly decreased the levels of phospho-Smad2 of A549 cells exposed to TGF-β1 (Fig. S2 in Supporting information), suggesting that TA has the potential to alleviate IPF by inhibiting the occurrence of EMT through inhibition of the TGF-β1/Smad signaling pathway.
Figure 1
Figure 1.
Effect of TA on the expression of EMT-related proteins and the migration of A549 cells. (A) Western blot analysis of EMT-related proteins. (B) Wound healing assay, scale bar: 500 µm. Data are presented as the mean ± standard deviation (SD) (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the TGF-β1 group; ###P < 0.001 and ####P < 0.0001 compared to the control group.
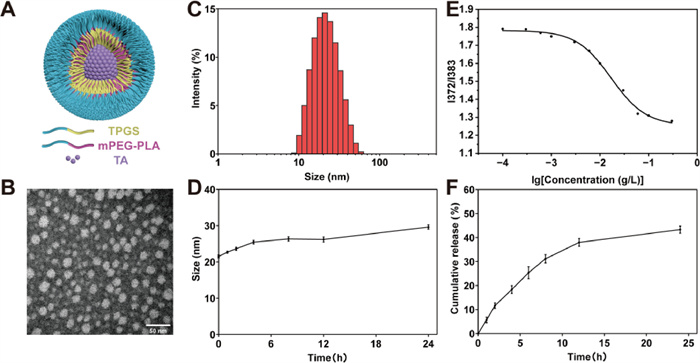
Although TA has been proven to have a favorable anti-EMT effect in vitro, its potential use in vivo is limited by its poor aqueous solubility. To improve the aqueous solubility and pulmonary accumulation of TA, an inhalable drug delivery system was developed to encapsulate TA into mPEG2000-PLA2000/TPGS mixed micelles. The diagrammatic structure of TA-PM is shown in Fig. 2A. Transmission electron microscope (TEM) revealed that TA-PM were monodispersed with a uniform spherical morphology and a particle size of just about 20 nm (Fig. 2B). Dynamic light scattering analysis showed that the average particle size of TA-PM was 20.62 ± 0.42 nm with a single-peak distribution (Fig. 2C) and a low polydispersity index (PDI) of 0.127 ± 0.02 (Table S1 in Supporting information). The small particle size of about 20 nm, along with the dense PEG shell, contributed to enhanced mucus penetration of TA-PM and reduced clearance by macrophages [42,43]. The zeta potential analysis revealed a zeta potential of −4.19 ± 0.10 mV for TA-PM (Table S1). The near-neutral surface charge made TA-PM more conducive to rapidly penetrating the mucus barrier [44]. In addition, the stability of TA-PM was measured in vitro, and no great change in particle size was observed within 24 h when TA-PM were incubated with phosphate buffered saline (PBS) at pH 7.4 and 37 ℃, indicating their superior stability (Fig. 2D). The encapsulation efficiency and drug loading of TA-PM were 92.38% ± 1.09% and 4.40% ± 0.05%, respectively, indicating that most TA can be encapsulated by the mixed micelles (Table S1). The CMC was a parameter indicative of the self-assembly ability of polymer materials [45]. With a low CMC, the polymer materials form micelles with a strong anti-dilution capacity, preventing depolymerization [46]. According to previous research, x0 is considered the common logarithm of CMC [47]. As shown in Fig. 2E, the mPEG2000-PLA2000/TPGS equal-mass mixture displayed an obvious lower CMC of 0.017 g/L than that of TPGS (~0.2 g/L). Then, a simulated lung fluid (SLF) [48] was used to explore the release behavior of TA-PM. And TA-PM released only 43.3% of TA within 24 h, indicating their slow-release performance, which could reduce the administration frequency and prevent potential toxicity caused by high concentrations (Fig. 2F). Collectively, the above characterization results indicated that TA-PM might have favorable pulmonary drug delivery performance.
Figure 2
Figure 2.
Characterization of TA-PM. (A) Diagrammatic structure of TA-PM. (B) TEM image of TA-PM, scale bar: 50 nm. (C) Particle size distribution of TA-PM. (D) Stability of TA-PM. (E) CMC of the mPEG2000-PLA2000/TPGS equal-mass mixture. (F) TA accumulative release curve from TA-PM in SLF. Data are presented as the mean ± SD (n = 3).
