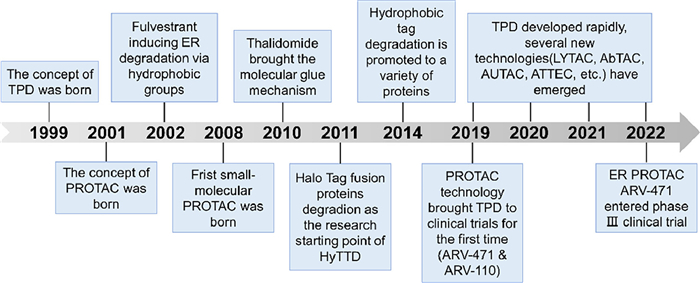
Figure 1.
The development timeline of TPD.

Traditional small-molecule drugs tend to regulate protein activity in an occupancy-driven manner, interacting with specific amino acids in particular spatial postures of functional protein domains. This mode of action has shown clinical benefits in many drugs which are usually divided into activators and inhibitors according to their different impacts on the function of proteins like receptors or enzymes. However, classical small-molecule drugs require a manageable active or allosteric site on the target protein and rely on high affinity or particular interaction with the target protein. These requirements restrict the application of small-molecule protein regulators in some undruggable or drug-resistant targets. Targeted protein degradation (TPD) has emerged as a viable alternative over the last two decades (Fig. 1) [1,2]. TPD involves hijacking endogenous degradation pathways to selectively deplete the pathogenic protein. Unlike protein function modulators, TPD does not require extremely high affinity nor precise action sites to achieve the degradation effects, due to its ability to degrade proteins in an event-driven manner. This mode of action fundamentally breaks the constraints of small molecule modulators in regard to undruggable or drug-resistant targets [3].
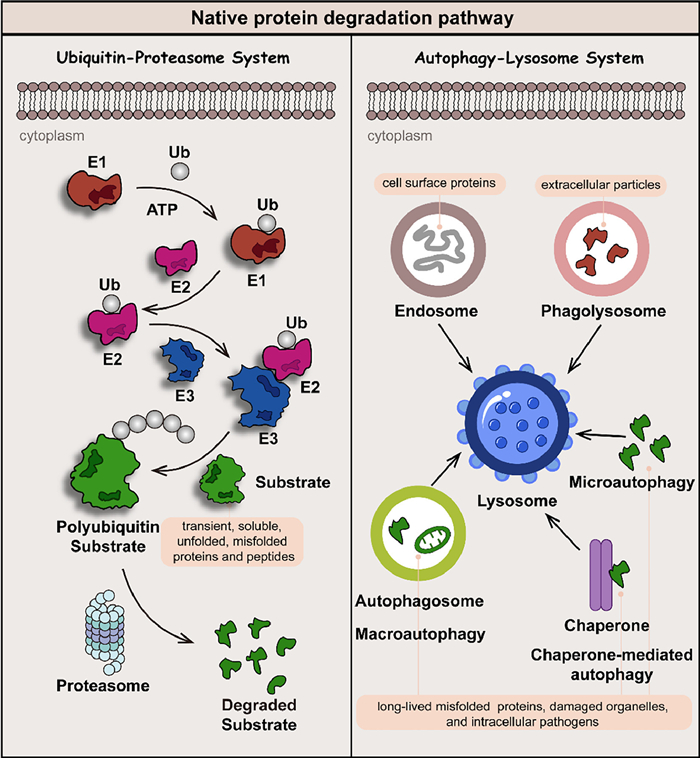
Native protein degradation is mainly based on two mechanisms: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (ALS) (Fig. 2) [4,5]. The proteasome in the UPS effectively degrades transient, soluble unfolded, or misfolded proteins and peptides, while the ALS is devoted to eliminating long-lived, insoluble protein aggregates and dysfunctional organelles. The ubiquitin consists of 76 amino acids, in which seven lysine residues (Lys 6, 11, 27, 29, 33, 48, and 63) are involved in the formation of the polyubiquitin chain. With the assistance of E1, E2, and E3 ligase, covalently polyubiquitination-tagged proteins can be degraded by lysosome (Lys 63-linked ubiquitin chains) or proteasome (Lys 11 and Lys 48-linked ubiquitin chains) pathway. Medicinal chemists utilize the ubiquitination process to guide the reduction of target protein levels by developing small-molecule drugs [6].
Proteolysis-targeting chimera (PROTAC) technology is a fast-evolving strategy for TPD in recent years [7–11]. PROTACs, as hetero-bifunctional molecular degraders, recruit both the target protein and the E3 ligase for protein-protein interaction (PPI) to induce ubiquitination of the target protein and subsequent degradation via the proteasome (Fig. 3). Accumulating studies have revealed the basic design strategy of PROTAC molecules. With the potent degradation efficacy of undruggable or drug-resistant targets, PROTAC technology quickly catches the attention of the industry [12]. Up to now, numerous PROTAC molecules for several pathogenic proteins have entered the clinical stage, among which estrogen receptor (ER) PROTAC ARV-471 has initiated phase Ⅲ clinical studies (NCT05909397). However, the large sheer size of PROTACs results in poor bioavailability and pharmacokinetic properties. Meanwhile, PROTACs can degrade the target protein under low binding affinity, and the resulting off-target side effects cannot be ignored [13,14]. Similar hetero-bifunctional molecules include lysosome-targeting chimera (LYTAC) [15,16], antibody-based PROTAC (AbTAC) [17], autophagy-targeting chimera (AUTAC) [18], autophagosome-tethering compound (ATTEC) [19] and so on, which have been introduced specifically in other reviews and will not be expanded here [20]. Monovalent molecular glues serve as low-molecular-weight and high-selectivity target protein degraders providing a more drug-like option (Fig. 3) [21]. Nevertheless, molecular glues, based on the degradation mechanism of target protein-E3 ligase interaction, have more stringent requirements for spatial conformation of both proteins, which badly limits its application scope and rational design.
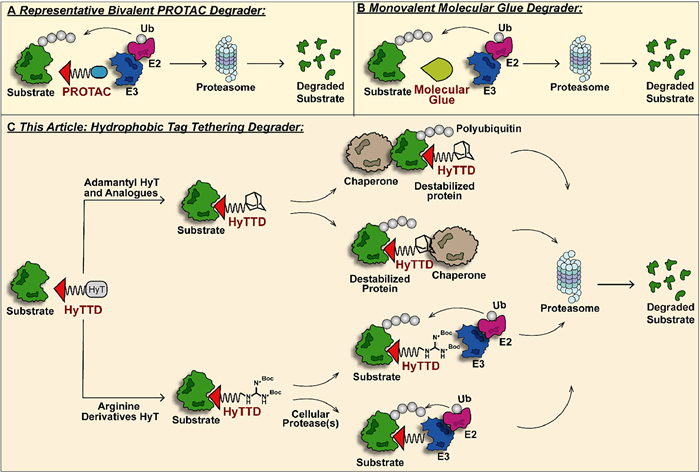
Hydrophobic tag tethering degrader (HyTTD), an alternative degradation paradigm with a novel mechanism, demonstrates biochemistry and medical value in recent years (Fig. 3) [22]. HyTTDs consist of two pharmacophores: a ligand of the target protein and a hydrophobic moiety to induce degradation. The prevailing hydrophobic tags that are frequently employed are adamantane and arginine derivatives, while norbornene, carborane, fluorene, pyrene, etc. have also exhibited potential for promoting the degradation of target proteins [23,24]. A hydrophobic tag is connected to the ligand of the target protein through a linker, simulating endogenous protein misfolding and triggering the degradation through the proteasome pathway. Compared to PROTAC, molecular glue, etc., the HyTTD technology is far underestimated. Likewise, HyTTDs also degrade drug-resistant or undruggable proteins via the event-drive manner at a catalytic dose. Benefiting from a more streamlined chemical structure, HyTTDs have superior bioavailability and pharmacokinetic parameters to PROTACs, especially when downregulating central nervous system-associated proteins [25]. For the construct of structure composition, HyTTDs are more likely to achieve rational design than molecular glues. The E3 ligase system-independent feature also makes HyTTDs more widely applicable than PROTACs and molecular glues. It is a remarkable fact that protein-protein interactions or protein complexes contribute a lot to the progression of the disease, however, PROTAC and molecular glue-based protein degraders often have poor efficacy on such protein complexes, while the accumulated evidence highlights that HyTTD technology complements this gap well [26]. Reported hydrophobic tags have a relatively universal degradation effect on a variety of pathogenic targets, which provides a reasonable reference for the discovery of novel protein degraders that have not been explored. However, the development of HyTTD technology is also experiencing challenges, including how to break through the limited biological activity and how to explore the detailed mechanism of the degradation effect for target proteins brought by HyTTDs.
The prominent hallmarks of HyTTDs have attracted extensive interest from academia and industry in recent years (Fig. S1 in Supporting information), but there is still a lot of space to progress. This article focuses on the hotspot HyTTD technology in the protein degradation field and comprehensively introduces the development history, the mechanism of action, and the application of HyTTDs. We comment on the current status of this technology and provide the perspective for the development direction of the HyTTD in degrading pathogenic targets to treat refractory diseases.
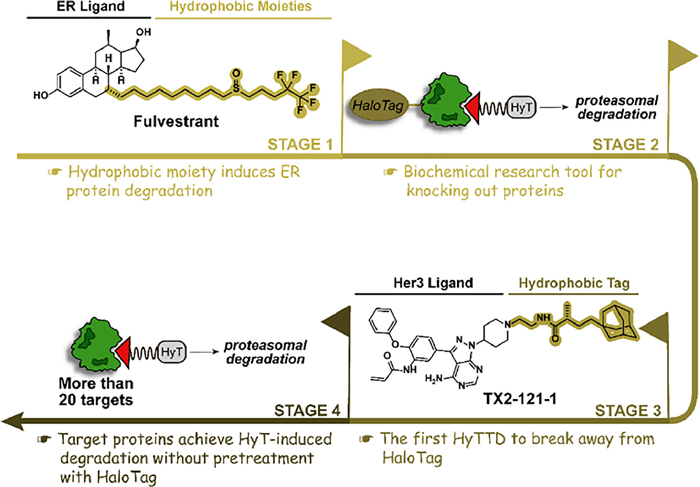
Researchers began to realize the association between hydrophobic structure and protein degradation can be traced back to the first selective estrogen receptor degrader (SERD) fulvestrant [27]. The hydrophobic alkyl structure in the fulvestrant subtly perturbs the ER protein, inducing the recognition of the normal ER protein as the misfolded protein and subsequently degrading ER by the proteasome pathway. To extend the hydrophobic structure-induced degradation to more proteins, the HaloTag fusion protein system was designed in 2011 [28,29]. After the fusion of the HaloTag protein and the target protein, the hydrophobic structure is covalently attached to the HaloTag protein and indirectly induces the degradation of the target protein. Adamantyl groups are the preferred hydrophobic tags with the best degradation activity due to their satisfying cell permeability and stability. For example, adamantyl group-medicated HaloTag 7 degradation by HyT13 and HyT36 provided a novel mechanism-based protein degradation strategy [28]. However, the HaloTag fusion protein system can only be utilized as a biochemical research tool for knocking out proteins while cannot achieve medicinal value. If the small molecule hydrophobic chain can directly mediate the degradation of the target protein without the assistance of the HaloTag, the application scope will be greatly broadened. In 2014, it is reported that TX2–121–1, an adamantane-conjugated small molecular, directly binds to Her3 protein and induces its degradation without pretreatment with HaloTag, which is a significant step in the transformation of the tool molecule to small molecule drugs labeled with hydrophobic tag [30]. Since then, HyTTDs have attracted widespread attention. Hydrophobic tags, which extend to a variety of chemical structures, have begun to exhibit degradation in an increasing number of proteins (Fig. 4). After nearly 10 years of development, the anaplastic lymphoma kinase (ALK) degrader Hyt-9 labeled with a norbornene degron achieved a tumor inhibition rate of 58.03% in a xenograft tumor model of 20 mg/kg oral administration, which confirms that HyTTDs possesses the druggable potential [23].
The unfolded protein response (UPR) is a quality control system that maintains cellular homeostasis [31]. Unfolded or misfolded proteins expose hydrophobic regions to recruit chaperones to interact with them, which can help proteins fold correctly or directly degrade proteins in the proteasome pathway. The concrete degradation mechanism of HyTTDs is still being explored. Up to now, it is known that HyTTDs mediate the specific binding of the hydrophobic tag to the target protein via a flexible linker, inducing the simulation of abnormal folding and aggregation of the target protein. The destabilized protein activates the UPR in cells inducing the subsequent proteasomal degradation. Adamantane and its derivatives hydrophobic tags tend to clear the protein of interest (POI) by interacting directly with the chaperone (such as HSP70) or initiating the interaction of the POI with the chaperone [32]. Arginine derivatives were originally designed based on the N-end rule pathway for protein degradation. The protein N-terminal arginine has been reported to be recognized as an "N-degron" by specific E3 ligase UBR1, UBR2, UBR4, and UBR5 named "N-recognin" [33]. This recognition, once occurring, mediates the entry of POI into the proteasome degradation pathway. However, recently published research revealed that arginine is not "N-degron" in the degradation process of nuclear receptor-binding SET domain-containing 2 (NSD2), but is inclined to play a role as a prodrug, releasing the long-chain alkyl amine structure after protease hydrolysis in vivo as a recognition site to mediate proteasome degradation (Fig. 3) [34]. As a field with great potential, more biochemistry strategies should be applied to uncover the essential mechanism of HyTTDs in the future.
Breast cancer leads to an increasing global incidence of 25% and cancer mortality of 16 %, which is the second dominant cause of cancer death among women [35]. ER, which regulates gene transcription by binding to endocrine estrogen (e.g., estradiol, estrone, estriol), is closely related to the development of breast cancer. As a subtype of ER, ERα is observed to be expressed in approximately 75% of breast cancer, which makes ERα a significant target for breast cancer therapy. Endocrine therapies effectively inhibit estrogen-ERα signaling by intercepting ERα activities or reducing circulating estrogen in early-stage ERα-positive breast cancer. However, endocrine-resistant breast cancers caused by ERα overexpression or ligand-independent mutations are a major obstacle to patient survival after long-term treatment (Fig. 5A)[36–38].
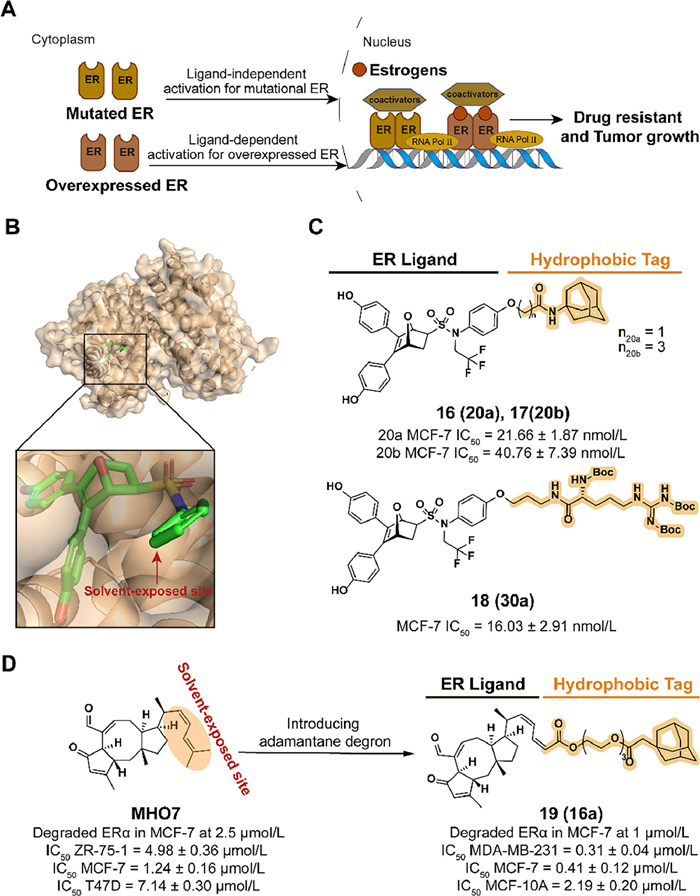
As a strategy with the ability to both antagonize ERα function and degrade ERα protein, SERDs provide a new pathway to overcome endocrine-resistant breast cancer. SERDs currently marketed and in clinical development are displayed in Table S1 (Supporting information). For the antiestrogens with fewer agonist activity, a series of 7α-estradiol derivatives with a long-chain alkyl was discovered [39]. Among the compounds, fulvestrant (1) displayed potent ER degradation and antiproliferation activity in MCF-7 cells and prolonged the median of progression-free survival (PFS) and overall survival (OS) (respectively, 10.6 and 43.2 months) in patients with ER-positive metastatic breast cancer in a retrospective study (NCT01509625). Moreover, treatment with ER degrader fulvestrant provided clinical benefit in patients who had received first-line hormonal therapy which dedicated to antagonizing ER function (e.g., tamoxifen). Mechanism studies revealed that Fulvestrant increases the exposure of hydrophobic surfaces by protruding the alkyl chain outside the ligand binding pocket and blocks coactivator binding which ultimately leads to protein degradation [40,41]. U.S. Food and Drug Administration (FDA) approved the fulvestrant for the treatment of hormone receptor-positive metastatic breast cancer in 2002 which demonstrated the breakthrough and recognition of ER degraders adopted the hydrophobic-based mechanism of protein degradation. Hence, FDA approved the fulvestrant for the treatment of hormone receptor-positive metastatic breast cancer in 2002. However, the drawbacks of reduced efficacy and pain of the intramuscular injection during fulvestrant treatment focus attention. This may be due to inadequate drug exposure caused by pharmacokinetic limitations in steroid derivatives with long hydrophobic chains, which stimulated the development of a new generation of oral SERDs [42]. To achieve the final point of high potency for oral administration, the new generation of SERDs starts mainly with two orientations, acrylic acid chains or basic amine chains.
SERDs containing acrylic acid side chains reposition helix 12 (H12) by interacting with its N terminus, which leads to an increase in the hydrophobic surface and subsequent protein degradation. Several compounds entered clinical trials (Table S1) but struggled to break through to Phase Ⅰ which was blamed on their poor tolerance in the gastrointestinal tract and difficulty in providing superior efficacy to fulvestrant [43]. However, breakthroughs have been seen in SERDs containing basic amine chains which perturb the ERα conformation and exhibit stronger and relatively stable ER degradation activities. Up to now, several basic SERDs have been rushed into clinical trials (Table S1). Among these candidates, elacestrant (2) was approved by the FDA on January 27, 2023, for the treatment of ER-positive, human epidermal growth factor receptor 2 (HER2)-negative, ESR1-mutated advanced or metastatic breast cancer. In a phase Ⅲ clinical trial that compared the efficacy and safety of elacestrant and standard of care with fulvestrant or aromatase inhibitors in breast cancer patients (NCT03778931), treatment with elacestrant provided a longer 6- or 12-month PFS rate and lower all-cause mortality for all patients [44]. Small molecules-induced hydrophobic exposure to ER initiates subsequent protein degradation, which motivates researchers to utilize hydrophobic groups in small molecules to achieve manageable TPD.
In 2021, Zhou et al. disclosed a type of ERα degrader labeled with hydrophobic tags in the patent (CN113773283A). Based on the ER binding mode of the antagonist 7-oxabicyclo[2.2.1]heptene sulfonamide (OBHSA) obtained previously [45], they synthesized a series of derivative compounds containing adamantyl or hydrophobic amino acid groups (Fig. 5B). The majority of these compounds exhibited the ability of ERα affinity and MCF-7 cell growth inhibition. As the optimal compounds, 16 (20a), 17 (20b), and 18 (30a) achieved target degradation activity equivalent to Fulvestrant at 5 µmol/L (Fig. 5C).
Based on the investigation of natural products, 6–epi-ophiobolin G (MHO7) downregulates ERα levels in a novel mechanism and is a promising SERD [46]. To improve the degradation activity of MHO7, the hydrophobic tagging strategy is applied. In 2022, Liang et al. synthesized compound 19 (16a), which combined MHO7 to an adamantyl group through a poly(ethylene glycol) (PEG) linker (Fig. 5D) [47]. The structure and activity relationship (SAR) studies demonstrated that a moderate length of linker, such as 3-PEG, is beneficial for the activity. In MCF-7 cells incubated with 19, an effective degradation of ERα and downregulation of the estrogen signaling pathway was observed. 19 displayed the ability to inhibit cell viability and induce cell apoptosis, which could be confirmed by phenotypes such as the induction of reactive oxygen species (ROS) generation and the decrease of mitochondrial membrane potential.
Glutathione S-transferases (GSTs) demonstrate cytoprotection from oxidative and environmental stress as phase Ⅱ enzymes involved in the deactivation of harmful electrophilic compounds by catalyzing the conjugation of glutathione (GSH). The level of GSTs is also related to the development and drug resistance of cancer. As a member of the GST isoenzymes family, the overexpression of GST pi (GSTP) can be detected in several human tumor cell lines [48,49]. Moreover, GSTP displays apoptosis resistance activity by forming the complex with Jun N-terminal kinase (JNK) to inhibit the release of JNK (Fig. 6A) [49]. Target GST degradation is a potential strategy to improve drug sensitivity and pro-apoptosis in tumor cells.
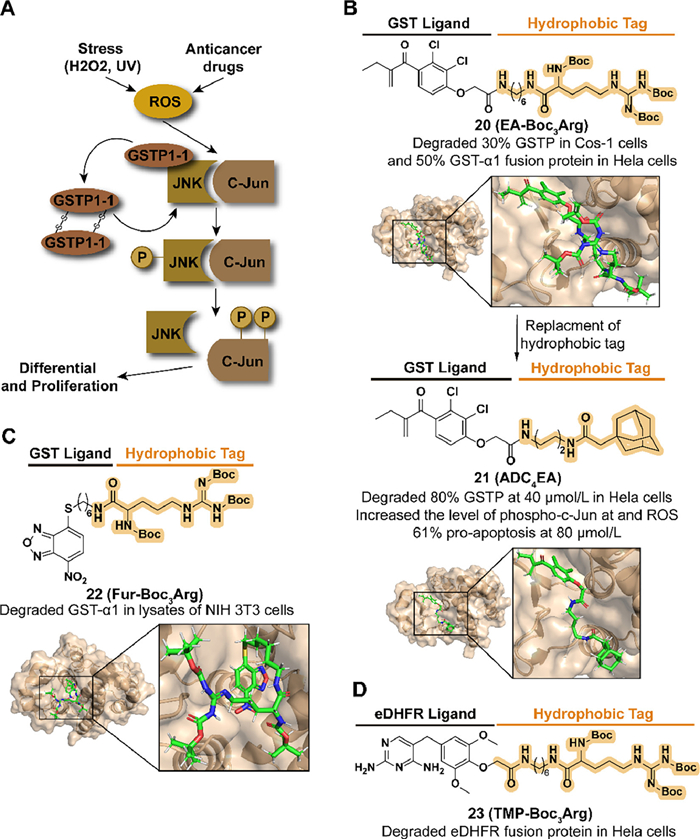
Based on the development of GST inhibitors, such as ethacrynic acid (EA) and thiobenzofurazan (Fur, also called NBDHEX) [50,51], Hedstrom's group combined the corresponding inhibitors with a tert–butyl carbamate-protected arginine (Boc3Arg) to synthesize the compound 20 (EA-Boc3Arg) and 22 (Fur-Boc3Arg) in 2012 (Figs. 6B and C) [52]. Both of them induced the degradation of modified GST-α1 in cell lysates and 20 displayed a dose-dependent degradation with approximately 50% in 10 µmol/L. A reasonable speculation for this degradability was that the Boc3Arg group provides a hydrophobic surface to induce unfolded protein degradation. Moreover, they also synthesized the compound 23 (TMP-Boc3Arg) which exhibited the ability to degrade ectopically expressed Escherichia coli dihydrofolate reductase (eDHFR) fusion proteins (Fig. 6D).
Encouraged by Hedstrom's study, in 2021, Li et al. synthesized a series of EA derivatives linked with the adamantyl group [53]. Among the compounds, 21 (ADC4EA) (Fig. 6B), which carried a linker with four carbon lengths, displayed the best GSTP degradation activity, over 80% at 40 µmol/L in HeLa cells. For JNK signaling regulation and oxidative stress, 21 results in the release of JNK, followed by phosphorylation of c-Jun and accumulation of ROS, which is not observed in EA-treated cells. Compared with the poor pro-apoptosis activity in EA, 21 induced 61% apoptosis in HeLa cells at 80 µmol/L.
Her kinase family members, such as epidermal growth factor receptor (EGFR), are involved in several cellular signaling processes, and their deregulation is observed in multiple cancers. As a member of Her, Eer3 is a significant heterodimeric partner for EGFR and co-regulates the subsequent phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling (Fig. 7A). Moreover, accumulating evidence proves that Her3 participates in the resistance of Eer2-medicated cancers [54]. However, there are few small-molecular kinase inhibitors discovered targeting Eer3 because the mutations in key residues of Eer3 result in weak kinase activity, which is classified as a "pseudokinase" [55]. Based on the previous exploration, Lim et al. reported the first Eer3 covalent ligand TX1-85-1 but with poor inhibition in Her3-dependent signaling and tumor growth [30]. The HyTTD strategy is considered to block the physiological functions of Eer3 by inducing target degradation.
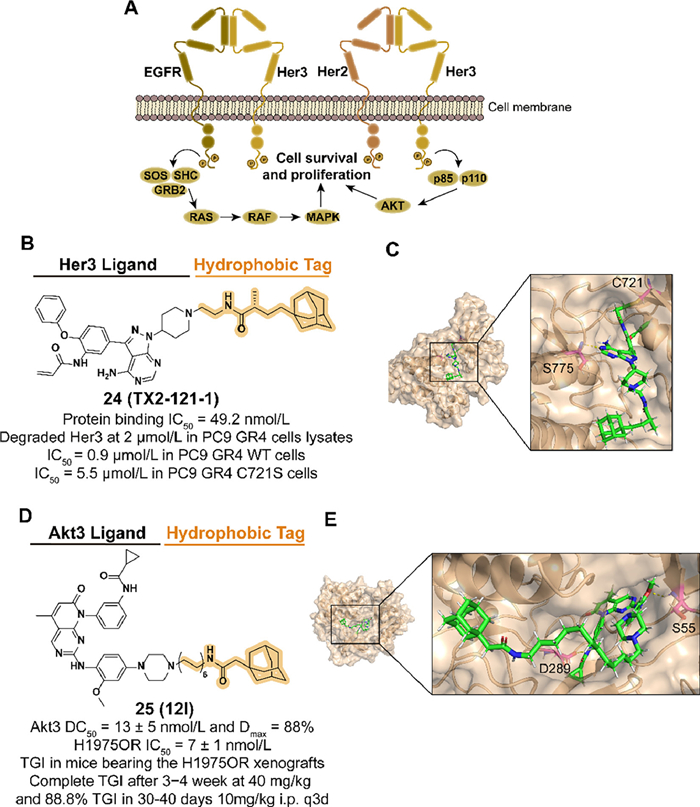
In 2014, they designed and synthesized the compound 24 (TX2-121-1) which combined TX1-85-1 and an adamantyl group with a flexible alkyl linker (Figs. 7B and C) [30]. 24 displayed effective degradation of Eer3 and inhibition of Eer3 downstream signaling. Antagonist activity of heterodimerization between Eer3 and relevant partners was also observed. 24 inhibited both wild-type and C721S mutated PC9 GR4 cells, which demonstrated that 24 was a potential small-molecular degrader against Eer3-dependent cancers.
After the emergence of acquired drug resistance to the first approved third-generation inhibitor osimertinib, non-small cell lung cancer (NSCLC) patients who have benefited from EGFR inhibitors therapy are facing an inevitable resistance problem [56,57]. Among multiple mechanisms of resistance, as a member of the serine/threonine-specific kinase family, upregulation of Akt3 isoform is associated with the development of many tumor types [58]. Given the unique functions among the three isoforms of Akt, it is advantageous to selectively target modulators of Akt3, which is challenging. Moreover, the noncatalytic function of Akt3 is considered to mediate tumor resistance, which has inspired the application of targeting Akt3 protein degradation [59].
To enhance the selectivity by targeting the allosteric pocket, Xu et al. obtained an allosteric Akt binder XTF-262 through virtual screening and synthesized the Akt3 degrader 25 (12l) which was introduced an adamantyl group with a linker in 2022 (Figs. 7D and E) [60]. Among a series of linkers in different types and lengths, a 12-carbon alkyl chain was selected for attachment to the piperazinyl group which is extended to the solvent-exposed surface. 25 displayed selective Akt3 degradation potency in a panel of NSCLC cells and made an anti-proliferation effect in H1975OR cells with osimertinib-induced resistance. In the H1975OR xenograft mice model, intraperitoneal administration of 25 with a dose of 10 or 20 mg/kg significantly inhibited tumor growth with acceptable safety data.
Androgen receptor (AR) can regulate relevant gene transcription by interacting with the corresponding ligand and forming dimer to bind to the DNA sequence in nuclear. However, AR-mediated gene expression also drives the development of prostate cancer. Although early treatment with a combination of androgen deprivation and AR antagonists was effective, drug resistance is inevitable after long-time administration. Due to overexpression of AR proteins, splicing variants, point mutations, and other mutations, traditional small-molecule AR antagonists fail in castration-resistant prostate cancer (CRPC) (Fig. 8A) [61–67]. With the development of TPD [68], the HyTTD strategy is used for selective AR degraders (SARD) design.
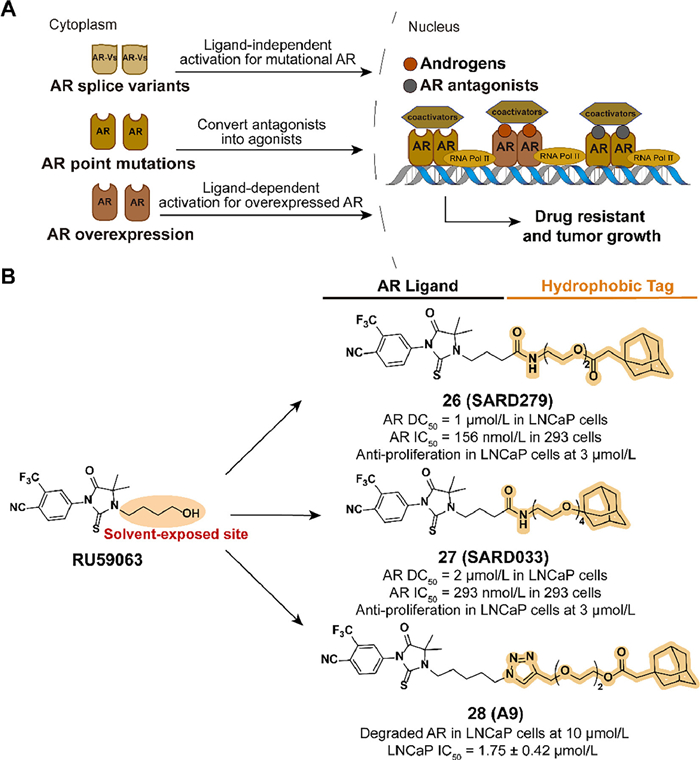
In 2015, Crews' group designed two small-molecule AR degraders, 26 (SARD279) and 27 (SARD033), which combined the agonist RU59063 and an adamantyl moiety through a PEG linker (Fig. 8B) [69]. Despite the binding affinity reduced compared to the parent compound, both SARD can selectively induce the degradation of AR in LNCaP cells. The AR gene expression and transactivation activities are blocked. AR-dependent LNCaP cell line is suppressed but AR-independent models are unaffected. Compared with traditional prostate cancer drug enzalutamide, HYTTD 26 demonstrates the ability against resistance mechanisms of AR mutants.
Inspired by Crews' study, Xie et al. reported a series of optimized SARD utilizing the hydrophobic tagging strategy in 2020 [70]. Based on the investigation of the SARs, they concluded that the introduction of the 1, 2, 3-triazole group is beneficial for the activity. Five carbons from imidazolidine to triazole group and 2-PEG linker may improve the pharmacological effects. Among the compounds, 28 (A9) exhibited excellent activity in blocking AR-dependent tumor cell proliferation, for the ability to induce the selective degradation of AR (Fig. 8B).
Plk1 is a member of the polo-like kinases (PLKs), which belong to the subfamily of serine–threonine protein kinases and participate in multiple significant physiological and oncogenic processes [49,71,72]. To obtain a higher selectivity than Plk1 catalytic domain-targeted inhibitors, the polo-box domain (PBD), which is involved in the protein-protein interaction function of Plk1, has become another target for the development of Plk1 inhibitors [73]. Based on the SAR of the natural product derivative poloxin, Berg's group discovered the optimized Plk1-PBD inhibitor poloxin-2 [74]. To further enhance the activity of blocking Plk1 function, a protein degradation approach by introducing the hydrophobic tag is applied.
SAR studies clarify that the iminoquinone ring and ester group are the main sites bound by the nucleophilic amino acid residues, benzoyl moiety is a suitable location for modification. In 2018, they fused an adamantyl group to the poloxin-2, which resulted in the development of 29 (Poloxin-2HT) (Fig. 9) [75]. Although the introduction of hydrophobic degron led to a decrease in the inhibition activity of Plk1 PBD, 29 effectively induced the degradation of Plk1 and the apoptosis of HeLa cells. By analyzing the chromosomal phenotype during mitosis, 29 displayed a dual ability to inhibit Plk1 PBD rapidly and subsequently block the function of Plk1.
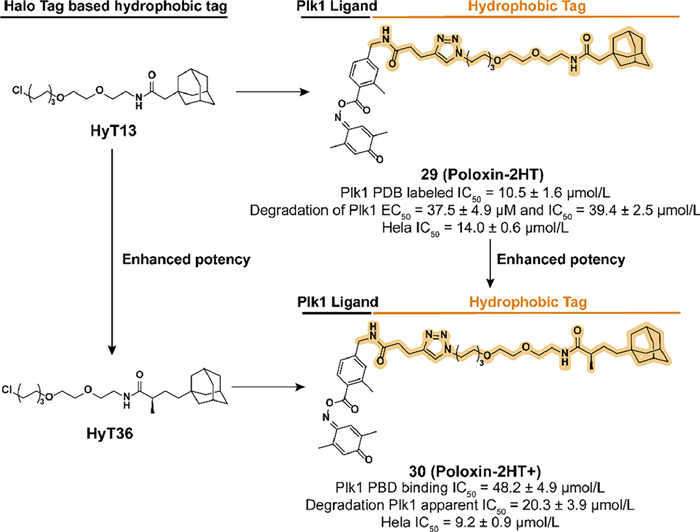
Inspired by the study of the HyTTD in Crews' group [28], HyT36 which has an additional methyl group compared to HyT13 is used in the activity optimization of the Plk1 degrader. Berg's group synthesized a new Plk1 PBD degrader 30 (Poloxin-2HT+) (Fig. 9) [76]. Compared to 29, although the introduction of HyT36 resulted in a further decrease in binding affinity, 30 displayed an enhanced ability to degrade Plk1 and induce apoptosis in HeLa cells. These studies also confirmed the superiority of HyT36 in designing Plk1 degraders compared to HyT13.
The transcription factor p53, as a tumor suppressor, plays a significant role in cell physiological processes and the mutation or functional absence of p53 has been observed in multiple human tumor types [77,78]. The overexpression of mouse double minute 2 homolog (MDM2), which negatively regulates the protein level of p53 as an E3 ubiquitin ligase, ubiquitinates p53 to induce proteasomal degradation and the subsequence development of cancers (Fig. 10A) [79,80]. The inhibitors of p53-MDM2 interaction bind with MDM2 to release and stabilize p53, resulting in the activation of the p53 pathway and inducing apoptosis in tumor cells [81]. The application of the hydrophobic tag provides a novel strategy to regulate p53-MDM2 interaction.
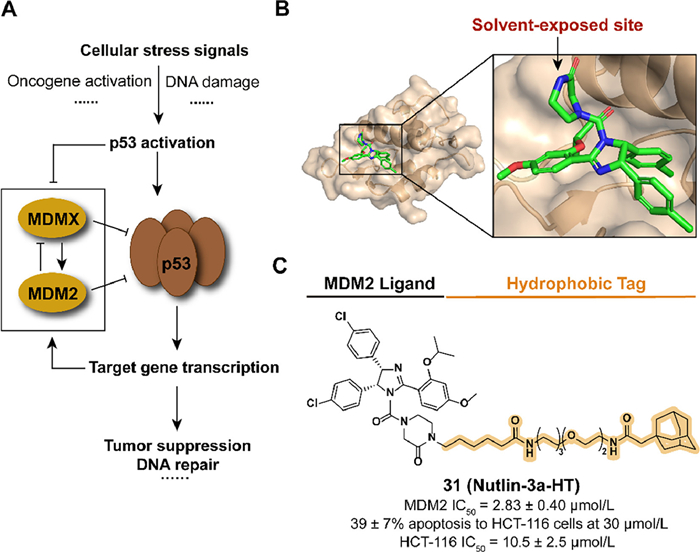
Inspired by the previous studies in poloxin-2, Berg's group constructed the compound 31 (Nutlin-3a-HT) by fusing the inhibitor Nutlin-3a and the degron adamantyl group through a linker, which is also an attempt to expand the application scope of HyTTD introducing the reversible inhibitor as a binder (Figs. 10B and C) [82,83]. Although the introduction of hydrophobic degron leads to a decrease in activity against MDM2, 31 overcome the obvious increase of both MDM2 and p53 induced by Nutlin-3a, which also displayed anti-tumor abilities in HCT-116 cells.
As a member of the poly-ADP-ribose polymerase (PARP) family, PARP1 plays a pivotal role in base excision repair (BER) of single-stranded DNA breaks by recognizing damaged DNA and activating the DNA repair system (Fig. 11A) [84,85]. Breast cancer gene 1/2 (BRCA1/2) participates in the DNA double-strand breaks and the mutation of BRCA1/2 may result in the development of ovarian and breast cancer [86,87]. PARP inhibition intensifies genomic instability in BRCA1/2-mutated cells and consequent cell apoptosis [88]. Hence, it is an effective therapeutic strategy in BRCA1/2-mutated cancer by targeting PARP1. Several PARP1 inhibitors have been approved in which the first approved molecule is olaparib from AstraZeneca [89], while there have not been any hydrophobic tag-tethered inhibitors developed for PARP1 degradation.
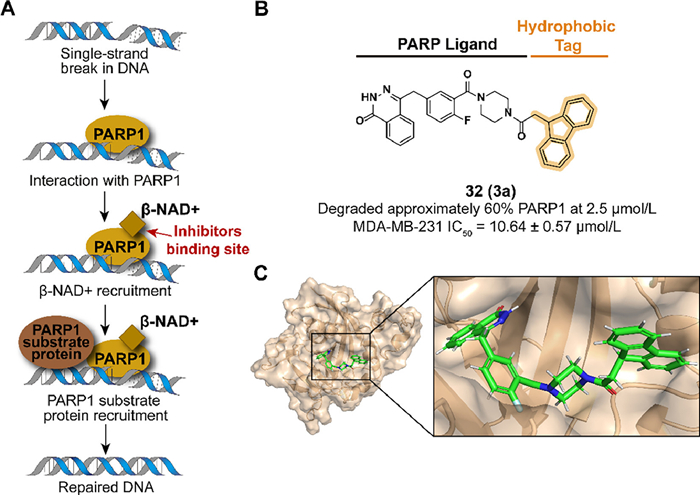
In 2020, Go et al. synthesized a PARP1 degrader 32 (3a) which used Olaparib as a PARP1 binder conjugated with a fluorene group (Figs. 11B and C) [90]. Because of the lack of installation space in the phthalazinone location, piperidine amine moiety was designed to link the hydrophobic tag. 32 displayed an effective degradation to PARP1 in the MDA-MB-231 cell line and similar results were observed in MDA-MB-468 at 48 h or HCC1937. In cell levels, 32 inhibited the viability and cell growth of both the absence or presence of BRCA1/2-mutated triple-negative breast cancer cells.
As a significant enzymatic subunit of the polycomb repressive complex (PRC2), enhancer of zeste homolog 2 (EZH2) is involved in the trimethylation activation of histone 3 lysine 27 (H3K27) which regulates gene expression through transcriptional repression [91,92]. The overexpression of EZH2 is observed in multiple cancers, such as breast cancer, and therefore a series of EZH2 inhibitors have been developed as an effective therapeutic strategy [93–95]. However, inhibition targeting the methyltransferase activity of EZH2 fails in triple-negative breast cancer (TNBC) treatment, which may be due to the noncatalytic functions of EZH2 in cancers [96,97]. Hence, EZH2 targeted degradation utilizing hydrophobic tagging is a promising strategy for treating TNBC by downregulating the protein levels of EZH2.
In 2020, Ma et al. synthesized the first selective EZH2 degrader, 33 (MS1943), which connected the piperazine moiety on the solvent-exposed side of C24 to an adamantyl group through a short linker (Figs. 12A and B) [98]. 33 exhibited an effective activity in reducing the protein level of EZH2 and suppressing cell proliferation in some TNBC cell lines. Oral and intraperitoneal pharmacokinetics (PK) studies and mouse xenograft model demonstrated promising anti-cancer bioactivity of 33 in vivo.
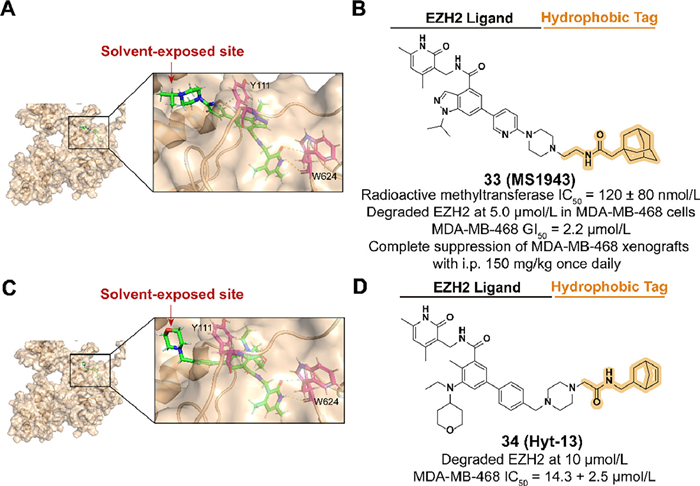
Based on the FDA-approved EZH2 inhibitor tazemetostat, Xie et al. synthesized the EZH2 degrader 34 (Hyt-13) which contained a norbornene moiety as the hydrophobic tag (Figs. 12C and D) [23]. 34 achieved the activity of degrading EZH2 at 10 µmol/L and enhanced antiproliferative activity in MDA-MB-468 cells.
As a transcription coactivator, steroid receptor coactivator-1 (SRC-1) is involved in regulating the transcription of targeted genes by interacting with hormone-activated nuclear receptors (NRs) (Fig. 13A) [99]. The abnormal expression of SRC-1 is related to a series of pathological processes, such as the development and migration of breast cancer [99–101]. Compared to genetic techniques, the chemical knockdown approach at the post-translational stage provides a potential therapeutic option to reduce abnormal proteins [102]. In previous studies, Choi et al. reported an SRC-1 degrader ND1-YL2 by combining a stapled peptide ligand YL2 with the N-degron peptide, but the poor cell permeability and stability limited the cellular activity of the ND1-YL2 [103]. To overcome these shortages, the hydrophobic tagging strategy is utilized to degrade SRC-1.
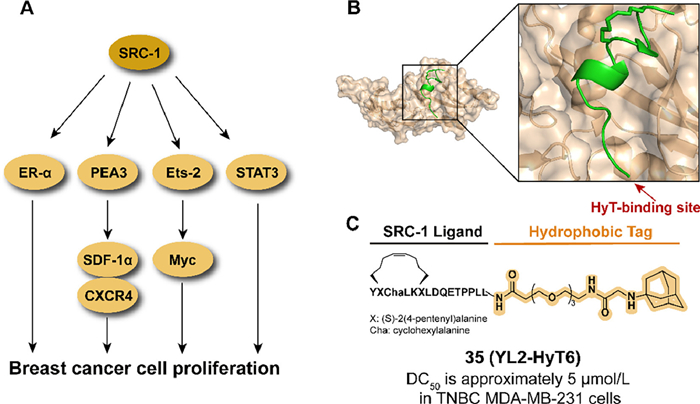
By analyzing the crystal structure of YL2 (Fig. 13B), the hydrophobic tag bound to the N-terminal of YL2 is tolerable in binding affinity. In 2021, they introduced an adamantyl group to replace the N-degron peptide of ND1-YL2, which resulted in synthesizing the series of YL2-HyT [104]. Among the compound, 35 (YL2-HyT6) displayed an improved degradation activity in the TNBC cell line compared to parental ND1-YL2 and the hydrophobic tag enhanced the cell permeability and stability of the compound (Fig. 13C). Moreover, 35 eliminated the migration and invasion of tumor cells by regulating the level of CSF-1 and E-cadherin rather than anti-proliferation.
Cyclin-dependent kinase (CDKs), which usually interacts with the regulatory subunit cyclins to exhibit enzymatic activity, plays a significant role in cell cycle transition and cell growth processes [105]. Hence, CDK and CDK-cyclin complexes are the potential therapeutic targets for anti-cancer and anti-viral strategies.
CDK4 and 6 (CDK4/6) form a complex with cyclin D and co-phosphorylate the tumor suppressor retinoblastoma (Rb) to regulate the G1-S phase in the cell cycle [106]. Therefore, as a target of cancer therapy, several CDK4/6 inhibitors are reported to limit the proliferation of tumor cells [107]. TNBC is considered an aggressive subtype of breast cancer with extreme malignancy, poor prognosis, and limited treatment. Although TNBC is insensitive to CDK4/6 inhibitors, a novel hydrophobic-tagged CDK4/6 inhibitor still be beneficial for blocking the proliferation of Rb wild-type TNBC [108,109].
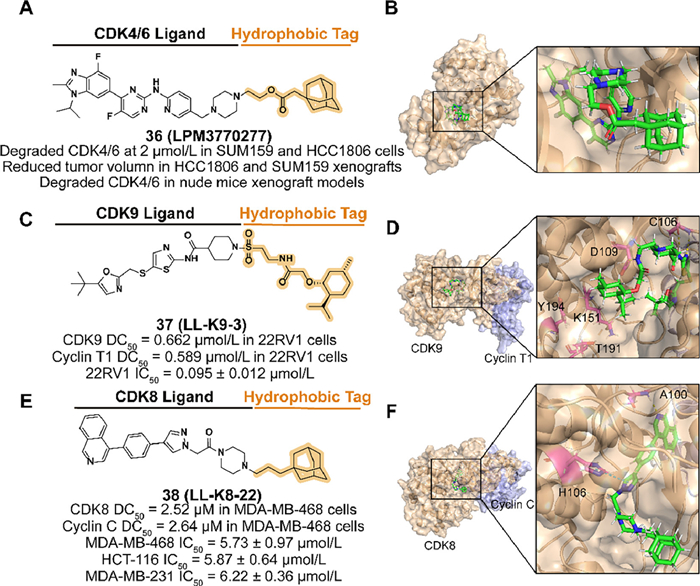
Qiu et al. designed a compound 36 (LPM3770277) (Figs. 14A and B), which is based on the active metabolite of CDK4/6 inhibitor abemaciclib, linked an adamantyl moiety with an ester linker [110]. Because of the steric hindrance of the hydrophobic tag, 36 displayed weaker binding affinity on CDK4 kinase than the parent. However, its effective degradation both in vitro and in vivo, reliable safety, and anti-cancer ability demonstrated that 36 is a promising compound against some types of TNBC.
As a significant transcription-associated CDK-cyclin pair, CDK9 is activated by the paired cyclin T1 and interacts with it to form the positive transcription elongation factor b (P-TEFb), which induces transcription complex assembly and elongates transcription by phosphorylating the C-terminal domain (CTD) of the RNAPII [111]. Hyperactivity of CDK9-cyclin T1 is associated with many types of cancers, so it is a promising strategy against relevant transcriptional-dependent cancers by inhibiting the complex [112–114]. Because of the similarity of kinase domains among CDKs and the lack of catalytic activity among cyclins, the development of direct targeting CDK9 or cyclin T1 selective inhibitors is challenging [115–117]. Among the technology of protein complex degradation, hydrophobic tagging degraders provide a novel idea to overcome the shortage of rapid monomeric CDKs degradation in PROTACs and the lack of structure-based design in molecular glues to degrade the complex effectively [21,118,119].
In 2022, Zhou's group reported a small-molecule degrader 37 (LL-K9-3) by utilizing the CDK9 inhibitor SNS032 linked with a (−)-menthoxyacetyl degron (Figs. 14C and D) [26]. Due to the polar residues surrounding the solvent-exposed piperidine of SNS032, they introduced a sulfonyl group to enhance the binding affinity. To the linker, increased length over C2 or flexible options are not beneficial to the activity. 37 degrade both CDK9 and cyclin T1 selectively and possibly synchronously in 22RV1 cells with dose-dependent and time-dependent effects. Moreover, 37 downregulated some factors related to the development of the CRPC and inhibited AR-medicated transcription, which is associated with the anti-proliferation and pro-apoptosis induced by 37.
CDK8 is another type of transcription-associated CDK, which phosphorylates the CTD of RNAP Ⅱ by forming a Mediator complex with cyclin C and MED12/MED13 [120]. The CDK8-cyclin C complex is associated with multiple signaling processes [121–123] and the complex is considered a significant anti-tumor target due to the overexpression observed in various cancer types [124–126]. To overcome the off-target effects in CDK8 inhibitors [127] and the lack of small-molecule modulators to target cyclin C directly, target protein degradation approaches are applied to inhibit tumor cell growth. As a feverish strategy, PROTAC degrader fails to affect cyclin C levels [128]. Hence, the HyTTD technology is used as a possible scheme to degrade the CDK8-cyclin C complex, which provides moderate degradation kinetics.
In 2023, Hu's group designed and synthesized a synchronous and selective CDK8-cyclin C complex degrader 38 (LL-K8-22) which attaches an adamantyl group to the CDK8/19 inhibitor BI-1347 (Figs. 14E and F) [129]. In the selection of linkers, alkyl linker with rigid piperazine is beneficial to the degradation activity. The molecular docking model between CDK-cyclin C and 38 demonstrated that the linker length contained three atoms effectively occupying the positively charged cavity of CDK8 resulting in a perturbation of protein stability, which may further lead to the degradation of the complex. 38 displayed the ability to degrade CDK8 and cyclin C involving no effect on gene transcriptome level and the anti-proliferation in several breast cancer cell lines.
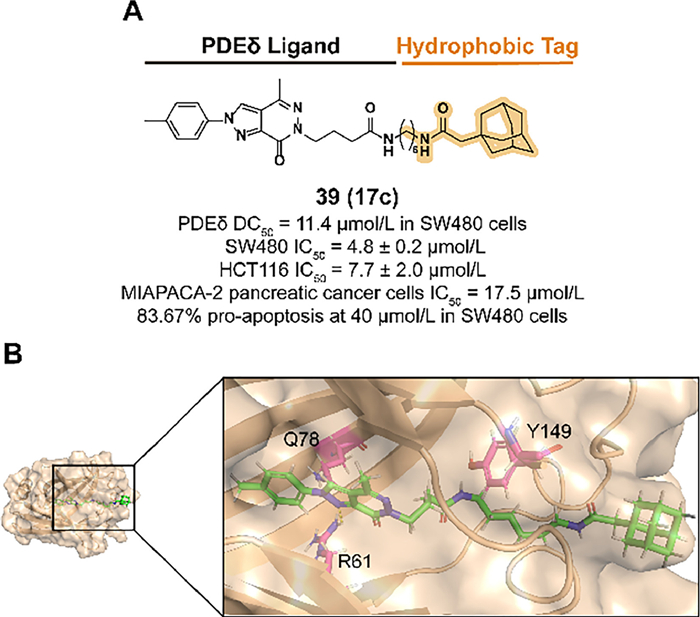
Kirsten rat sarcoma 2 viral oncogene homolog (KRAS) mutants can be observed in many types of cancers, such as pancreatic cancer and colon cancer [130,131]. Mutated KRAS contributes to the continuous binding between GTP and KRAS, which eventually results in the development and abnormal proliferation of cancers [132]. Phosphodiesterase-δ (PDEδ) is involved in the pro-solubilization and membrane-localized transport processes in the pharmacological ability of KRAS [133]. PDEδ-targeted therapy is a promising strategy against cancer proliferation. However, the treatment efficacy of KRAS-PDEδ inhibitors is not satisfied due to the poor anti-cancer activity [134]. Hence, a new attempt to degrade KRAS-PDEδ by introducing a hydrophobic tag has been reported.
Previous studies demonstrated that the alternative of phenylamine in the PDEδ inhibitor deltazinone with amphetamine is tolerable in binding affinity [135]. In 2022, Gou et al. synthesized compound 39 (17c) which fused the previous structure with an adamantyl degron (Figs. 15A and B) [136]. A rigid linker such as the phenyl group or a linker with improper length decreased the binding activity. 39 displayed a desirable activity to degrade PDEδ in SW480 cells and effectively blocked the cell proliferation in some types of tumor cell lines.
ALK activated by locus translocation and mutation results in the production of ALK fusions which are involved in the development of a variety of cancer types (Fig. 16A) [137–140]. Moreover, the low expression of ALK in adult tissues, which is presumed to reduce side effects in inhibiting ALK functions, makes it a potential therapeutic target [141]. Although several ALK small-molecule inhibitors were approved for clinical treatment, such as alectinib, the reduction of efficacy caused by subsequent drug resistance is challenging [142]. Inspired by the previous studies on PROTAC Q2 [143], Xie et al. introduced a hydrophobic tag to alectinib derivative A3 to improve the physicochemical and PK properties of ALK degraders.
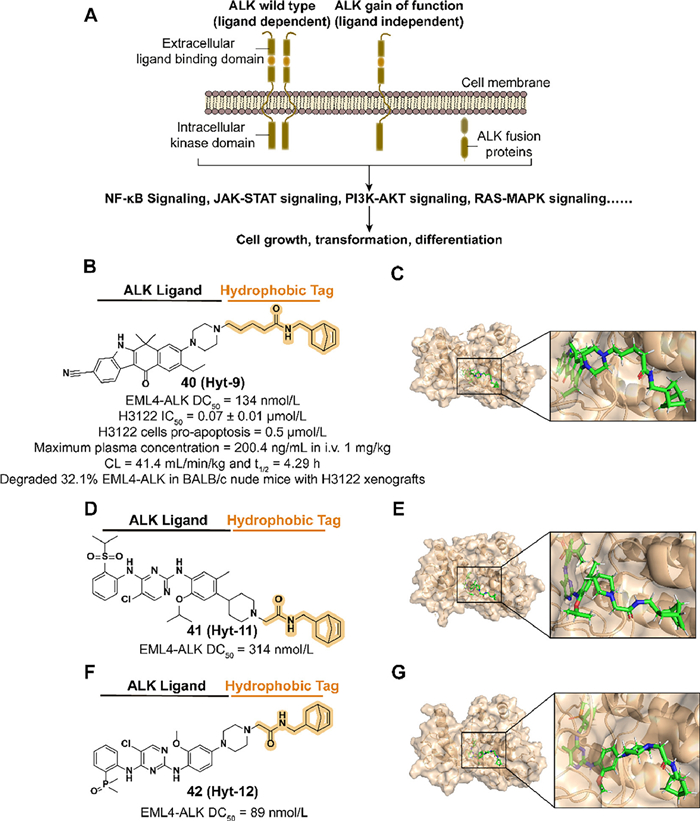
In 2023, they synthesized the compound 40 (Hyt-9) which combined the norbornene degron to A3 with an alkyl acid linker (Figs. 16B and C) [23]. The change of activity in utilizing hydrogenated norbornene and extended linkers demonstrated that the alkenyl group in norbornene and the length of the linker are essential to the potency. 40 displayed a potent ability to degrade ALK fusion proteins rapidly which provides an opportunity to overcome drug resistance medicated by upregulating protein expression in inhibitor therapy. Mechanism studies suggested that the norbornene degron medicated proteasome-associated degradation by recruiting HSP70 rather than direct binding. The activities of antiproliferation and enhanced downregulation of ALK levels and tumor growth inhibition were observed in 40 treated H3122 cells and xenograft tumor models. They also synthesized two more compounds, 41 (Hyt-11) and 42 (Hyt-12), which fused norbornene degron and two ALK inhibitors ceritinib and brigatinib respectively to expand the application of this hydrophobic tag (Figs. 16D–G). Both compounds achieve a dose-dependent degradation to EML4-ALK fusion protein.
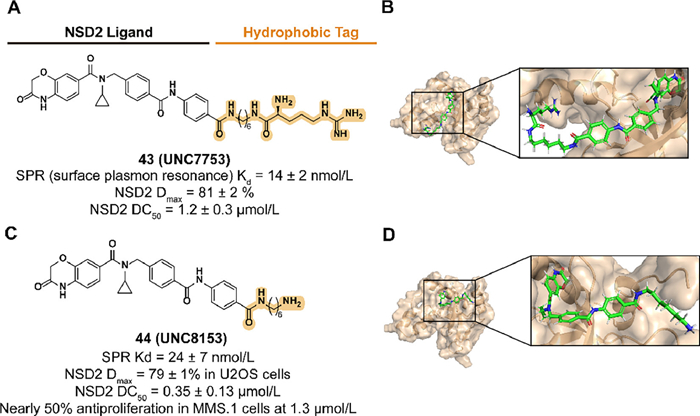
As an important protein in epigenetic regulation, NSD2 is involved in activating gene transcription through demethylating lysine 36 of histone 3 (H3K36me2) [144]. The upregulation of H3K36me2 induced by abnormally increased expression and mutations of NSD2 is associated with the development of the tumor [145,146]. However, the small molecules which can block the demethylation of H3K36me2 by targeting the catalytic function of NSD2 are lacking [147]. Inspired by the previous studies in NSD2 antagonists, Hanley et al. reported the compound UNC6934 which targeted the PWWP1 domain of NSD2 and disturbed the binding between NSD2 and nucleosomes with H3K36me2 [148]. To further counteract the catalytic ability of NSD2 and downregulate the protein levels, a targeted protein degradation strategy is utilized.
Based on the protein degradation strategy of the N-end rule pathway, they synthesized a series of UNC6934 derivatives attached to arginine or truncated side chains in 2023 [34]. Previous studies demonstrated that the attachment of arginine to the solvent-exposed pyrimidine side via a 6-carbon alkyl amine linker provides the compounds with the degradation of NSD2. Subsequent truncation studies of the potential compound 43 (UNC7753) led to 44 (UNC8153) with an enhanced degradation activity and validated the result as a prodrug design, which also raised questions about the mechanism of 43-medicated degradation (Fig. 17). 44 displayed selective NSD2 degradation and H3K36me2 levels downregulation activities in several cell lines with NSD2 overexpression or NSD2 E1099K mutation and altered the adhesion phenotype of KMS11 cells.
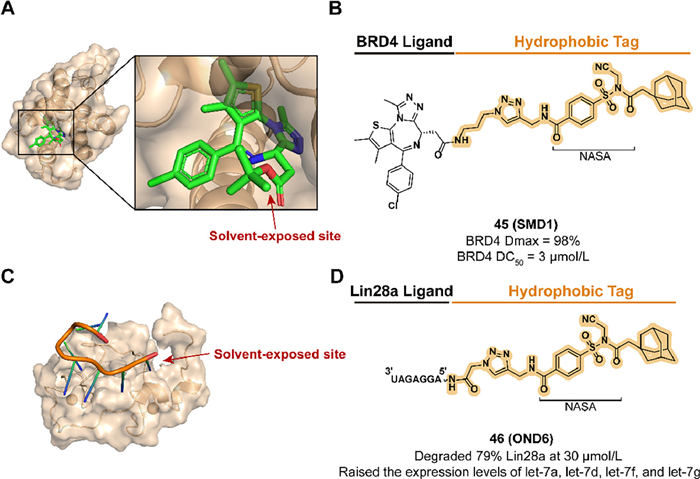
As a prospective target, DNA and RNA binding proteins (DBPs and RBPs) exhibit an indispensable role in regulating gene expression. With the development of TPD, this class of proteins which was once considered undruggable due to the lack of small-molecule ligand binding pockets finds a therapeutic breakthrough [149]. Although the classical bifunctional molecule PROTAC has been demonstrated in this field, the requirement for the expression and distribution of specific E3 ligases limits its application [13,150]. To extend the HyTTD scheme in the absence of high-affinity DBPs and RBPs, Wang et al. utilized the strategy of irreversible HyT-labeled target proteins by constructing a reactive electrophile linker for the first time [151,152].
With the discovery of the section N-acyl-N-alkyl sulfonamide (NASA) which contains rapid reaction kinetics and high labeling efficiency, they developed two degraders targeting DBP or RBP in 2023. Based on the non-covalent ligand JQ1, compound 45 (SMD1) modified JQ1 with a reactive NASA, and an adamantyl group was synthesized to target BRD4, a member of the bromodomain and extra-terminal (BET) family, and demonstrated a tag-translocated behavior in buffer system containing BRD4 (Figs. 18A and B) [153–155]. Because of the preferred labeling to lysine residues, the BRD4 BD1-JQ1 crystal structure elucidated possible binding sites K102 and K111. 45 displayed persistent and reversible BRD4 degradation activity in MV4-11 cells and lacked the hook effect that may occur in PROTACs at high concentrations. Lin28a is an RNA-binding protein that promotes tumorigenesis and progression by inhibiting let-7 miRNA production [156]. As a compound that could be taken up and effectively resistant to RNase A, the reactive degrader 46 (OND6), which contains an RNA binder [157] and an adamantyl tag, exhibited selective Lin28a downregulation in K562 cells and upregulated the expression of four let-7 family members (Figs. 18C and D). They further broadened the application of the HYTTD strategy by introducing RNA as a targeting ligand.
The microtubule-associated protein Tau exerts a microtubule (MT) stabilization function in the axons of neurons and also plays a significant role in the development of Alzheimer's disease (AD). Several pathological conditions cause disturbances in the balance of Tau binding to MTs, such as hyperphosphorylation of Tau, which may lead to an abnormal increase in free Tau concentration and subsequent Tau aggregation and the formation of neurofibrillary tangles (NFT) to damage neurons (Fig. 19A) [158]. The reduction of Tau levels can decrease aggregation and resistance to memory impairment [159,160]. Considering the poor safety and stability of genetic knockdown [161], the utilization of chemical methods to achieve the degradation of the non-enzyme protein Tau is promising.
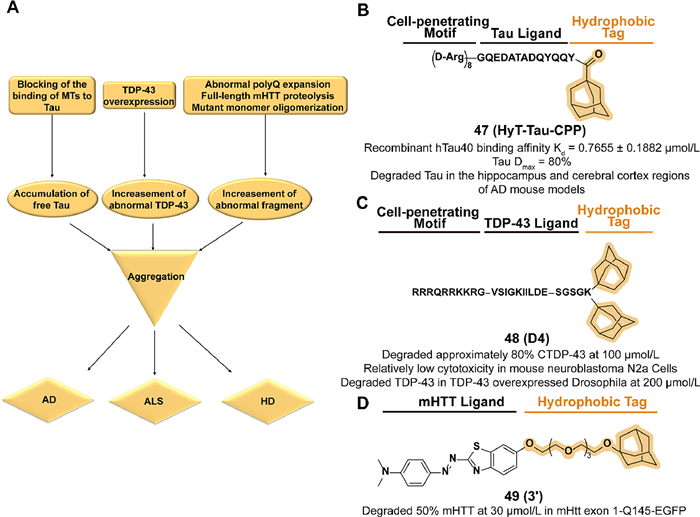
Chen's group synthesized a tri-functional compound 47 (HyT-Tau-CPP) which fused an adamantyl group with a cell-penetrating peptide on the Tau-recognition motif in 2017 (Fig. 19B) [162]. Supported by the binding affinity assay and cell-penetration assay, 47 displayed a dose-dependent and time-dependent Tau degradation potency in Tau-EGFP overexpressed cell lines. The little cytotoxicity in N2a cells indicated the safety of 47 and exerted downregulation of Tau levels in hippocampus and cerebral cortex regions in subsequent AD mouse model assay.
TAR DNA binding protein-43 (TDP-43) is a widely expressed RNA/DNA-binding protein that is associated with neuronal and embryonic development-related mRNA regulation [163–166]. As a key pathogenic protein of amyotrophic lateral sclerosis (ALS), overexpression of wild-type TDP-43 may cause accumulation and aggregation in the cytoplasm, resulting in the loss of function and generation of toxicity (Fig. 19A) [167,168]. Moreover, growing studies demonstrate that an increase in the concentration of pathogenic TDP-43 monomers accelerates the aggregation process [169]. Hence, considering the advantageous safety profile of chemical strategy to reduce undruggable proteins, downregulation of the level of TDP-43 by utilizing protein degradation techniques has clinical potential.
In 2019, Chen's group reported a series of multifunctional peptides which combined the four groups of cell-penetrating, TDP-43 recognition motif, linker, and hydrophobic motif in turn [170]. Among the compounds, 48 (D4) which contains two hydrophobic adamantyl groups, displayed the degradation potency to intracellular CTDP-43 in a stable 162–414 CTDP-43-EGFP A315T(CTDP-43) overexpressed cell line (Fig. 19C). Relatively low normal cytotoxicity and a reduction of CTDP-43-induced cytotoxicity were observed in D4-treated cells. Moreover, 48 decreased the level of TDP-43 in the drosophila model.
An excessive repetition appears in the autosomal inherited CAG trinucleotide of the Huntingtin gene on chromosome 4, resulting in the expression of the mutant Huntingtin (mHTT) protein [171]. Its abnormal polyglutamine (polyQ) expansion in N-terminal produces cytotoxic aggregation after intracellular full-length protein proteolysis or mutant monomer oligomerization, which is responsible for the development of Huntingtin disease (HD) (Fig. 19A) [172–174]. Moderate clearance of mHTT is considered to be a rational approach to curtail and slow the progression of HD [175]. However, because the biological function of such protein is independent of its aggregating, the traditional drug design model based on the function of disease-related proteins is not effective [176].
Inspired by the previous studies of SNIPER 2 [177], Hirai et al. developed the first small-molecule aggregation degrader 49 (3′) conjugated with aggregate binder moiety and an adamantyl group through an ethylene glycol linker (Fig. 19D) [25]. In a constructed HTT C-terminally EGFP-fused exon 1-expressing HeLa cells model with extended 145-CAG repeat (mHtt exon 1-Q145-EGFP), 49 displayed a dose-dependent degradation activity of mHtt exon 1-Q145-EGFP and there is no effect on a non-pathogenic repeat threshold of Htt exon 1-Q23-EGFP. In immobilized artificial membrane (IAM) column analysis, 49 possessed a better index of membrane permeability (PI) than SNIPER and achieved blood-brain barrier crossing and active uptake in the brain in subsequent tests in vivo. However, Caco-2 permeability experiments suggest that 49 may not be suitable for oral administration.
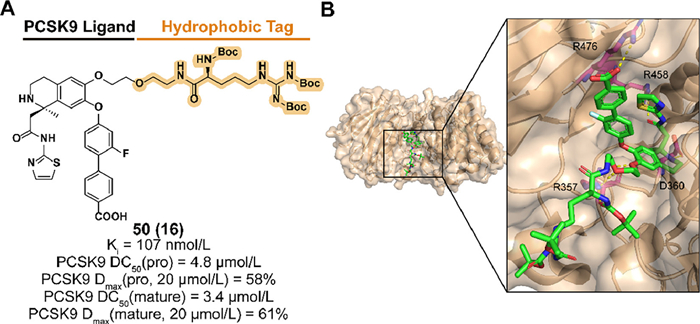
The serine protease proprotein convertase subtilisin kexin 9 (PCSK9) regulates blood low-density lipoprotein (LDL) uptake levels by binding to cell surface LDL receptors (LDLR) and targeting the complex for lysosomal degradation. Reducing LDL in blood by blocking the interaction between PCSK9 and LDLR provides a therapeutic approach to decreasing the risk of cardiovascular disease [178]. However, the flatness of interacting surfaces and the nonessential catalytic function of PCSK9 for LDLR downregulation make challenges to the development of small-molecule inhibitors [179,180].
Based on the structure of the PCSK9 binder obtained from previous screening and optimizing, Petrilli et al. designed and synthesized compound 50 (named 16 in the original research article) which introduced a Boc3Arg degron to C6 position of the binder (Figs. 20A and B) [181]. 50 exhibited effective affinity to PCSK9 and target degradation abilities of both pro and mature forms in PCSK9-overexpressed HEK293 cells [182]. Insignificant effects on cell viability indicated the safety of blood LDL regulation.
The TPD technology, rooted in the principles of chemical biology, is dedicated to the eradication of functional proteins in vivo, with a particular emphasis on small molecule degraders. This innovative approach exhibits considerable therapeutic value, boasting advantages such as precise regulation, efficient degradation, and extensive applicability in combatting drug resistance and undruggable pathogenic targets. Consequently, TPD technology emerges as an extensively adaptable paradigm within target-driven drug discovery, enabling the engagement of formerly intractable proteins and propelling the development of therapeutics for previously untreatable diseases.
HyTTDs mimic the unfolded or misfolded state of proteins in vivo by spatially approaching POI through hydrophobic small molecules and then activating the UPR system to provoke POI degradation via the proteasome pathway. HyTTD technology is a biotechnological tool with great potential and has received extensive attention in drug discovery and basic biological research fields. Compared with PROTAC technology and molecular glue technology, which are in the limelight in drug discovery, the HyTTD is still an impressive protein degradation paradigm to be reckoned with. Firstly, highly specific. Through POI ligand optimization, HyTTDs can precisely degrade the target protein without affecting the function and stability of other proteins. It makes the HyTTD a precise protein regulatory tool for targeted intervention in specific biological processes. Secondly, efficient degradation. HyTTDs quickly and efficiently degrade the target protein into fragments or eliminate it, blocking its biological function. Compared with traditional protein inhibitors or RNAi, the degradation effect of HyTTDs is more thorough and enduring. Thirdly, expansive applicability. Due to HyTTDs acting through native protein clearance pathways and independent of specific E3 ligases, HyTTDs can be applied to proteins of various types containing enzymes, signaling proteins, or receptor proteins on cell membranes. Also, high biocompatibility. Benefiting from the streamlined molecular structure, better bioavailability makes HyTTDs possess high potential and application value in drug development.
However, further expansion and enhancement of research on HyTTDs is warranted. Firstly, it is imperative to improve the biological activity of HyTTDs. Through an analysis of the structure-activity relationship of existing HyTTDs, it is discernible that the length and composition of hydrophobic groups exert a significant influence on their activity. Consequently, optimizing the length of hydrophobic fragments or discovering hydrophobic tags with innovative structures holds promise for augmenting their degradation efficacy and catering to clinical requirements. Secondly, it is crucial to unravel the specific mechanism of action underlying HyTTDs. Although numerous HyTTDs have been developed, the investigation into their mechanism of action remains largely superficial, leaving several knowledge gaps that need to be addressed. Thus, forthcoming research should concentrate on employing biochemical techniques to delve into the intricate mechanisms involved in HyTTDs-induced protein degradation, thereby furnishing a robust theoretical foundation for the rational design of HyTTDs. Thirdly, additional efforts should be made to expand the therapeutic scope of HyTTDs. Currently, HyTTDs have witnessed considerable development in the field of oncology, while research on neurodegenerative diseases and cardiovascular diseases remains relatively limited. Moreover, there exists significant research potential in the realms of immunity, hematologic disorders, and so on. Furthermore, future endeavors should encompass the integration of precision treatment concepts. By optimizing the length and structural composition of POI ligands and hydrophobic components, HyTTDs can exhibit high selectivity and precisely degrade target proteins without compromising the functionality and stability of normal proteins. This capability will position HyTTDs as an exceptionally precise protein regulation tool capable of modulating specific biological processes while safeguarding normal physiological processes [150,183].
This article presents a systematic and comprehensive review of the development of HyTTD technology, encompassing an in-depth analysis of HyTTDs as disclosed in both scholarly publications and patents. Moreover, the critical appraisal summarizes the strengths and limitations prevalent in this burgeoning field, while forecasting future directions in the form of novel structural advancements within HyTTDs. By illuminating these perspectives, this review aims to inspire researchers and accelerate the translational efforts of HyTTDs to uncover their full medical potential, ultimately resulting in clinical benefits for patients.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 82103978 and 81874286), the Natural Science Foundation of Jiangsu Province (No. BK20210423), "Double-First-Class" University Project (Nos. CPU 2018PZQ02 and CPU 2018GY07).
Supplementary material associated with this article can be found, in the online version, at doi:
T.K. Neklesa, J.D. Winkler, C.M. Crews, Pharmacol. Ther. 174 (2017) 138–144. doi: 10.1016/j.pharmthera.2017.02.027
A.J. Tao, G.E. Gadbois, S.A. Buczynski, et al., Curr. Opin. Chem. Biol. 67 (2022) 102114. doi: 10.1016/j.cbpa.2021.102114
M.P. Kannan, S. Sreeraman, C.S. Somala, et al., Fut. Med. Chem. 15 (2023) 867–883. doi: 10.4155/fmc-2023-0072
M.H. Glickman, A. Ciechanover, Physiol. Rev. 82 (2002) 373–428. doi: 10.1152/physrev.00027.2001
K. Cassidy, H. Zhao, Biochemistry 62 (2023) 580–587. doi: 10.1021/acs.biochem.1c00330
L.M. Luh, U. Scheib, K. Juenemann, et al., Angew. Chem. Int. Ed. 59 (2020) 15448–15466. doi: 10.1002/anie.202004310
X. Li, Y.C. Song, J. Hematol. Oncol. 13 (2020) 50. doi: 10.1002/jeq2.20016
Y.H. Sun, X. Luo, Z.M. Yang, et al., Chin. Chem. Lett. 34 (2023) 107924. doi: 10.1016/j.cclet.2022.107924
M. He, C.G. Cao, Z.H. Ni, et al., Signal Transduct. Target. Ther. 7 (2022) 181. doi: 10.1038/s41392-022-00999-9
A.P. Chen, Y. Zhong, Y.X. Liu, et al., Chin. Chem. Lett. 34 (2023) 107923. doi: 10.1016/j.cclet.2022.107923
C.L. Pu, S.R. Wang, L. Liu, et al., Chin. Chem. Lett. 34 (2023) 107927. doi: 10.1016/j.cclet.2022.107927
Z.Y. Hu, C.M. Crews, ChemBioChem 23 (2022) e202100270. doi: 10.1002/cbic.202100270
K. Li, C.M. Crews, Chem. Soc. Rev. 51 (2022) 5214–5236. doi: 10.1039/d2cs00193d
M. Bekes, D.R. Langley, C.M. Crews, Nat. Rev. Drug Discov. 21 (2022) 181–200. doi: 10.1038/s41573-021-00371-6
G. Ahn, S.M. Banik, C.L. Miller, et al., Nat. Chem. Biol. 17 (2021) 937–946. doi: 10.1038/s41589-021-00770-1
K. Wang, A.L. Yu, K.W. Liu, et al., Adv. Sci. 10 (2023) 2300288. doi: 10.1002/advs.202300288
A.D. Cotton, D.P. Nguyen, J.A. Gramespacher, et al., J. Am. Chem. Soc. 143 (2021) 593–598. doi: 10.1021/jacs.0c10008
D. Takahashi, J. Moriyama, T. Nakamura, et al., Mol. Cell 76 (2019) 797–810. doi: 10.1016/j.molcel.2019.09.009
Z.Y. Li, C.G. Zhu, Y. Ding, et al., Autophagy 16 (2020) 185–187. doi: 10.1080/15548627.2019.1688556
L. Zhao, J. Zhao, K.H. Zhong, et al., Signal Transduct. Target. Ther. 7 (2022) 113. doi: 10.1007/978-981-16-9515-5_9
G.Q. Dong, Y. Ding, S.P. He, et al., J. Med. Chem. 64 (2021) 10606–10620. doi: 10.1021/acs.jmedchem.1c00895
J.K. Gao, J.T. Zhang, X.L. Han, et al., Curr. Med. Chem. 30 (2023) 3137–3155. doi: 10.2174/0929867329666220930120328
S.W. Xie, F.Y. Zhan, J.J. Zhu, et al., Angew. Chem. Int. Ed. 135 (2023) e202217246. doi: 10.1002/ange.202217246
Y. Asawa, K. Nishida, K. Kawai, et al., Bioconjug. Chem. 32 (2021) 2377–2385. doi: 10.1021/acs.bioconjchem.1c00431
K. Hirai, H. Yamashita, S. Tomoshige, et al., ACS Med. Chem. Lett. 13 (2022) 396–402. doi: 10.1021/acsmedchemlett.1c00500
J.C. Li, T. Liu, Y.L. Song, et al., J. Med. Chem. 65 (2022) 11034–11057. doi: 10.1021/acs.jmedchem.2c00257
H.K. Patel, T. Bihani, Pharmacol. Ther. 186 (2018) 1–24. doi: 10.1016/j.pharmthera.2017.12.012
H.S. Tae, T.B. Sundberg, T.K. Neklesa, et al. ChemBioChem 13 (2012) 538–541. doi: 10.1002/cbic.201100793
T.K. Neklesa, D.J. Noblin, A.P. Kuzin, et al. ACS Chem. Biol. 8 (2013) 2293–2300. doi: 10.1021/cb400569k
T. Xie, S.M. Lim, K.D. Westover, et al., Nat. Chem. Biol. 10 (2014) 1006–1012. doi: 10.1038/nchembio.1658
G.E. Karagöz, D. Acosta-Alvear, P. Walter, Cold Spring Harbor Perspect. Biol. 11 (2019) a033886. doi: 10.1101/cshperspect.a033886
A.C. Lai, C.M. Crews, Nat. Rev. Drug Discov. 16 (2017) 101–114. doi: 10.1038/nrd.2016.211
Y. Shi, M.J.C. Long, M.M. Rosenberg, et al., ACS Chem. Biol. 11 (2016) 3328–3337. doi: 10.1021/acschembio.6b00656
R.P. Hanley, D.Y. Nie, J.R. Tabor, et al., J. Am. Chem. Soc. 145 (2023) 8176–8188. doi: 10.1021/jacs.3c01421
R.K. Rej, J.E. Thomas, R.K. Acharyya, et al., J. Med. Chem. 66 (2023) 8339–8381. doi: 10.1021/acs.jmedchem.3c00136
L. Wang, A. Sharma, Chem. Med. Chem. 15 (2020) 2072–2097. doi: 10.1002/cmdc.202000473
X. Lin, H. Xiang, G.S. Luo, Eur. J. Med. Chem. 206 (2020) 112689. doi: 10.1016/j.ejmech.2020.112689
S. Arnesen, Z. Blanchard, M.M. Williams, et al. Cancer Res. 81 (2021) 539–551. doi: 10.1158/0008-5472.can-20-1171
A.E. Wakeling, J. Bowler, J. Endocrinol. 112 (1987) R7–R10. doi: 10.1677/joe.0.112R007
L. Wang, A. Sharma, Chem. Soc. Rev. 51 (2022) 8149–8159. doi: 10.1039/d2cs00117a
B.M. Wittmann, A. Sherk, D.P. McDonnell, Cancer Res. 67 (2007) 9549–9560. doi: 10.1158/0008-5472.CAN-07-1590
T. Downton, F.A. Zhou, D. Segara, et al., Drug Des. Dev. Ther. 16 (2022) 2933–2948. doi: 10.2147/dddt.s380925
M. Mottamal, B.R. Kang, X.Y. Peng, et al., ACS Omega 6 (2021) 9334–9343. doi: 10.1021/acsomega.0c06362
F.C. Bidard, V.G. Kaklamani, P. Neven, et al., J. Clin. Oncol. 40 (2022) 3246–3256. doi: 10.1200/jco.22.00338
Y.Y. Li, S.L. Zhang, J. Zhang, et al., Eur. J. Med. Chem. 172 (2019) 48–61. doi: 10.1016/j.ejmech.2019.03.058
Y. Zhao, C.X. Zhao, J. Lu, et al., Pharmacol. Res. 146 (2019) 104294. doi: 10.1016/j.phrs.2019.104294
J.J. Liang, W.L. Yu, L. Yang, et al., Eur. J. Med. Chem. 229 (2022) 114081. doi: 10.1016/j.ejmech.2021.114081
J.D. Hayes, J.U. Flanagan, I.R. Jowsey, Annu. Rev. Pharmacol. Toxicol. 45 (2005) 51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857
V. Adler, Z.M. Yin, S.Y. Fuchs, et al., EMBO J. 18 (1999) 1321–1334. doi: 10.1093/emboj/18.5.1321
J. Ploemen, B. Vanommen, J.J.P. Bogaards, et al., Xenobiotica 23 (1993) 913–923. doi: 10.3109/00498259309059418
G. Ricci, F. De Maria, G. Antonini, et al., J. Biol. Chem. 280 (2005) 26397–26405. doi: 10.1074/jbc.M503295200
M.J.C. Long, D.R. Gollapalli, L. Hedstrom, Chem. Biol. 19 (2012) 629–637. doi: 10.1016/j.chembiol.2012.04.008
X.N. Li, Z.R. Lu, C. Wang, et al., ACS Med. Chem. Lett. 12 (2021) 720–725. doi: 10.1021/acsmedchemlett.0c00627
R. Mishra, H. Patel, S. Alanazi, et al., Oncol. Rev. 12 (2018) 45–62.
N. Jura, Y.B. Shan, X.X. Cao, et al., Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 21608–21613. doi: 10.1073/pnas.0912101106
M.J. Niederst, H.C. Hu, H.E. Mulvey, et al., Clin. Cancer Res. 21 (2015) 3924–3933. doi: 10.1158/1078-0432.CCR-15-0560
K.S. Thress, C.P. Paweletz, E. Felip, et al., Nat. Med. 21 (2015) 560–562. doi: 10.1038/nm.3854
C. Stottrup, T. Tsang, Y.R. Chin, Mol. Cancer Ther. 15 (2016) 1964–1974. doi: 10.1158/1535-7163.MCT-15-0748
J. Levenga, H. Wong, R.A. Milstead, et al., eLife 6 (2017) e30640. doi: 10.7554/eLife.30640
F. Xu, X. Zhang, Z.P. Chen, et al., J. Med. Chem. 65 (2022) 14032–14048. doi: 10.1021/acs.jmedchem.2c01246
J.D. Joseph, N. Lu, J. Qian, et al., Cancer Discov. 3 (2013) 1020–1029. doi: 10.1158/2159-8290.CD-13-0226
R.R. McKay, L. Kwak, J.P. Crowdis, et al., Clin. Cancer Res. 27 (2021) 3610–3619. doi: 10.1158/1078-0432.ccr-20-4616
A.W. Wyatt, M. Annala, R. Aggarwal, et al., J. Natl. Cancer Inst. 110 (2017) djx118.
G.N. Brooke, C.L. Bevan, Curr. Genom. 10 (2009) 18–25. doi: 10.2174/138920209787581307
M.D. Balbas, M.J. Evans, D.J. Hosfield, et al., eLife 2 (2013) e00499. doi: 10.7554/eLife.00499
S. Prekovic, T. Van den Broeck, S. Linder, et al. Endocr. Relat. Cancer 25 (2018) R545–R557. doi: 10.1530/erc-17-0136
S.R. Viswanathan, G. Ha, A.M. Hoff, et al. Cell 174 (2018) 433–447. doi: 10.1016/j.cell.2018.05.036
S. Ha, G.S. Luo, H. Xiang, J. Med. Chem. 65 (2022) 16128–16154. doi: 10.1021/acs.jmedchem.2c01487
J.L. Gustafson, T.K. Neklesa, C.S. Cox, et al. Angew. Chem. Int. Ed. 54 (2015) 9659–9662. doi: 10.1002/anie.201503720
H. Xie, J.J. Liang, Y.L. Wang, et al., Eur. J. Med. Chem. 204 (2020) 112512. doi: 10.1016/j.ejmech.2020.112512
M. Bollen, D.W. Gerlich, B. Lesage, Trends Cell Biol. 19 (2009) 531–541. doi: 10.1016/j.tcb.2009.06.005
K. Strebhardt, A. Ullrich, Nat. Rev. Cancer 6 (2006) 321–330. doi: 10.1038/nrc1841
J.P. Yuan, M. Sanhaji, A. Kramer, et al., Am. J. Pathol. 179 (2011) 2091–2099. doi: 10.1016/j.ajpath.2011.06.031
A. Scharow, M. Raab, K. Saxena, et al., ACS Chem. Biol. 10 (2015) 2570–2579. doi: 10.1021/acschembio.5b00565
S. Rubner, A. Scharow, S. Schubert, et al., Angew. Chem. Int. Ed. 57 (2018) 17043–17047. doi: 10.1002/anie.201809640
S. Rubner, S. Schubert, T. Berg, Org. Biomol. Chem. 17 (2019) 3113–3117. doi: 10.1039/c9ob00080a
D. Hamroun, S. Kato, C. Ishioka, et al., Hum. Mutat. 27 (2006) 14–20. doi: 10.1002/humu.20269
D. Lane, A. Levine, Cold Spring Harbor Perspect. Biol. 2 (2010) a000893.
M. Wade, Y.C. Li, G.M. Wahl, Nat. Rev. Cancer 13 (2013) 83–96. doi: 10.1038/nrc3430
H.J. Li, X.Y. Chen, M.H. Wu, et al., Chin. Chem. Lett. 33 (2022) 1254–1258. doi: 10.1016/j.cclet.2021.08.130
T. Thompson, C. Tovar, H. Yang, et al., J. Biol. Chem. 279 (2004) 53015–53022. doi: 10.1074/jbc.M410233200
F. Nietzold, S. Rubner, T. Berg, Chem. Commun. 55 (2019) 14351–14354. doi: 10.1039/c9cc07795b
T.K. Neklesa, H.S. Tae, A.R. Schneekloth, et al., Nat. Chem. Biol. 7 (2011) 538–543. doi: 10.1038/nchembio.597
I. Faraoni, G. Graziani, Cancers 10 (2018) 487. doi: 10.3390/cancers10120487
X.W. Zhang, C.L. Zhang, L. Tang, et al., Chin. Chem. Lett. 31 (2020) 136–140. doi: 10.1016/j.cclet.2019.04.045
A. Shailani, R.P. Kaur, A. Munshi, Med. Oncol. 35 (2018) 18. doi: 10.1007/s12032-018-1085-8
K.B. Kuchenbaecker, J.L. Hopper, D.R. Barnes, et al., J. Am. Med. Assoc. 317 (2017) 2402–2416. doi: 10.1001/jama.2017.7112
H. Farmer, N. McCabe, C.J. Lord, et al., Nature 434 (2005) 917–921. doi: 10.1038/nature03445
P.C. Fong, T.A. Yap, D.S. Boss, et al., J. Clin. Oncol. 28 (2010) 2512–2519. doi: 10.1200/JCO.2009.26.9589
A. Go, J.W. Jang, W. Lee, et al., Eur. J. Med. Chem. 204 (2020) 112635. doi: 10.1016/j.ejmech.2020.112635
R. Margueron, D. Reinberg, Nature 469 (2011) 343–349. doi: 10.1038/nature09784
A. Kuzmichev, K. Nishioka, H. Erdjument-Bromage, et al., Genes Dev. 16 (2002) 2893–2905. doi: 10.1101/gad.1035902
K.H. Kim, C.W.M. Roberts, Nat. Med. 22 (2016) 128–134. doi: 10.1038/nm.4036
S. Varambally, S.M. Dhanasekaran, M. Zhou, et al., Nature 419 (2002) 624–629. doi: 10.1038/nature01075
C.G. Kleer, Q. Cao, S. Varambally, et al., Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 11606–11611. doi: 10.1073/pnas.1933744100
J. Kim, Y. Lee, X.D. Lu, et al., Cell Rep. 25 (2018) 2808–2820. doi: 10.1016/j.celrep.2018.11.035
E. Curry, I. Green, N. Chapman-Rothe, et al., Clin. Epigenet. 7 (2015) 84. doi: 10.1186/s13148-015-0118-9
A.Q. Ma, E. Stratikopoulos, K.S. Park, et al., Nat. Chem. Biol. 16 (2020) 214–222. doi: 10.1038/s41589-019-0421-4
J.M. Xu, R.C. Wu, B.W. O'Malley, Nat. Rev. Cancer 9 (2009) 615–630. doi: 10.1038/nrc2695
M. Spears, S. Oesterreich, I. Migliaccio, et al., Breast Cancer Res. Treat. 131 (2012) 463–472. doi: 10.1007/s10549-011-1426-1
C.A. Walsh, L. Qin, J.C.Y. Tien, et al., Int. J. Biol. Sci. 8 (2012) 470–485. doi: 10.7150/ijbs.4125
P.M. Cromm, C.M. Crews, Cell Chem. Biol. 24 (2017) 1181–1190. doi: 10.1016/j.chembiol.2017.05.024
Y. Lee, J. Heo, H. Jeong, et al., Angew. Chem. Int. Ed. 59 (2020) 17548–17555. doi: 10.1002/anie.202005004
S.R. Choi, H.M. Wang, M.H. Shin, et al., Int. J. Mol. Sci. 22 (2021) 6407. doi: 10.3390/ijms22126407
P. Gupta, S. Narayanan, D.H. Yang, CDK inhibitors as sensitizing agents for cancer chemotherapy, in: Z.S. Chen, D.H. Yang (Eds.), Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy, Academic Press, New York, 2019, pp. 125–149.
B. O'Leary, R.S. Finn, N.C. Turner, Nat. Rev. Clin. Oncol. 13 (2016) 417–430. doi: 10.1038/nrclinonc.2016.26
B. Laderian, T. Fojo, Semin. Oncol. 44 (2017) 395–403. doi: 10.1053/j.seminoncol.2018.03.006
U.S. Asghar, A.R. Barr, R. Cutts, et al., Clin. Cancer Res. 23 (2017) 5561–5572. doi: 10.1158/1078-0432.CCR-17-0369
A. Fassl, C. Brain, M. Abu-Remaileh, et al., Sci. Adv. 6 (2020) eabb2210. doi: 10.1126/sciadv.abb2210
J.H. Qiu, X.F. Bai, W.J. Zhang, et al., Front. Pharmacol. 13 (2022) 853993. doi: 10.3389/fphar.2022.853993
A.T. Anshabo, R. Milne, S.D. Wang, et al., Front. Oncol. 11 (2021) 678559. doi: 10.3389/fonc.2021.678559
R. Mandal, S. Becker, K. Strebhardt, Cancers 13 (2021) 2181. doi: 10.3390/cancers13092181
C.H. Huang, A. Lujambio, J. Zuber, et al., Genes Dev. 28 (2014) 1800–1814. doi: 10.1101/gad.244368.114
A. Richters, S.K. Doyle, D.B. Freeman, et al., Cell Chem. Biol. 28 (2021) 134–147. doi: 10.1016/j.chembiol.2020.10.001
E.A. Musgrove, C.E. Caldon, J. Barraclough, et al., Nat. Rev. Cancer 11 (2011) 558–572. doi: 10.1038/nrc3090
T.Z. Wu, Z. Qin, Y.C. Tian, et al., J. Med. Chem. 63 (2020) 13228–13257. doi: 10.1021/acs.jmedchem.0c00744
C. Sanchez-Martinez, M.J. Lallena, S.G. Sanfeliciano, et al., Bioorg. Med. Chem. Lett. 29 (2019) 126637. doi: 10.1016/j.bmcl.2019.126637
C.M. Olson, B.S. Jiang, M.A. Erb, et al., Nat. Chem. Biol. 14 (2018) 163–170. doi: 10.1038/nchembio.2538
X.Q. Qiu, Y.Q. Li, B. Yu, et al., Eur. J. Med. Chem. 211 (2021) 113091. doi: 10.1016/j.ejmech.2020.113091
B.L. Allen, D.J. Taatjes, Nat. Rev. Mol. Cell Biol. 16 (2015) 155–166. doi: 10.1038/nrm3951
Nitulescu, I.I., S.C. Meyer, Q.J. Wen, et al., EBioMedicine 26 (2017) 112–125. doi: 10.1016/j.ebiom.2017.11.013
D.C. Porter, E. Farmaki, S. Altilia, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 13799–13804. doi: 10.1073/pnas.1206906109
M.Q. Chen, J.X. Liang, H. Ji, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 10208–10213. doi: 10.1073/pnas.1710467114
D.Z. Xu, C.F. Li, X. Zhang, et al., Nat. Commun. 6 (2015) 6641. doi: 10.1038/ncomms7641
J. Bragelmann, N. Klumper, A. Offermann, et al., Clin. Cancer Res. 23 (2017) 1829–1840. doi: 10.1158/1078-0432.CCR-16-0094
J.X. Liang, M.Q. Chen, D. Hughes, et al., Cancer Res. 78 (2018) 6594–6606. doi: 10.1158/0008-5472.can-18-1583
M.Q. Chen, J. Li, J.X. Liang, et al., Cells 8 (2019) 1413. doi: 10.3390/cells8111413
I. Menzl, T.H. Zhang, A. Berger-Becvar, et al., Nat. Commun. 10 (2019) 4741. doi: 10.1038/s41467-019-12656-x
M.Y. Wang, R.K. Lin, J.C. Li, et al., J. Med. Chem. 66 (2023) 4932–4951. doi: 10.1021/acs.jmedchem.2c02045
A.D. Cox, S.W. Fesik, A.C. Kimmelman, et al., Nat. Rev. Drug Discov. 13 (2014) 828–851. doi: 10.1038/nrd4389
J. Hu, P.C. Zhu, Y.M. Li, et al., Chin. Chem. Lett. 29 (2018) 1043–1050. doi: 10.1016/j.cclet.2018.05.035
J.L. Bos, H. Rehmann, A. Wittinghofer, Cell 129 (2007) 865–877. doi: 10.1016/j.cell.2007.05.018
F.Q. Chen, M.P. Alphonse, Y. Liu, et al., Curr. Top. Med. Chem. 19 (2019) 2098–2113. doi: 10.2174/1568026619666190902151307
P. Martin-Gago, E.K. Fansa, C.H. Klein, et al., Angew. Chem. Int. Ed. 56 (2017) 2423–2428. doi: 10.1002/anie.201610957
P. Martin-Gago, E.K. Fansa, A. Wittinghofer, et al., Biol. Chem. 398 (2017) 535–545. doi: 10.1515/hsz-2016-0272
M.L. Guo, S.P. He, J.F. Cheng, et al., ACS Med. Chem. Lett. 13 (2022) 298–303. doi: 10.1021/acsmedchemlett.1c00670
R.H. Palmer, E. Vernersson, C. Grabbe, et al., Biochem. J. 420 (2009) 345–361. doi: 10.1042/BJ20090387
M. Soda, Y.L. Choi, M. Enomoto, et al., Nature 448 (2007) 561–566. doi: 10.1038/nature05945
C.M. Coffin, A. Patel, S. Perkins, et al., Mod. Pathol. 14 (2001) 569–576. doi: 10.1038/modpathol.3880352
R.D. Gascoyne, L. Lamant, J.I. Martin-Subero, et al. Blood 102 (2003) 2568–2573. doi: 10.1182/blood-2003-03-0786
T. Iwahara, J. Fujimoto, D.Z. Wen, et al., Oncogene 14 (1997) 439–449. doi: 10.1038/sj.onc.1200849
X.T. Kong, P.C. Pan, H.Y. Sun, et al., J. Med. Chem. 62 (2019) 10927–10954. doi: 10.1021/acs.jmedchem.9b00446
S.W. Xie, Y. Sun, Y.L. Liu, et al., J. Med. Chem. 64 (2021) 9120–9140. doi: 10.1021/acs.jmedchem.1c00270
A.J. Kuo, P. Cheung, K.F. Chen, et al., Mol. Cell 44 (2011) 609–620. doi: 10.1016/j.molcel.2011.08.042
D. Sengupta, L.Y. Zeng, Y.M. Li, et al., Mol. Cell 81 (2021) 4481–4492. doi: 10.1016/j.molcel.2021.08.034
S. Yuan, R. Natesan, F.J. Sanchez-Rivera, et al., Cancer Discov. 10 (2020) 854–871. doi: 10.1158/2159-8290.cd-19-1299
N.P. Coussens, S.C. Kales, M.J. Henderson, et al., J. Biol. Chem. 293 (2018) 13750–13765. doi: 10.1074/jbc.ra118.004274
D. Dilworth, R.P. Hanley, R.F. de Freitas, et al., Nat. Chem. Biol. 18 (2022) 56–63. doi: 10.1038/s41589-021-00898-0
J.H. Bushweller, Nat. Rev. Cancer 19 (2019) 611–624. doi: 10.1038/s41568-019-0196-7
M. Schapira, M.F. Calabrese, A.N. Bullock, et al., Nat. Rev. Drug Discov. 18 (2019) 949–963. doi: 10.1038/s41573-019-0047-y
T. Tamura, I. Hamachi, J. Am. Chem. Soc. 141 (2019) 2782–2799. doi: 10.1021/jacs.8b11747
Y. Wang, J. Zhang, J. Deng, et al., CCS Chem. 5 (2023) 2207–2214. doi: 10.31635/ccschem.023.202302873
W.K. Jin, H.D. Tan, J.H. Wu, et al., Drug Discov. Today 27 (2022) 246–256. doi: 10.1016/j.drudis.2021.08.007
C. -Y. Yang, C. Qin, L. Bai, et al., Drug Discov. Today: Technol. 31 (2019) 43–51. doi: 10.1016/j.ddtec.2019.04.001
P. Filippakopoulos, J. Qi, S. Picaud, et al., Nature 468 (2010) 1067–1073. doi: 10.1038/nature09504
J. Balzeau, M.R. Menezes, S.Y. Cao, et al., Front. Genet. 8 (2017) 31.
A. Ghidini, A. Clery, F. Halloy, et al., Angew. Chem. Int. Ed. 60 (2021) 3163–3169. doi: 10.1002/anie.202012330
C. Ballatore, V.M.Y. Lee, J.Q. Trojanowski, Nat. Rev. Neurosci. 8 (2007) 663–672. doi: 10.1038/nrn2194
K.A. Vossel, K. Zhang, J. Brodbeck, et al., Science 330 (2010) 198. doi: 10.1126/science.1194653
L.M. Ittner, Y.D. Ke, F. Delerue, et al., Cell 142 (2010) 387–397. doi: 10.1016/j.cell.2010.06.036
D. Bumcrot, M. Manoharan, V. Koteliansky, et al. Nat. Chem. Biol. 2 (2006) 711–719. doi: 10.1038/nchembio839
N. Gao, T.T. Chu, Q.Q. Li, et al., RSC Adv. 7 (2017) 40362–40366. doi: 10.1039/C7RA05347A
C.F. Sephton, B. Cenik, B.K. Cenik, et al., Biol. Chem. 393 (2012) 589–594. doi: 10.1515/hsz-2012-0115
T. Geuens, D. Bouhy, V. Timmerman, Hum. Genet. 135 (2016) 851–867. doi: 10.1007/s00439-016-1683-5
M. Polymenidou, C. Lagier-Tourenne, K.R. Hutt, et al., Nat. Neurosci. 14 (2011) 459–468. doi: 10.1038/nn.2779
C.F. Sephton, C. Cenik, A. Kucukural, et al., J. Biol. Chem. 286 (2011) 1204–1215. doi: 10.1074/jbc.M110.190884
S.J. Barmada, G. Skibinski, E. Korb, et al., J. Neurosci. 30 (2010) 639–649. doi: 10.1523/JNEUROSCI.4988-09.2010
H. Ederle, D. Dormann, FEBS Lett. 591 (2017) 1489–1507. doi: 10.1002/1873-3468.12646
H. Wils, G. Kleinberger, J. Janssens, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 3858–3863. doi: 10.1073/pnas.0912417107
N. Gao, Y.P. Huang, T.T. Chu, et al., Bioorg. Chem. 84 (2019) 254–259. doi: 10.1016/j.bioorg.2018.11.042
M.E. Macdonald, C.M. Ambrose, M.P. Duyao, et al., Cell 72 (1993) 971–983. doi: 10.1016/0092-8674(93)90585-E
G.P. Bates, R. Dorsey, J.F. Gusella, et al., Nat. Rev. Dis. Primers 1 (2015) 15005. doi: 10.1038/nrdp.2015.5
C.A. Ross, S.J. Tabrizi, Lancet Neurol. 10 (2011) 83–98. doi: 10.1016/S1474-4422(10)70245-3
F. Saudou, S. Humbert, Neuron 89 (2016) 910–926. doi: 10.1016/j.neuron.2016.02.003
B. Boland, W.H. Yu, O. Corti, et al., Nat. Rev. Drug Discov. 17 (2018) 660–688. doi: 10.1038/nrd.2018.109
J.J. Hu, Y.F. Zhao, Y.M. Li, Chin. Chem. Lett. 34 (2023) 107623. doi: 10.1016/j.cclet.2022.06.046
S. Tomoshige, S. Nomura, K. Ohgane, et al., Angew. Chem. Int. Ed. 56 (2017) 11530–11533. doi: 10.1002/anie.201706529
E.M. Roth, M.H. Davidson, Rev. Cardiovasc. Med. 19 (2018) S31–S46.
N.G. Seidah, A. Prat, A. Pirillo, et al., Cardiovasc. Res. 115 (2019) 510–518. doi: 10.1093/cvr/cvz003
H.J. Kwon, T.A. Lagace, M.C. McNutt, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 1820–1825. doi: 10.1073/pnas.0712064105
W.L. Petrilli, G.C. Adam, R.S. Erdmann, et al., Cell Chem. Biol. 27 (2020) 32–40. doi: 10.1016/j.chembiol.2019.10.002
T.S. Fisher, P. Lo Surdo, S. Pandit, et al., J. Biol. Chem. 282 (2007) 20502–20512. doi: 10.1074/jbc.M701634200
M. Naito, N. Ohoka, N. Shibata, et al., Front. Chem. 7 (2019) 849. doi: 10.3389/fchem.2019.00849

Figure 2 Native protein degradation is mainly based on two mechanisms: the UPS and ALS. Transient, soluble unfolded or misfolded proteins and peptides are usually labeled by polyubiquitin and degraded by the proteasome in UPS, while long-lived, insoluble protein aggregates and dysfunctional organelles are degraded in ALS via endosome, phagolysosome, microautophagy, chaperone-mediated autophagy, and macroautophagy pathways.

Figure 3 (A) The mechanism of bivalent PROTAC-induced protein degradation. The rigid or flexible chemical component-linked target protein ligand and E3 ligase ligand recruited spatial proximity of the two proteins, respectively, and mediated the UPS degradation of the target protein. (B) Monovalent molecular glue initiates UPS degradation of the target protein with a leaner structure. (C) The representative mechanism of HyTTDs-induced protein degradation. Adamantane and its derivatives can trigger the degradation of POI by binding directly to chaperone proteins or by inducing the POI binding to chaperone proteins, while arginine derivatives or their hydrolysates tend to recruit E3 ligase and induce ubiquitin-proteasome pathway degradation of POI.

Figure 4 The development history of HyTTDs. The ER protein degradation brought by the hydrophobic moiety of fulvestrant serves as an origin for the emergence of HyTTDs. After that, HyTTDs gradually overcome the limitations of HaloTag-pretreated proteins and moved from biochemical experimental technology to promising drug candidates for the degradation of multiple pathogenic targets.

Figure 5 (A) Mechanism of ER antagonist drugs-resistant breast cancer after long-term endocrine therapies. Ligand-dependent ER overexpression and ligand-independent ER mutation result in the form of drug resistance and the progression of breast cancer. (B) Crystal structure of the ERα ligand-binding domain (LBD) in complex with oxabicyclic heptane sulfonamide (OBHS-N) (PDB ID: 5KCC). In the docking model, the ligand compounds are shown in the following form: carbon atoms are shown in green, hydrogen atoms in white, oxygen atoms in red, nitrogen atoms in blue, and sulfur atoms in yellow, chlorine atoms in dark green and phosphorus atoms in orange. The following docking models all follow this pattern. (C) Chemical structure and biological activity data of compounds 16–18. (D) Chemical structure and biological activity data of compounds MHO-7 and 19.

Figure 6 (A) Mechanism of GSTP-medicated drug resistance and proliferation in cancers. Upon external stress or drug stimulation, phosphorylated JNK releases from the complex and subsequently phosphorylates C-Jun, ultimately leading to cell proliferation. (B) Chemical structure and biological activity data of compounds 20, 21 and predicted binding models of human GST P1-1 in complex with compounds 20, 21 (PDB ID: 2GSS). Compound 20 obtained an enhanced biological activity by replacing Boc3Arg with adamantyl degron. (C) Chemical structure and biological activity data of compound 22 and predicted binding models of human GST P1-1 in complex with compound 22 (PDB ID: 6GSS). (D) Chemical structure and biological activity data of compound 23.

Figure 7 (A) Mechanism of Her3-medicated cell survival and proliferation. Her2-Her3 complex binds to ligands and regulates cell growth through signaling pathway transduction. (B) Chemical structure and biological activity data of compound 24. (C) Predicted binding models of the catalytically inactive kinase domain of the human Her3 in complex with compound 24 (PDB ID: 3KEX). Hydrogen bonding of 24 with S775 is shown by yellow dashed lines and covalent bonding with C721 is shown in the figure. In the docking model, the residues are shown in the following form: carbon atoms are shown in pink, hydrogen atoms in white, oxygen atoms in red, nitrogen atoms in blue and hydrogen bonds are shown in yellow dashed lines. The following docking models all follow this pattern. (D) Chemical structure and biological activity data of compound 25 (TGI: tumor growth inhibition). (E) Predicted binding models of the pH domain of human Akt3 protein kinase in complex with compound 25 (PDB ID: 2 × 18). Hydrogen bonding of 25 with S55 and D289 is shown.

Figure 8 (A) Mechanism of AR-medicated cell survival and proliferation. Overexpression of AR proteins, splicing variants, and mutations allow tumor cells to avoid the inhibitory effects of traditional small-molecule AR antagonists, which results in the development of drug resistance. (B) Chemical structures and biological activity data of compounds 26–28 modified from RU59063.

Figure 9 Chemical structure and biological activity data of compound 29 and compound 30 which are based on the optimization of HyT13 and HyT36. The addition of the α-position methyl group to the carbonyl group allows 30 to obtain better biological activities compared with 29.

Figure 10 (A) Mechanism of p53-medicated gene expression regulation. Activation of p53 induced the negative-feedback regulation of ubiquitin ligase MDM2, which results in the degradation of p53. (B) Crystal structure of the humanized Xenopus Mdm2 with Nutlin-3a (PDB ID: 4J3E). (C) Chemical structure and biological activity data of compound 31.

Figure 11 (A) Mechanism of PARP1-medicated gene repair process. PARP1 recognizes single-stranded DNA damage, which subsequently recruits associated proteins and activates the DNA repair system. (B) Chemical structure and biological activity data of compound 32. (C) Predicted binding models of the constitutively active PARP1 in complex with compound 32 (PDB ID: 5DS3).

Figure 12 (A) Predicted binding models of the PRC2 in complex with compound C24 (PDB ID: 5LS6). The hydrogen bonding of C24 with W624 and π–π bonding of Y111 are shown. π–π bonds are shown in blue dashed lines. The following docking models all follow this pattern. (B) Chemical structure and biological activity data of compound 33. (C) Predicted binding models of the PRC2 in complex with compound tazemetostat (PDB ID: 5LS6). The hydrogen bonding of tazemetostat with W624 is shown. (D) Chemical structure and biological activity data of compound 34.

Figure 13 (A) Mechanism of SRC-1-medicated cancer cell proliferation. SRC-1 promotes the development of breast cancer by activating transcription factors and nuclear receptors which medicate multiple downstream signaling molecules. (B) Crystal structure of the Nco-A1 PAS-B domain with YL-2 (PDB ID: 5Y7W). YL-2 is shown in green. (C) Chemical structure and biological activity data of compound 35.

Figure 14 (A) Chemical structure and biological activity data of compound 36. (B) Predicted binding models of the human CDK6 in complex with compound 36 (PDB ID: 5L2S). (C) Chemical structure and biological activity data of compound 37. (D) Predicted binding models of the CDK9 and cyclin T in complex with compound 37 (PDB ID: 4BCH). Hydrogen bonding of 37 with C106 and D109 is shown and K151, T191, and Y194 may be partially obscured by 37. Cyclin T is shown in purple. (E) Chemical structure and biological activity data of compound 38. (F) Predicted binding models of the human CDK8 and cyclin C in complex with compound 38 (PDB ID: 6QTG). Hydrogen bonding of 38 with A100 and π–π bonding of H106 is shown.

Figure 15 (A) Chemical structure and biological activity data of compound 39. (B) Predicted binding models of the PDEδ in complex with compound 39 (PDB ID: 5E80). Hydrogen bonding of 39 with R61 is shown.

Figure 16 (A) Mechanism of ALK-medicated signaling pathways. ALK regulates cellular activities through various pathways such as nuclear factor-kappa B (NF-κB), PI3K/Akt. ALK translocations at pathogenic sites express ALK fusion proteins, which may be associated with tumor development. (B) Chemical structure and biological activity data of compound 40. (C) Predicted binding models of the human anaplastic lymphoma kinase in complex with compound 40 (PDB ID: 3AOX). (D) Chemical structure and biological activity data of compound 41. (E) Predicted binding models of the anaplastic lymphoma kinase in complex with compound 41 (PDB ID: 4MKC). (F) Chemical structure and biological activity data of compound 42. (G) Predicted binding models of the anaplastic lymphoma kinase in complex with compound 42 (PDB ID: 6MX8).

Figure 17 (A) Chemical structure and biological activity data of compound 43. (B) Predicted binding models of the Histone-lysine N-methyltransferase NSD2-PWWP1 in complex with compound 43 (PDB ID: 6XCG). (C) Chemical structure and biological activity data of compound 44. (D) Predicted binding models of the Histone-lysine N-methyltransferase NSD2-PWWP1 in complex with compound 44 (PDB ID: 6XCG).

Figure 18 (A) Crystal structure of the first bromodomain of human BRD4 in complex with the inhibitor JQ1 (PDB ID: 3MXF). (B) Chemical structure and biological activity data of compound 45. (C) Solution structure of the Lin28-ZnF domains bound to AGGAGAU of pre-let-7 miRNA (PDB ID: 2LI8). (D) Chemical structure and biological activity data of compound 46.

Figure 19 (A) Mechanism summary of the HyTTD strategy cases in neurodegenerative diseases. Disruption of the binding balance between MT and Tau proteins, overexpression of TDP43, and aberrant expansion of ployQ in HTT protein cause the accumulation and aggregation of the corresponding pathogenic monomers, which ultimately leads to the corresponding neurological diseases. (B) Chemical structure and biological activity data of compound 47. (C) Chemical structure and biological activity data of compound 48. (D) Chemical structure and biological activity data of compound 49.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: