Department of Obstetrics and Gynecology, The Third Affiliated Hospital of Southern Medical University, Guangzhou 510630, China
b.
Department of Obstetrics and Gynecology, Huizhou Central People’s Hospital, Huizhou 516001, China
c.
Affiliated Foshan Maternity & Child Healthcare Hospital, Southern Medical University (Foshan Maternity & Child Healthcare Hospital), Foshan 528000, China
d.
Guangdong Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China
e.
Department of Nursing, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China
f.
Department of Laboratory Medicine, Dongguan Institute of Clinical Cancer Research, The Tenth Affiliated Hospital of Southern Medical University (Dongguan People's Hospital), Dongguan 523018, China
*Corresponding authors. E-mail addresses: nfyykyb@126.com (S. Shi)
Received Date:
24 August 2023 Accepted Date:
20 October 2023 Revised Date:
17 October 2023 Available Online:
15 August 2024
Abstract:
Recently, the utilization of nonsteroidal anti-inflammatory drugs (NSAIDs) to sensitize cisplatin (CDDP) has gained substantial traction in the treatment of ovarian cancer (OC). However, even widely employed NSAIDs such as celecoxib and naproxen carry an elevated risk of cardiovascular events, notably thrombosis. Furthermore, the diminished sensitivity to CDDP therapy in OC is multifactorial, rendering the application of NSAIDs only partially effective due to their cyclooxygenase-2 (COX-2) inhibiting mechanism. Hence, in this study, reactive oxygen species (ROS)-responsive composite nano-hydrangeas loaded with the Chinese medicine small molecule allicin and platinum(Ⅳ) prodrug (DTP@AP NPs) were prepared to achieve comprehensive chemosensitization. On one front, allicin achieved COX-2 blocking therapy, encompassing the inhibition of proliferation, angiogenesis and endothelial mesenchymal transition (EMT), thereby mitigating the adverse impacts of CDDP chemotherapy. Simultaneously, synergistic chemosensitization was achieved from multifaceted mechanisms by decreasing CDDP inactivation, damaging mitochondria and inhibiting DNA repair. In essence, these findings provided an optimized approach for synergizing CDDP with COX-2 inhibitors, offering a promising avenue for enhancing OC treatment outcomes.
The highly aggressive and lethal nature of ovarian cancer (OC) makes it a killer of women's health [1]. The first-line treatment approach involves surgery combined with platinum (Pt)-containing chemotherapy, particularly cisplatin (CDDP)-based regimens [2,3]. Although CDDP-based chemotherapy initially achieves remission in most OC patients, prolonged treatment can lead to reduced tumor cell sensitivity and treatment failure [4]. Notably, CDDP-killed OC cells were found to stimulate macrophage-derived proinflammatory cytokines to upregulate cyclooxygenase-2 (COX-2) [5], which attenuates the therapeutic effects of CDDP by promoting tumorigenesis, anti-apoptosis, angiogenesis and endothelial mesenchymal transition (EMT) [6–11]. COX-2 levels are closely associated with OC staging and prognosis [12,13], thus making COX-2 a potential therapeutic target [5,14]. Increasing evidences suggest that combination therapy using CDDP and nonsteroidal anti-inflammatory drugs (NSAIDs) may be a powerful strategy for sensitizing tumor cells [15–17]. However, NSAIDs in common use (celecoxib and naproxen) pose an increased risk of thrombotic cardiovascular events even within 1 week of use [18], thus limiting the beneficiary population. Furthermore, the diminished CDDP sensitivity in OC results from various factors, and the application of NSAIDs only results in a limited sensitizing effect by inhibiting COX-2. Its high toxic side effects, detoxification of thiol-containing proteins represented by glutathione (GSH) and enhanced DNA repair are problems that must be overcome for the clinical application of CDDP. Therefore, the quest for a therapeutic agent that can concurrently substitute NSAIDs for COX-2 blockade could potentially circumvent the deficiencies of CDDP and enhance its clinical effectiveness.
Traditional Chinese medicine has become a research boom for its multi-targeting properties, low toxicity and synergistic potential with chemotherapy [19]. Allicin, the active ingredient in garlic, is of great interest owing to its anti-proliferative and cytotoxic effects [20]. In addition to inhibiting COX-2 [21] and inducing apoptosis, allicin blocks cell cycle progression and promotes the reactive oxygen species (ROS) production resulting in comprehensive antitumor effects beyond conventional NSAIDs [20,22]. Nonetheless, allicin's chemical instability has hindered its clinical application [23]. Emerging nano-delivery systems for special tumor microenvironments (high ROS, GSH and low pH) [24,25] are now available to address the shortcomings of allicin and CDDP in clinical applications, and this combination therapy has the potential to optimize the application of CDDP in the treatment of OC.
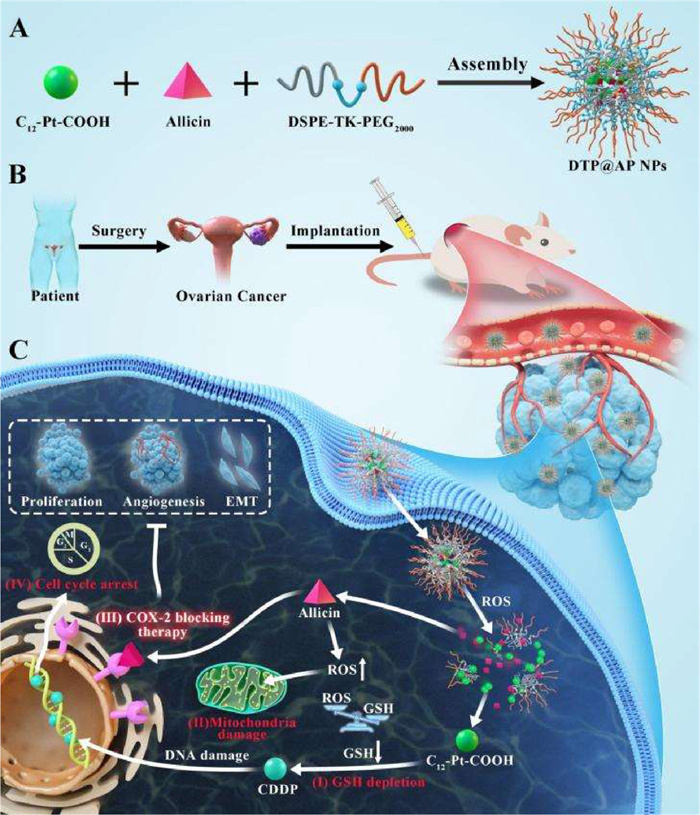
In the present study, tetravalent Pt prodrugs and allicin were co-loaded in 1,2-distearoyl-sn‑glycero-3-phosphoethanolamine-thioketal-N-[methoxy(polyethylene glycol)-2000] (DSPE-TK-PEG2000) ROS-responsive composite nano-hydrangeas (DTP@AP NPs). The shell formed by hydrophilic PEG2000 effectively avoided mononuclear phagocyte system recognition [26] and enriched the tumor region through enhanced permeability and retention (EPR) effect. Upon internalization by tumor cells, the thioketal (TK) bonds within the nano-hydrangeas respond to high ROS concentrations in tumor tissues [27], triggering drug release for synergistic chemosensitization (Scheme 1). DTP@AP NPs enhanced CDDP cytotoxicity through four mechanisms: (ⅰ) Consuming GSH to reduce CDDP detoxification; (ⅱ) inducing ROS production to dysfunctionalize mitochondria; (ⅲ) blocking COX-2 involving inhibition of cell proliferation, angiogenesis and EMT, to reverse the negative effects of CDDP; and (ⅳ) inhibiting cell cycle progression to suppress DNA repair.
Scheme 1
Scheme 1.
(A) Synthesis of DTP@AP NPs. (B) Illustration of establishing a PDX model of ovarian cancer. (C) Schematic representation of the mechanisms of DTP@AP NPs-mediated chemosensitization: consuming GSH (ⅰ) to reduce CDDP detoxification; inducing ROS (ⅱ) to dysfunctionalize mitochondria; blocking COX-2 (ⅲ) involving inhibition of cell proliferation, angiogenesis and EMT, to reverse the negative effects of CDDP; and inhibiting cell cycle progression (ⅳ) to suppress DNA repair.
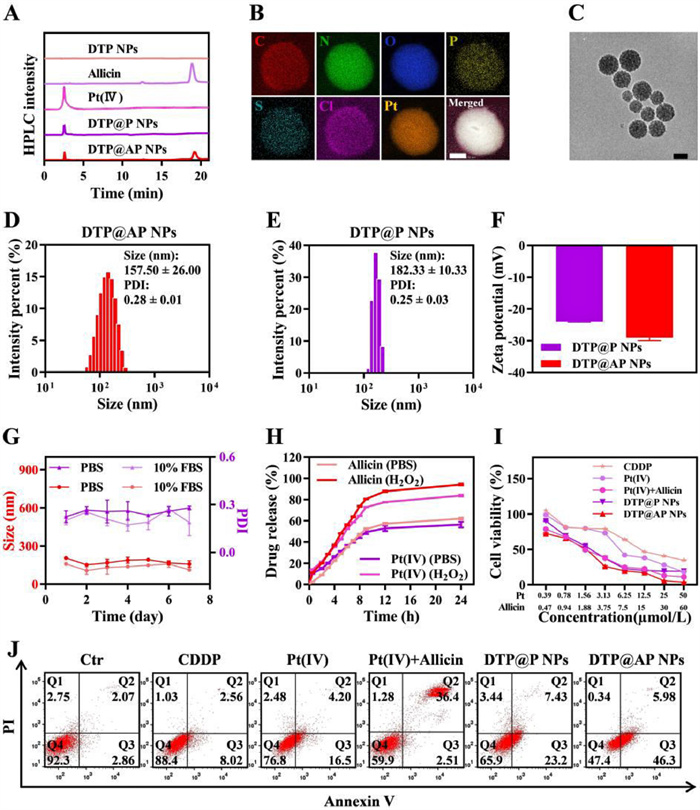
First, the DSPE-TK-PEG2000 and Pt(Ⅳ) were synthesized as Schemes S1 and S2 (Supporting information), and the successful synthesis of DSPE-TK-PEG2000 and Pt(Ⅳ) was verified using 1H NMR (Figs. S1 and S2 in Supporting information). Subsequently, Pt(Ⅳ) and allicin loaded DTP NPs (DTP@AP NPs), as well as DTP@P NPs, were fabricated using the nano-precipitation method. High-performance liquid chromatography (HPLC) profiles (Fig. 1A) revealed that the NPs were loaded with Pt(Ⅳ) and allicin. The homogeneous distributions of Cl, Pt, P, N, O, S and C in DTP@AP NPs were confirmed by transmission electron microscope (TEM) elemental mapping (Fig. 1B), further illustrating that Pt(Ⅳ) was encapsulated in the NPs. The encapsulation efficiencies of Pt(Ⅳ) and allicin in the optimized DTP@AP NPs were 49.78% and 40.39%, respectively, while the molar ratio of Pt(Ⅳ)/allicin was approximately 1:1.5 (Table S1 in Supporting information). TEM images revealed (Fig. 1C) that the DTP@AP NPs were uniformly distributed spherical morphology, which looked like blooming hydrangeas, and thus named as nano-hydrangeas. The dynamic light scattering (DLS) results indicated (Figs. 1D–F) that the hydrodynamic size of the DTP@P NPs was 182.33 ± 10.33 nm with −23.93 ± 0.23 mV of the zeta potential, and the DTP@AP NPs was 157.50 ± 26.00 nm with −29.03 ± 0.91 mV of the zeta potential. Furthermore, the particle size changes of DTP@AP NPs were evaluated and there no significant changes in size and polydispersity index (PDI) within 7 d (Fig. 1G). The above results indicate that DTP@AP NPs with appropriate size and negative potential prevent nanoparticles (NPs) aggregation and promote the retention of NPs in tissues [28].
Figure 1
Figure 1.
Characterizations and cytotoxicity of NPs. (A) HPLC profiles of Pt(Ⅳ) and allicin in DTP@AP NPs. (B) TEM elemental mappings (scale bar: 50 nm) and (C) TEM image (scale bar: 200 nm) of DTP@AP NPs. (D, E) Size distribution and (F) zeta potential of NPs. (G) The size and PDI variation of DTP@AP NPs in PBS buffer and 10% fetal bovine serum (FBS) within 7 days. (H) Cumulative Pt/allicin release of DTP@AP NPs after incubation with 100 µmol/L H2O2 at 37 ℃. (Ⅰ) Cell viability of SKOV3 cells after treatment with DTP@AP NPs, DTP@P NPs, allicin+Pt(Ⅳ), Pt(Ⅳ) and CDDP for 48 h. (J) Cell apoptosis in SKOV3 cells after different treatments. Data were given as mean±standard deviation (SD) (n = 3). PI, propidium iodide.
TK bonds were placed in the NPs to confer ROS responsiveness [27]. Consequently, the ROS-triggered drug release profile of DTP@AP NPs was investigated. The NPs were added to a 100 µmol/L H2O2 solution to simulate the oxidized tumor environment (Fig. 1H). In the absence of H2O2, the drug release was gradual, and after 6 h, the release of allicin and Pt in phosphate buffer saline (PBS) solution was about 35.00%, while in H2O2 solution the release rate increased to more than 50.00%. After 24 h, 62.12% of allicin and 56.47% of Pt were released in PBS solution, while 94.24% of allicin and 83.77% of Pt were released in H2O2 solution. Additionally, DLS and TEM results indicated a loss of particle size uniformity for DTP@AP NPs after 24 h of incubation with 100 µmol/L H2O2 (Fig. S3 in Supporting information). These findings underscored the ROS-responsive nature of DTP@AP NPs.
Given that intracellularization is a key step in nanodrugs reaching their intracellular targets [29], rhodamine B (Rh B) was encapsulated into DTP NPs (DTP@Rh B NPs) which were observed under confocal laser scanning microscopy (CLSM). As shown in Fig. S4 (Supporting information), DTP@Rh B NPs exhibited time-dependent cellular uptake. Further quantification using flow cytometry (FCM) (Fig. S5 in Supporting information) demonstrated a nearly 1.5-fold increase in uptake by SKOV3 cells after 6 h of DTP@Rh B NPs treatment compared to 1 h, confirming efficient uptake of DTP@AP NPs.
The cytotoxicity of different preparations was examined via MTT assay. Concentrations of DTP NPs ranging from 10 µg/mL to 1280 µg/mL exhibited over 90% cell viability, indicating their biocompatibility (Fig. S6 in Supporting information), consistent with prior reports [30]. The combination strategy exhibited notable tumor cell-killing efficacy (Fig. 1I, Fig. S7 and Table S2 in Supporting information). Further analysis by FCM revealed apoptotic rates of 10.58%, 20.70%, 38.91%, 30.63% and 52.28% in SKOV3 cells following CDDP, Pt(Ⅳ), allicin+Pt(Ⅳ), DTP@P NPs and DTP@AP NPs treatment, respectively (Fig. 1J and Fig. S8 in Supporting information), consistent with the MTT assay results. Thus, it can be concluded that DTP@AP NPs can achieve synergistic chemosensitization through multiple mechanisms.
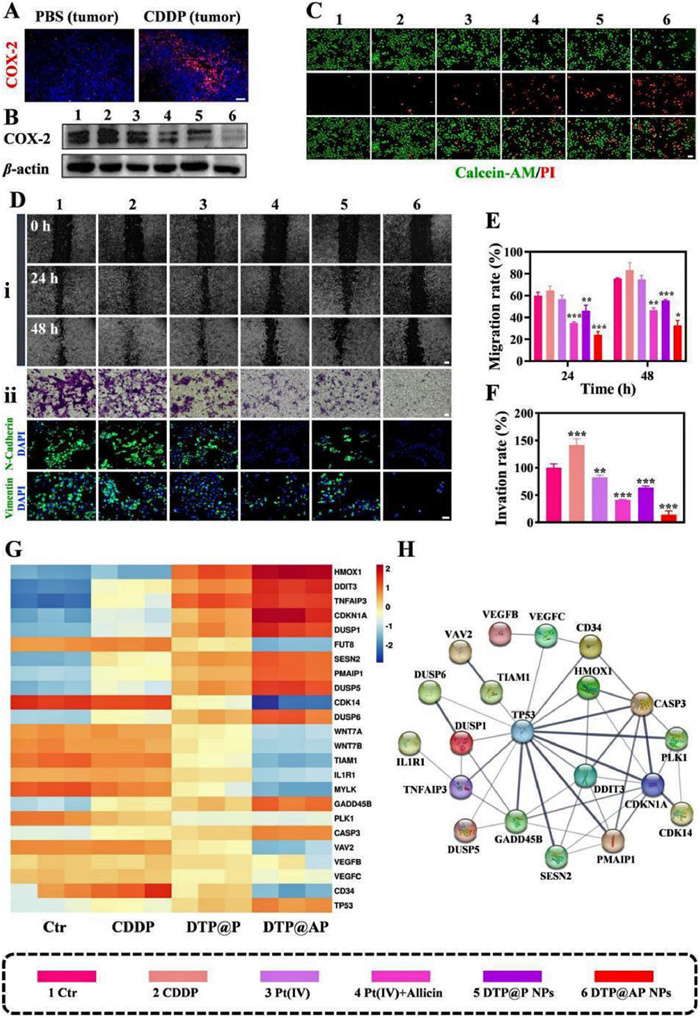
The aforementioned results underscored the synergistic antitumor activity of DTP@AP NPs. Subsequent direct evidence of COX-2 expression was presented. The findings revealed enhanced COX-2 expression in tumor tissues of nude mice after CDDP administration (Fig. 2A), a phenomenon replicated in SKOV3 cells, while DTP@AP NPs effectively counteracted COX-2 expression (Fig. 2B and Fig. S9 in Supporting information). COX-2 plays a pivotal role in driving proliferation, EMT and angiogenesis, collectively contributing to the tumorigenic microenvironment [5]. In the cell live/dead staining analysis demonstrated robust red fluorescence in cells subjected to the combination strategy, with particularly pronounced effects in DTP@AP NPs, indicating a synergistic inhibitory impact on proliferation (Fig. 2C). EMT activation is a key process in tumor cell metastasis, enhancing the migration and invasive capacity of tumor cells [31]. The anti-migration potential of DTP@AP NPs was then evaluated by the wound-healing assay. The combinational strategy significantly inhibited tumor cell migration, with an approximately 1.6-fold increase in the inhibition efficiency of DTP@AP NPs compared to CDDP alone (Figs. 2D(ⅰ) and E). As expected, DTP@AP NPs exhibited the worst ability to penetrate the chamber and matrigel (Figs. 2D(ⅱ) and F). EMT activation is accompanied by augmented interstitial properties, and the interstitial markers neural cadherin (N-cadherin) and vimentin [32] were efficiently inhibited after treatment with DTP@AP NPs (Fig. 2D). COX-2 expression has been reported to be positively associated with the expression of matrix metalloproteinase-9 (MMP-9) and vascular endothelial growth factor (VEGF) [33]. MMP-9, by degrading the extracellular matrix and basement membrane, and VEGF, by regulating angiogenesis, together foster EMT [9,14]. Western blot assay showed that DTP@AP NPs effectively suppressed MMP-9 and VEGF expression (Fig. S10 in Supporting information). These observations implied that DTP@AP NPs inhibited proliferation, angiogenesis and EMT following COX-2 blocking treatment.
Figure 2
Figure 2.
Antitumor mechanisms of DTP@AP NPs in vitro. (A) Immunofluorescence of COX-2 in tumors (PBS, CDDP). scale bar: 10 µm. (B) Western blot of COX-2 protein in SKOV3 cells after various treatments for 48 h. (C) The cell live/dead staining of SKOV3 cells treated with different formulations for 48 h, scale bar: 10 µm. (D) Wound-healing (ⅰ) (scale bar: 20 µm), invasion (ⅱ) (scale bar: 200 µm), and immunofluorescence assays (scale bar: 50 µm) of vimentin and N-cadherin in SKOV3 cells. (E, F) Quantitative analysis of cells migration and invasion rate. (G) A heat map indicating the transcription of the interest gene. (H) Protein–protein interaction (PPI) network. Data were given as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001 vs. Ctr.
GSH is pivotal in CDDP detoxification [34]. Consequently, cellular GSH levels after diverse treatments were evaluated (Fig. S11A in Supporting information), revealing DTP@AP NPs achieved a more impressive GSH depletion. GSH depletion is known to increase DNA damage [35]. Western blot analysis (Fig. S10) showed that DTP@AP NPs effectively increased the level of γ-H2A.X, a hallmark of DNA damage [16]. Allicin is recognized for elevating intracellular ROS levels [36]. After incubation with CDDP, ROS levels were not enhanced, while the combined strategy significantly improved intracellular ROS levels (Fig. S11B in Supporting information). Supraphysiological levels of ROS induce mitochondrial dysfunction, further promoting apoptosis [37]. The mitochondrial apoptotic pathway is one of the key pathways through which ROS drive apoptosis [38]. It causes a decrease in mitochondrial membrane potential (MMP) and opens the mitochondrial permeability transition pore by mediating the expression of Bcl-2 and Bax, which ultimately activates the caspase family cascade [39]. JC-1 staining illustrated significantly reduced MMP in cells treated with DTP@AP NPs (Fig. S11C in Supporting information), validated by FCM (Figs. S11D and E in Supporting information). Western blot results (Fig. S10) revealed that CDDP treatment alone upregulated cleaved caspase-3 (CASP3) to induce apoptosis and activated anti-apoptotic Bcl-2 protein, dampening the chemotherapeutic effect-consistent with the initial observation of CDDP activating COX-2, thereby upregulating the Bcl-2 gene [11]. Conversely, DTP@AP NPs elevated apoptotic proteins Bax and cleaved caspase-3 expression while inhibiting Bcl-2. These results collectively suggested that DTP@AP NPs might induce apoptosis in SKOV3 cells through the mitochondrial apoptosis pathway. Despite the high toxicity of chemotherapy, tumor cells may activate various DNA repair mechanisms [40]. Therefore, blocking the cell cycle during chemotherapy can enhance therapeutic efficacy [41]. Subsequent cell-cycle assays revealed that DTP@AP NPs significantly arrested the cell cycle in S phase (44.77%) and G2/M phase (23.30%) (Figs. S11F and G in Supporting information), effectively boosting chemotherapeutic efficacy. In conclusion, DTP@AP NPs synergistically contributed to antitumor effects by GSH depletion, ROS production, mitochondrial damage and cell cycle blockade.
To further investigate the potential cytotoxic mechanisms of CDDP, DTP@P NPs and DTP@AP NPs, whole-genome RNA-sequencing of SKOV3 cells exposed to various treatments was performed. The transcription of 10, 731 genes was detected in SKOV3 cells. Among these genes, 202, 101, 132 and 247 gene transcripts were uniquely transcribed in the cells treated with the control (Ctr), CDDP, DTP@P NPs and DTP@AP NPs, respectively (Fig. S12A in Supporting information). In comparison to the Ctr group, 246, 860 and 1828 genes were upregulated (red dots), and 720, 1176 and 2822 genes were downregulated in the CDDP, DTP@P NPs and DTP@AP NPs groups, respectively (Fig. S12B in Supporting information). Multiple signaling pathways were significantly affected as shown by Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis (Fig. S12C in Supporting information). Specifically, compared with the Ctr group, CDDP alone had a significant effect on inflammatory pathways in addition to inducing apoptosis, including the tumor necrosis factor (TNF) signaling pathway, interleukin-17 (IL-17) signaling pathway and nuclear factor-kappa B (NF-κB) signaling pathway. Activation of the NF-κB signaling pathway, as an example, promotes tumor cell progression by upregulating pro-inflammatory factors such as COX-2 [42]. DTP@AP NPs mainly affected the Rap1 signaling pathway, pathways in cancer and transcriptional misregulation in cancer. Accordingly, the expression of heme oxygenase 1 (HMOX1), DNA damage-inducible transcript 3 (DDIT3), tumor necrosis factor alpha-induced protein 3 (TNFAIP3), cyclin dependent kinase inhibitor 1A (CDKN1A), dual specificity phosphatase 1 (DUSP1), sestrin 2 (SESN2), phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), DUSP5, DUSP6, growth arrest and DNA damage inducible beta (GADD45B), tumor protein p53 (TP53) and CASP3 were upregulated, and the expression of T-lymphoma invasion and metastasis (TIAM1), interleukin 1 receptor type 1 (IL1R1), polo-like kinase 1 (PLK1), VEGFB, VEGFC and cluster of differentiation 34 (CD34) genes were downregulated in the DTP@AP NPs treatment group (Fig. 2G). It was shown that upregulated DUSP1, DUSP5 and DUSP6 played a protective role in the inflammatory response by negatively regulating the mitogen-activated protein kinase (MAPK) pathway, which subsequently promoted apoptosis, inhibited EMT and slowed cancer progression [43,44]. In addition, TNFAIP3 negatively regulated the NF-κB pathway as an inflammation inhibitor [45], while downregulated IL1R1 suppressed the tumor inflammatory phenotype [46]. Downregulated VEGFB, VEGFC and CD34 inhibited angiogenesis [47,48]. The overexpressed cellular stress transcription factor DDIT3 promoted apoptosis through the mitochondrial apoptotic pathway [49]. TIAM1 promoted tumor cell survival by antagonizing Bcl-2 conformational changes [50]. Upregulated CDKN1A and downregulated PLK1 played a role in inhibiting cell-cycle progression [51,52]. Finally, Fig. 2H shows the protein-protein interactions. In summary, CDDP treatment alone created a tumorigenic inflammatory environment, while DTP@AP NPs inhibited the tumor inflammatory phenotype and a range of tumorigenic activities, including apoptosis resistance, angiogenesis and EMT.
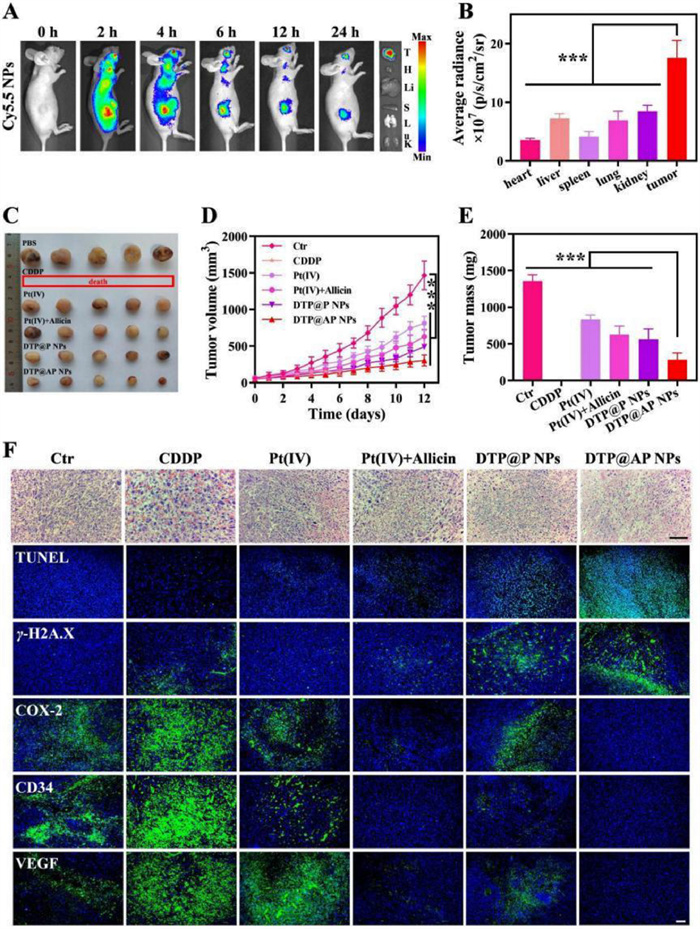
The excellent antitumor effects of DTP@AP NPs in vitro prompted us to conduct further in vivo studies. Animal experiments were carried out in the guidelines assessed and approved by the Ethics Committee of Southern Medical University (Guangdong, China). Initially, the hemolytic activity of NPs was assessed using the hemolysis assay. The hemolytic activity of DTP@P NPs and DTP@AP NPs was 5.66% ± 3.01% and 1.06% ± 0.43%, respectively (Fig. S13 in Supporting information), indicating the low hemolytic toxicity of NPs. To reveal the biodistribution of NPs, Cy5.5-labeled DTP@AP NPs were injected into tumor carrying mice intravenously. Consequently, the fluorescence signals in vivo and in vitro were mainly clustered at the tumor sites (Figs. 3A and B), which was attributed to EPR effect-mediated tumor targeting. The therapeutic effect of DTP@AP NPs was next evaluated in an ovarian cancer patient-derived tumor xenograft (PDX) model. The tumor-bearing mice were treated with DTP@AP NPs, DTP@P NPs, allicin+Pt(Ⅳ), Pt(Ⅳ), CDDP or PBS. Except for the PBS group, all treatments were effective at inhibiting tumor growth. In particular, DTP@AP NPs showed the most pronounced inhibition (Figs. 3C–E), suggesting that synergistic chemosensitization was effective. Hematoxylin and eosin staining (H&E) staining and terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) analysis further showed that DTP@AP NPs caused the greatest tumor cell death (Fig. 3F). The considerable increase in γ-H2A.X and the dramatic decrease in COX-2 revealed the mechanism of enhanced antitumor efficacy of DTP@AP NPs in vivo (Fig. 3F). The overexpression of COX-2 promoted angiogenesis, while both angiogenic markers VEGF and CD34 were significantly decreased after treatment with DTP@AP NPs (Fig. 3F). Finally, a safety assessment was performed. Following CDDP treatment, the entire group of mice died within one week, confirming its serious side effects, proven by the significant weight loss and liver injury (Fig. S14 in Supporting information). In contrast, DTP@AP NPs had negligible effects on the remaining biological indicators tested, except for causing slight weight loss, indicating their safety.
Figure 3
Figure 3.
Biodistribution and antitumor therapy of DTP@AP NPs in vivo. (A) Ex vivo fluorescence biodistribution of tumors and major organs. (B) The corresponding average radiance analysis at 24 h post-injection (n = 3). (C) Tumor image, (D) tumor growth curves and (E) tumor weight of mice (n = 5). (F) H&E (scale bar: 100 µm) and immunofluorescence staining of tumor sections (scale bar: 10 µm). ***P < 0.001. Data were given as mean ± SD.
In summary, the study presented a nanomedicine approach utilizing ROS-responsive nano-hydrangeas containing allicin and Pt(Ⅳ) to enhance chemosensitization in OC treatment. DTP@AP NPs effectively addressed COX-2 upregulation-induced limitations of CDDP therapy while offering multifaceted mechanisms for promoting apoptosis. Due to effective chemosensitization and tumor accumulation, DTP@AP NPs markedly suppressed tumor growth and minimized the toxic effects of CDDP, with promising applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Guangdong Basic and Applied Basic Research Foundation of China (No. 2021A1515011050), President Foundation of the Third Affiliated Hospital of Southern Medical University (No. YM202202), the Health Economics Association Research Program of Guangdong Province (No. 2022-WJZD-20), and the Higher Education Teaching Management Association Curriculum Thinking and Administration Program of Guangdong Province (No. X-KCSZ2021082).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109241.
[1]
X. Cai, S. Shi, G. Chen, et al., Acta Biomater. 158 (2022) 560–570. doi: 10.3390/ma15020560
Scheme 1
(A) Synthesis of DTP@AP NPs. (B) Illustration of establishing a PDX model of ovarian cancer. (C) Schematic representation of the mechanisms of DTP@AP NPs-mediated chemosensitization: consuming GSH (ⅰ) to reduce CDDP detoxification; inducing ROS (ⅱ) to dysfunctionalize mitochondria; blocking COX-2 (ⅲ) involving inhibition of cell proliferation, angiogenesis and EMT, to reverse the negative effects of CDDP; and inhibiting cell cycle progression (ⅳ) to suppress DNA repair.
Figure 1
Characterizations and cytotoxicity of NPs. (A) HPLC profiles of Pt(Ⅳ) and allicin in DTP@AP NPs. (B) TEM elemental mappings (scale bar: 50 nm) and (C) TEM image (scale bar: 200 nm) of DTP@AP NPs. (D, E) Size distribution and (F) zeta potential of NPs. (G) The size and PDI variation of DTP@AP NPs in PBS buffer and 10% fetal bovine serum (FBS) within 7 days. (H) Cumulative Pt/allicin release of DTP@AP NPs after incubation with 100 µmol/L H2O2 at 37 ℃. (Ⅰ) Cell viability of SKOV3 cells after treatment with DTP@AP NPs, DTP@P NPs, allicin+Pt(Ⅳ), Pt(Ⅳ) and CDDP for 48 h. (J) Cell apoptosis in SKOV3 cells after different treatments. Data were given as mean±standard deviation (SD) (n = 3). PI, propidium iodide.
Figure 2
Antitumor mechanisms of DTP@AP NPs in vitro. (A) Immunofluorescence of COX-2 in tumors (PBS, CDDP). scale bar: 10 µm. (B) Western blot of COX-2 protein in SKOV3 cells after various treatments for 48 h. (C) The cell live/dead staining of SKOV3 cells treated with different formulations for 48 h, scale bar: 10 µm. (D) Wound-healing (ⅰ) (scale bar: 20 µm), invasion (ⅱ) (scale bar: 200 µm), and immunofluorescence assays (scale bar: 50 µm) of vimentin and N-cadherin in SKOV3 cells. (E, F) Quantitative analysis of cells migration and invasion rate. (G) A heat map indicating the transcription of the interest gene. (H) Protein–protein interaction (PPI) network. Data were given as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001 vs. Ctr.
Figure 3
Biodistribution and antitumor therapy of DTP@AP NPs in vivo. (A) Ex vivo fluorescence biodistribution of tumors and major organs. (B) The corresponding average radiance analysis at 24 h post-injection (n = 3). (C) Tumor image, (D) tumor growth curves and (E) tumor weight of mice (n = 5). (F) H&E (scale bar: 100 µm) and immunofluorescence staining of tumor sections (scale bar: 10 µm). ***P < 0.001. Data were given as mean ± SD.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