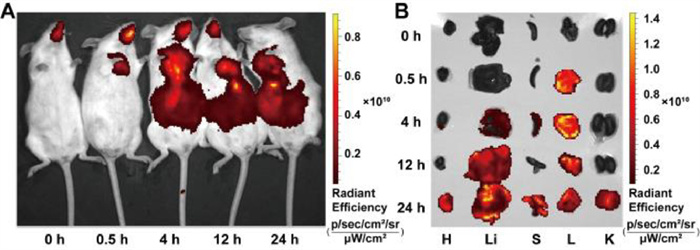
To evaluate the biodistribution of TA-PM via pulmonary administration, the lipophilic carbocyanine dye 1, 1′-dioctadecyl-3, 3, 3′, 3′-tetramethylindotricarbocyanine iodide (DiR) was encapsulated into TA-PM. After pulmonary administration of DiR labeled TA-PM, in vivo imaging in mice was recorded and the major organs (heart, liver, spleen, lungs, and kidneys) were excised for ex vivo imaging at 0, 0.5, 4, 12, and 24 h. As shown in Fig. 3A, the fluorescence signal in the lungs increased gradually within the first 4 h and then gradually decreased, but remained noticeable even at 24 h (Fig. 3A). In addition, the DiR fluorescence mainly accumulated in the lungs within 4 h and subsequently accumulated in the liver (Fig. 3A). The shifts for ex vivo fluorescence signals were almost consistent with those for in vivo (Figs. 3A and B). Taken together, DiR labeled TA-PM mostly accumulated in the lungs following pulmonary administration, indicating their high pulmonary deposition and retention.
Figure 3
Figure 3.
Biodistribution of DiR labeled TA-PM. (A) In vivo imaging system imaging of mice at various time points following pulmonary administration of DiR labeled TA-PM. (B) Optical ex vivo fluorescence images derived from excised tissues, from left to right, are the resected heart (H), liver (Li), spleen (S), lungs (L), and kidneys (K).
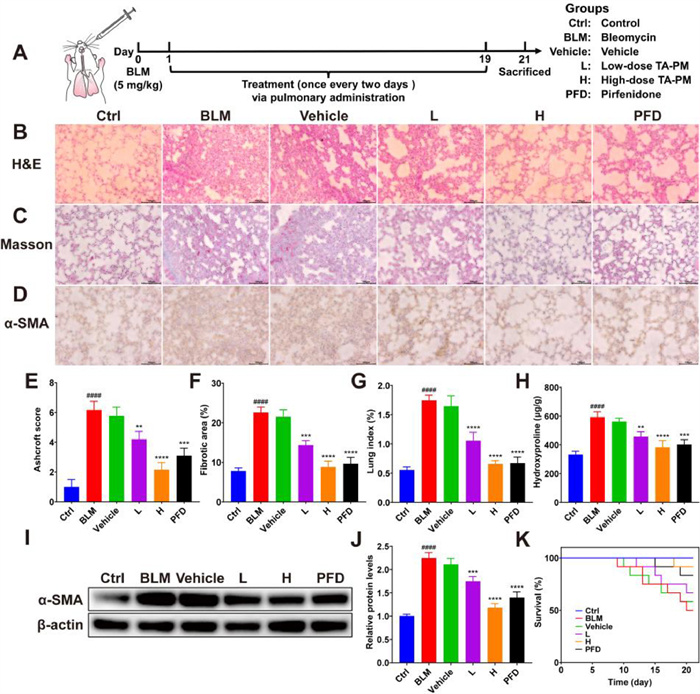
An IPF model was constructed through the pulmonary administration of BLM, and the IPF mice were treated with TA-PM to evaluate whether TA-PM could prevent the development of IPF (Fig. 4A). C57BL/6 mice were randomly divided into 6 groups (n = 12), including control group (Ctrl), BLM group (BLM), BLM + empty mixed micelles group (Vehicle), BLM + low-dose TA-PM group (L) at a dosage of 2.5 mg/kg of TA, BLM + high-dose TA-PM group (H) at a dosage of 5 mg/kg of TA, and BLM + 100 mg/kg PFD group (PFD). All groups, except the PFD group, received pulmonary administration, while the PFD group received oral administration as PFD is an oral medication in the clinic, and the administration frequency for each group was once every two days. All mice were sacrificed 21 days after BLM administration to collect lung tissues for further analysis. Hematoxylin and eosin (H&E) staining was carried out to analyze the pathological changes in lung tissues. As shown in Fig. 4B, the control group exhibited a clear alveolar structure without inflammation, while the BLM group and vehicle (empty mixed micelles) group showed severe alveolar damage and massive inflammatory cell infiltration. After treatments with TA-PM and PFD, the alveolar structure tended to be normal and inflammation was reduced, especially with the most significant effect of high-dose TA-PM treatment, and the Ashcroft scores of different groups showed the same trend (Figs. 4B and E). Masson staining showed a severe collagen deposition in the alveolar wall of the BLM group compared to the control group, suggesting obvious pulmonary fibrosis (Fig. 4C). In contrast, the collagen deposition was attenuated in the TA-PM and PFD groups, while no clearly attenuated phenomena were observed in the vehicle group (Figs. 4C and F). Compared to the low-dose TA-PM group, the PFD group and the high-dose TA-PM group had lower collagen deposition (Figs. 4C and F). Excessive deposition of myofibroblasts was a hallmark of IPF, and α-SMA was the characteristic protein of myofibroblasts [49]. The expression of α-SMA in lung tissues was detected by immunohistochemistry and Western blot analysis. The results showed that BLM significantly increased the expression of α-SMA compared to the control group, and the change was significantly reversed by TA-PM and PFD treatments, but not by vehicle treatment (Figs. 4D, I and J). Notably, high-dose TA-PM treatment exhibited a superior inhibition effect on the expression levels of α-SMA, demonstrating a positive anti-fibrotic effect of TA-PM (Figs. 4D, I and J).
Figure 4
Figure 4.
Anti-fibrotic effect of TA-PM in vivo. (A) Schematic diagram of animal model construction. (B) H&E staining, (C) Masson staining, and (D) immunohistochemical staining of α-SMA in lung tissue sections, scale bar: 100 µm. (E) Ashcroft score. (F) Fibrosis degree analysis of Masson staining. (G) Lung index. (H) The levels of hydroxyproline in lung tissues. (I) Western blot analysis of α-SMA in lung tissues. (J) The relative protein levels of α-SMA in lung tissues. (K) Survival curve of mice with different treatments, n = 12. Data are presented as the mean ± SD (n = 3). **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the BLM group; ####P < 0.0001 compared to the control group.
The lung index usually reflects the degree of pulmonary edema [50]. As shown in Fig. 4G, BLM treatment significantly increased the lung index, while TA-PM and PFD treatments significantly suppressed the lung index elevation, with high-dose TA-PM treatment achieving a better effect than that of low-dose TA-PM and PFD treatments, but vehicle treatment had no significant effect. The levels of hydroxyproline reflect the levels of collagen in lung tissues [51]. The hydroxyproline levels of the BLM group were much higher than those of the control group, and vehicle treatment did not significantly affect the hydroxyproline levels, while TA-PM and PFD treatments significantly decreased the hydroxyproline levels, with the lowest hydroxyproline levels in the high-dose TA-PM group (Fig. 4H). After 21 days of treatment, the body weight of the BLM group and the vehicle group significantly decreased (Fig. S3 in Supporting information). Nonetheless, the body weight loss was remarkably suppressed when TA-PM and PFD treatments were applied to the mice, indicating that these interventions could help mitigate the impact of BLM on body weight. Moreover, it is worth mentioning that high-dose TA-PM treatment showed the greatest reduction in body weight loss caused by BLM (Fig. S3). In addition, BLM treatment decreased the survival rate to 50% within 21 days, while high-dose TA-PM treatment notably increased the survival rate to 91.7%, which was higher than other treatment groups (Fig. 4K).
Numerous cytokines, including the fibrogenic cytokine TGF-β1 and the inflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-β (IL-1β), have been implicated in the pathogenesis of IPF [52]. Among these cytokines, TGF-β1, the most potent fibrogenic cytokine, has been found to have a strong stimulatory effect on collagen deposition [52,53]. TNF-α and IL-1β, on the other hand, play crucial roles in the inflammatory response and collagen deposition [54]. To further evaluate the anti-fibrotic and anti-inflammatory effects of TA-PM, we measured the levels of TGF-β1, TNF-α, and IL-1β. As shown in Fig. 5A, the levels of these three cytokines in the BLM group increased significantly compared to the control group, and vehicle treatment did not significantly reduce these three cytokines. In contrast, TA-PM and PFD treatments significantly reduced these three cytokines. Notably, high-dose TA-PM treatment showed the best inhibition effect, highlighting the superior anti-fibrotic and anti-inflammatory properties of TA-PM (Fig. 5A).
Figure 5
Figure 5.
Effect of TA-PM on the fibrogenic cytokine, inflammatory cytokines, and oxidation stress in vivo. (A) The levels of TGF-β1, TNF-α, and IL-1β tested by enzyme-linked immunosorbent assay (ELISA). (B) The levels of oxidative stress indicators GSH, SOD, and MDA. Data are presented as the mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the BLM group; ###P < 0.001, ####P < 0.0001 compared to the control group.
Oxidative stress, which shows an imbalance between oxidants and antioxidants, plays a major role in the development and progression of IPF [55]. We measured the levels of oxidative stress indicators such as glutathione (GSH), SOD, and malondialdehyde (MDA). As shown in Fig. 5B, compared to the control group, BLM treatment significantly increased the levels of MDA and decreased the levels of antioxidants GSH and SOD, and vehicle treatment reversed these alterations without any significant difference, even though TPGS could be used as an antioxidant. In contrast, TA-PM and PFD treatments significantly reversed the alterations, especially high-dose TA-PM treatment restored the oxidative stress indicators nearly back to the levels of the control group, suggesting that TA-PM could be potential antioxidants for IPF treatment (Fig. 5B). mPEG-PLA and TPGS have been widely utilized for micelle preparation due to their low toxicity and high biocompatibility [27,56]. Moreover, TA has been reported to have excellent biosafety [57,58]. As expected, H&E staining results of major organs such as heart, liver, spleen, and kidneys showed that TA-PM inhalation did not cause any conspicuous histopathological lesions and was safe (Fig. S4 in Supporting information).
In summary, our study showed that TA could inhibit EMT by inhibiting the TGF-β1/Smad signaling pathway in vitro. In addition, TA-PM that were well designed in the nanometer range and suitable for pulmonary administration were developed to solve the hydrophobic problem of TA. More importantly, inhaled TA-PM, which promoted pulmonary deposition and retention of TA and demonstrated high biocompatibility, eventually exhibited a desirable anti-fibrotic effect on the BLM-induced IPF mice via anti-inflammatory and antioxidant effects. Collectively, this study supports the idea that pulmonary administration of TA-PM may be a promising strategy for IPF treatment.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (No. 31872754), Fundamental Research Funds for the Central Universities (No. 201964018).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109248.
[1]
F.J. Martinez, H.R. Collard, A. Pardo, et al., Nat. Rev. Dis. Primers 3 (2017) 17074. doi: 10.1038/nrdp.2017.74
Z. Krajcovicova, A. Vachálková, K. Horváthová, Neoplasma 51 (2004) 407–414.
Figure 1
Effect of TA on the expression of EMT-related proteins and the migration of A549 cells. (A) Western blot analysis of EMT-related proteins. (B) Wound healing assay, scale bar: 500 µm. Data are presented as the mean ± standard deviation (SD) (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the TGF-β1 group; ###P < 0.001 and ####P < 0.0001 compared to the control group.
Figure 2
Characterization of TA-PM. (A) Diagrammatic structure of TA-PM. (B) TEM image of TA-PM, scale bar: 50 nm. (C) Particle size distribution of TA-PM. (D) Stability of TA-PM. (E) CMC of the mPEG2000-PLA2000/TPGS equal-mass mixture. (F) TA accumulative release curve from TA-PM in SLF. Data are presented as the mean ± SD (n = 3).
Figure 3
Biodistribution of DiR labeled TA-PM. (A) In vivo imaging system imaging of mice at various time points following pulmonary administration of DiR labeled TA-PM. (B) Optical ex vivo fluorescence images derived from excised tissues, from left to right, are the resected heart (H), liver (Li), spleen (S), lungs (L), and kidneys (K).
Figure 4
Anti-fibrotic effect of TA-PM in vivo. (A) Schematic diagram of animal model construction. (B) H&E staining, (C) Masson staining, and (D) immunohistochemical staining of α-SMA in lung tissue sections, scale bar: 100 µm. (E) Ashcroft score. (F) Fibrosis degree analysis of Masson staining. (G) Lung index. (H) The levels of hydroxyproline in lung tissues. (I) Western blot analysis of α-SMA in lung tissues. (J) The relative protein levels of α-SMA in lung tissues. (K) Survival curve of mice with different treatments, n = 12. Data are presented as the mean ± SD (n = 3). **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the BLM group; ####P < 0.0001 compared to the control group.
Figure 5
Effect of TA-PM on the fibrogenic cytokine, inflammatory cytokines, and oxidation stress in vivo. (A) The levels of TGF-β1, TNF-α, and IL-1β tested by enzyme-linked immunosorbent assay (ELISA). (B) The levels of oxidative stress indicators GSH, SOD, and MDA. Data are presented as the mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the BLM group; ###P < 0.001, ####P < 0.0001 compared to the control group.

 DownLoad:
DownLoad:


 下载:
下载:






 下载:
下载:
