Guangzhou Key Laboratory for Clean Energy and Materials/Key Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Guangzhou University, Guangzhou 510006, China
b.
State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Advanced Catalytic Engineering Research Center of the Ministry of Education, Hunan University, Changsha 410082, China
Received Date:
15 August 2023 Accepted Date:
25 September 2023 Revised Date:
04 September 2023 Available Online:
15 August 2024
Abstract:
Urea is extensively used in agriculture and chemical industry, and it is produced on an industrial scale from CO2 and Haber–Bosch NH3 under relatively high temperature and high pressure conditions, which demands high energy input and generates masses of carbon footprint. The conversion of CO2 and N sources (such as NO2−, NO3−, and N2) through electrocatalytic reactions under ambient conditions is a promising alternative to realize efficient urea synthesis. Of note, the design of electrocatalyst is one of the key factors that can improve the efficiency and selectivity of C–N coupling reactions. Defect engineering is an intriguing strategy for regulating the electronic structure and charge density of electrocatalysts, which endows electrocatalysts with excellent physicochemical properties and optimized adsorption energy of the reaction intermediates to reduce the kinetic barriers. In this minireview, recent advances of defect engineered electrocatalysts in urea electrosynthesis from CO2 and various N reactants are firstly introduced. Mechanistic discussions of C–N coupling in these advances are presented, with the aim of directing future investigations on improving the urea yield. Finally, the prospects and challenges of defect engineered electrocatalysts for urea synthesis are discussed. This overview is expected to provide in-depth understanding of structure–reactivity relationship and shed light on future electrocatalytic C–N coupling reactions.
Organonitrogen compounds such as amides, amines and urea, are widely used to manufacture fine chemicals, pharmaceutical intermediates, agrochemicals and fertilizers [1–3]. Organonitrogen compounds conventionally originate from the reaction between C and N sources under harsh reaction conditions, thus aggravating the energy crisis and environmental concerns [4–6]. Undoubtedly, energy-saving and environmentally friendly synthetic method is urgently required to replace traditional methods.
N-integrated electrocatalytic CO2 reduction strategies for organonitrogen compounds synthesis have some notable benefits compared with the current industrial routes. First, electrocatalytic method is a clean and sustainable synthetic route occurs in which hazardous redox reagents are replaced by an electricity [7–9]. Most of the electrocatalysts work under room temperature and standard pressure conditions, thus electrocatalytic reaction is easy to control. Second, CO2 is used as the C source in these reactions, which could meet the demands of carbon neutrality [10–12]. Third, other N sources aside from NH3 can be used, such as NO, NO2−, NO3−, and N2 [13–15]. Currently, over half of global NH3 production is consumed in the synthesis of organonitrogen compounds, and NH3 is typically produced via the energy-intensive and fossil-fuel based Haber–Bosch process [16–18].
Urea, a typical organonitrogen compound, is extensively used in agriculture and chemical industry [19,20]. Urea is produced on an industrial scale from CO2 and Haber–Bosch NH3 under relatively high temperature and high pressure conditions, which demands high energy input and generates masses of carbon footprint [6,21]. In the past decades, the synthesis of urea from CO2 through relatively mild electrochemical reactions has aroused extensive attention [11–13,22]. Great efforts have been made to improve the efficiency and selectivity of C–N coupling reactions toward urea synthesis, but the reaction rates and energy utilization efficiencies are unsatisfactory, and the reaction selectivity is also hindered by the diverse reaction pathways and complex reaction mechanisms [23,24]. In spite of that, researchers have never stopped to pursuing more efficient urea synthesis, especially the design of electrocatalysts.
According to the second law of thermodynamics, it is not hard to know that defects extensively exist in nanomaterials [25–27]. Defects can modulate the distribution of the electronic structure and disturb the periodic crystal structure of nanomaterials, thereby enhancing the electrocatalytic reaction activity by precise control of the species, concentration and location [28–30]. In recent years, various defect engineered electrocatalysts have developed for improved electrocatalytic synthesis of urea, and some typical examples have been mentioned upon in several recent reviews [11–13,22]. A more comprehensive understanding of defective electrocatalysts in terms of their intrinsic properties, catalytic mechanisms, and interface reaction processes is essential to identify the relationship between the defect sites and electrocatalytic reaction activity, facilitating the preparation of electrocatalysts for effective synthesis of urea.
2.
Defective engineered electrocatalysts for urea synthesis
The "one-step" conversion of C and N sources via electrocatalytic reactions under ambient conditions is a promising alternative to complex industrial method for efficient urea synthesis. In recent years, a series of robust electrocatalysts have been developed for electrocatalytic coupling of CO2 and NO2–/NO3– or N2 into urea [10–12,23]. Numerous works proved the defects in electrocatalysts could significantly enhance the electrocatalytic reaction activity [27,30,31], hence defective engineered electrocatalytic C–N coupling reactions for urea synthesis have aroused enormous interests. Herein, defective engineered electrocatalysts for urea synthesis under ambient conditions via the electrocatalytic coupling of CO2 and NO2−/NO3− or N2 will be discussed in detail.
2.1
C–N coupling of CO2 and NO2−/NO3− for urea synthesis
Human activities discharge plenty of nitrogen-containing substances into surface water in the form of NO2–/NO3–, posing severe risks to public health. Traditional catalytic conversion of harmful NO2–/NO3– into harmless nitrogen molecules fails to realize the product upgrading and the maximum use of energy and resources. Recent studies indicate the electrocatalytic coupling of CO2 and NO2–/NO3– into urea is an ideal alternative, which simultaneously realize the utilization of CO2, control of water pollution and acquirement of high value-added products.
In the early stages, Furuya et al. [32,33] used Ni-phthalocyanine (Ni-Pc) electrocatalysts for simultaneous reduction of CO2 and NO2– to produce urea, an optimal current efficiency of 40% was obtained at −1.5 V vs. standard hydrogen electrode (SHE), but the complex reaction pathways of CO2 reduction reaction (CO2RR) allowed very few active species to combine with N-containing intermediates to complete the C–N coupling reaction, and the competitive hydrogen evolution reaction (HER) also limits the reaction efficiency. Subsequent studies have shown that some transition metal oxides and p-block-elements based electrocatalysts can be candidates for this reaction because their inherent weak H adsorption abilities can restrain the HER [22,34]. Especially, inspired by the defect engineering strategy, a series of robust defective electrocatalysts have been developed to simultaneously activate N- and C-containing intermediates and reduce the HER for more effective urea synthesis.
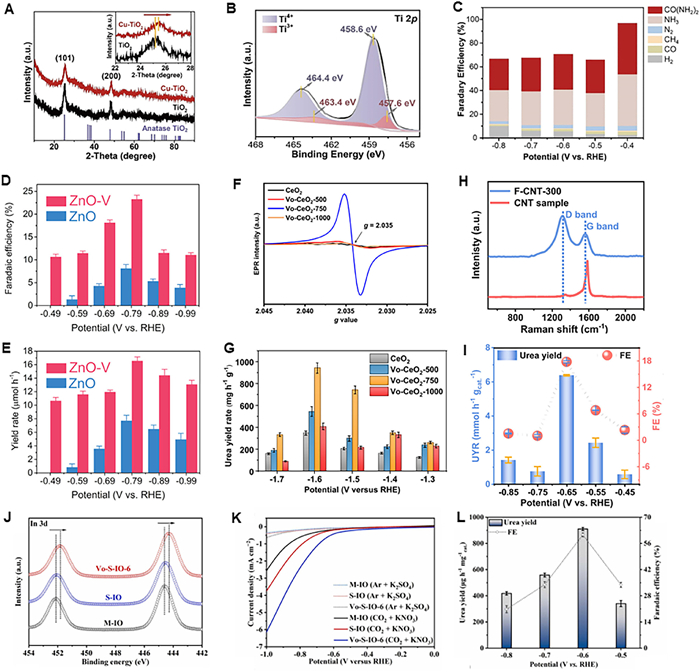
Cao et al. [35] designed O vacancy (VO)-riched Cu-doped anatase TiO2 nanotubes catalysts (Cu–TiO2) to improve the electrocatalytic co-reduction of CO2 and NO2–. A little red shift of (101) plane was observed in Cu–TiO2, indicating Cu doping reduced the TiO2 lattice constant (Fig. 1A). The remarkably increase in Ti3+/Ti4+ ratio indicated Cu doping contributed to the occurrence of VO on Cu–TiO2 (Fig. 1B). The VO facilitated the adsorption and activation of NO2– and cooperated with high CO2RR active Cu sites to realize efficient urea synthesis, and a Faraday efficiency (FE) of 43.1% was obtained at a low overpotential of −0.4 V vs. reversible hydrogen electrode (RHE) (Fig. 1C). The potentials mentioned in this work are relative to RHE, unless otherwise stated. Meng et al. [36] found VO–ZnO porous nanosheets display an excellent performance in electrocatalytic synthesis of urea from CO2 and NO2–. The experimental results indicated CO2 and NO2– filled the surface vacancy for accelerating urea synthesis, and the VO–ZnO displays a ~3- and 2-fold enhancement of the FEurea and urea yield rate (rurea), respectively, compared with that of pure ZnO at an applied potential of −0.79 V (Figs. 1D and E). Lv et al. [37] reported the intriguing electrocatalytic urea synthesis process on a defect engineered indium oxyhydroxide catalyst with VO (VO–InOOH) using CO2 and NO3– as reactants. VO–InOOH exhibited much better performance compared with pristine InOOH in terms of both urea yield (592.5 vs. 378.4 µg h–1 mgcat.–1) and FEurea (51.0% vs. 26.3%), indicating the presence of VO resulted in the improvement of C–N coupling efficiency. Wei et al. [38] used the VO-riched CeO2 (VO–CeO2) as the electrocatalyst for urea synthesis with CO2 and NO3– as reactants. Experimental results indicated the urea production was tightly associated with the VO concentration, and CeO2 treated with 750 ℃ (VO–CeO2-750), the one had the highest VO contents (Fig. 1F), exhibited the highest urea yield of 943.6 mg h−1 g−1 at −1.66 V, which was almost three times as much as that of pristine CeO2 (Fig. 1G). Beside to metal-based electrocatalysts, a novel metal-free F-riched carbon nanotubes (F-NCT) electrocatalyst was developed for urea synthesis with CO2 and NO3– as reactants by Liu et al. [39]. The F-doping introduced lots of defects in CNT according to the Raman spectra (Fig. 1H), and a high rurea of 6.36 mmol h–1 g–1 with a corresponding FEurea of 18.0% was achieved for F-CNT catalyst mediated urea synthesis at –0.65 V (Fig. 1I). The rurea was 3.8 times as high as that obtained from CNT catalyzed urea synthesis, proving the defects introduced by F doping facilitate the C–N coupling reaction to form urea. Recently, Li et al. [40] integrated facet and defect engineering strategy to develop a highly efficient VO-rich indium hydroxide nanocube catalyst with dominant facets (denoted as VO-S-IO-6) for urea synthesis from the electrocatalytic co-reductive coupling of NO3– and CO2. The In 3d orbital of VO-S-IO-6 shifted to lower binding energy compared with M-IO and S-IO, implying the increase of the local electron density of In caused by delocalized electrons after oxygen escaping (Fig. 1J). Linear cyclic scanning (LSV) measurements proved VO-S-IO-6 had the best performance toward electrocatalytic co-reduction of NO3– and CO2 (Fig. 1K), and a high rurea of 910.4 µg h–1 mg–1 and FEurea of 60.6% was achieved at −0.6 V (Fig. 1L), indicating the introduction of VO contributed to the C–N coupling toward urea synthesis.
Figure 1
Figure 1.
Defective electrocatalysts mediated coupling of CO2 and NO2–/NO3– for urea synthesis. (A) XRD patterns of Cu–TiO2 and TiO2; (B) XPS spectra of Ti 2p orbital on Cu–TiO2. (C) FE of the urea synthesis on Cu–TiO2. (A–C) Reproduced with permission [35]. Copyright 2020, Elsevier Inc. (D) FEurea and (E) rurea under the given potentials over ZnO-V. (D, E) Reproduced with permission [36]. Copyright 2021, Cell Press. (F) EPR spectra CeO2, VO-CeO2-500, VO-CeO2-750, and VO-CeO2-1000. (G) rurea of CeO2, VO-CeO2-500, VO-CeO2-750, and VO-CeO2-1000 at various applied potentials. (F, G) Reproduced with permission [38]. Copyright 2022, American Chemical Society. (H) Raman spectra of F-CNT-300 and CNT sample. (I) Urea yield rates and FE values at different potentials for F-CNT-300 in CO2 saturated 0.1 mol/L KNO3 electrolyte with CO2 flow. (H, I) Reproduced with permission [39]. Copyright 2022, Elsevier Inc. (J) In 3d XPS spectra of M-IO, S-IO, and VO-S-IO-6. (K) LSV curves recorded on the M-IO, S-IO, and VO-S-IO-6 during urea electrosynthesis and the electroreduction of VO-S-IO-6 in electrolyte without NO3– or CO2. (L) Yields and FEs of urea during urea electrosynthesis with VO-S-IO-6 catalyst at different potentials after collecting –7.65 C of charge. (J–L) Reproduced with permission [40]. Copyright 2023, Elsevier Inc.
N2 is the most abundant and cheapest N source on the earth, and the report on electrocatalytic N2 reduction to NH3 inspired researchers to synthesize urea in N2-integrated electrocatalytic CO2 reduction in spite of the inertness and high bonding energy of the N≡N bonds [41,42]. Compared with the NO2–/NO3–-integrated electrocatalytic CO2 reduction for urea synthesis, the N2-integrated electrocatalytic CO2 reduction one not only lowers the cost of raw material but also circumvents the use of energy-intensive fossil-fuel [10–12].
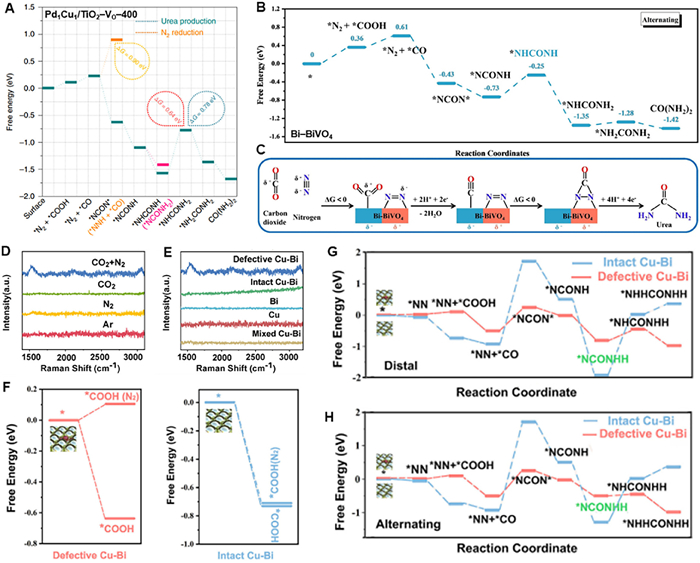
Kayan et al. [43] firstly used aqueous solution as the proton source to explore the co-reduction of N2 and CO2 for the production of urea on a polypyrrole-coated Pt electrode under a mixed atmosphere of 30 bar N2 and 30 bar CO2, and an optimized urea formation rate of 31.8 µg h–1 cm–2 and of FEurea of 6.9% at −0.325 V vs. NHE were achieved. The replace of ambient working condition to high pressure one favors to save energy and minimize the requirement of synthesis equipment. Inspired by the fact that metal alloying and defect engineering can promote the electronic interactions and provide more active sites for electrocatalytic reaction [44–47]. Wang’s group [48] developed the electrocatalyst of PdCu alloy nanoparticles on VO-riched defective TiO2 nanosheets (Pd1Cu1/TiO2–VO) for the efficient electrocatalytic synthesis of urea via the coupling of N2 with CO2 under ambient conditions. The interaction between TiO2–VO-400 (TiO2 with treatment at 400 ℃) with the most abundant defect sites and PdCu alloy was verified by X-ray photoelectron spectroscopy (XPS) analysis (Figs. 2A and B). The temperature programmed desorption (TPD) spectra illustrated that the proposed electrocatalyst displayed a stronger chemical adsorption ability for CO2 and N2 due to the strong electronic interaction between the bimetal alloys and the TiO2-VO-400 (Fig. 2C). Operando synchrotron radiation−Fourier transform infrared spectroscopy (SR–FTIR) analysis proved the formation of urea (Figs. 2D and E), and a desirable rurea of 3.36 mmol g–1 h–1 and FE of 8.92% at –0.4 V were obtained (Figs. 2F and G). Also, higher rurea and FEurea of Pd1Cu1/TiO2-VO-400 than that of Pd1Cu1/TiO2-VO were identified. On the basis of this work, Pan et al. [49] further found Pd1Cu1/TiO2-VO had better performance than Pd1/TiO2-VO on electrocatalytic co-reduction of N2 and CO2 for urea synthesis, indicating the important role of dual-atom Pd1Cu1 site. Wu et al. [50] reported the defective bimetallic Cu–Bi catalyst for urea electrosynthesis. HR-TEM images showed the bimetallic Cu–Bi had ordered regions consisting of missing-atom defects (Fig. 2H), and the EPR analysis also proved the presence of higher concentration of unsaturated Bi sites with unpaired electrons (Fig. 2I). Significantly, defective Cu–Bi catalyst offered a maximum urea concentration of 0.45 ± 0.06 mg/L and the largest faradaic efficiency of 8.7% ± 1.7% at −0.4 V (Fig. 2J), and meanwhile, it exhibited much higher active in producing urea compared with monometallic (Cu and Bi) and bimetallic (Cu–Bi mixture and intact Cu–Bi) catalysts (Fig. 2K). This work provides insights into intriguing relationships between defective structure and catalytic activity, shedding light on the rational design of defective electrocatalysts for producing urea from N2 and CO2 molecules.
Figure 2
Figure 2.
Defective electrocatalysts mediated coupling of CO2 and N2 for urea synthesis. (A) Pd 3d XPS spectra and (B) Cu 2p XPS spectra of Cu/TiO2, Pd1Cu1/TiO2 and Pd1Cu1/TiO2-400. (C) Competitive chemisorption of N2 (red) and CO2 (blue) on TiO2-400 and Pd1Cu1/TiO2-400. (D) Infrared signal in the range of 1100–1800 cm–1 under various potentials for Pd1Cu1/TiO2-400 during the electrocoupling of N2 and CO2. (E) Isotope-labeling infrared signals in the range of 1100–1800 cm–1 at −0.40 V for Pd1Cu1/TiO2-400 during the electrocoupling of 15N2 and/or 13CO2 processes. (F) Urea generation with CO2 and N2 as feeding gases. (G) The FEurea and the total current densities for all products at various potentials. (A–G) Reproduced with permission [48]. Copyright 2020, Nature Publishing. (H) TEM image from a double–corrected spherical aberration electron microscope. (I) Comparison of EPR spectra for intact and defective Cu–Bi. (J) The concentrations and faradaic efficiencies of urea. (K) Comparison of the urea concentrations by defective Cu–Bi (blue), the mixture of Cu–Bi (green), Cu (red), intact Cu–Bi (yellow), and Bi (gray) catalysts. (H–K) Reproduced with permission [50]. Copyright 2022, Cell Press.
N2-integrated electrocatalytic CO2 reduction for urea synthesis has huge application prospects, but the exceedingly sluggish kinetics of N2 reduction limits the overall rate of urea production [11,12,22]. Current works indicate the rurea and FEurea are unsatisfactory (Table 1) [2,23,35–40,48–70]. Of note, current works about defective electrocatalysts mediated coupling of CO2 and N2 for urea synthesis are scarce, and the development of more robust defective electrocatalysts is bound to push this field forward.
Table 1
Table 1.
Electrocatalytic urea synthesis from N-integrated CO2 reduction.
3.
Mechanism and role of defect in electrocatalytic synthesis of urea
3.1
Mechanism for C–N coupling of CO2 and NO2−/NO3−
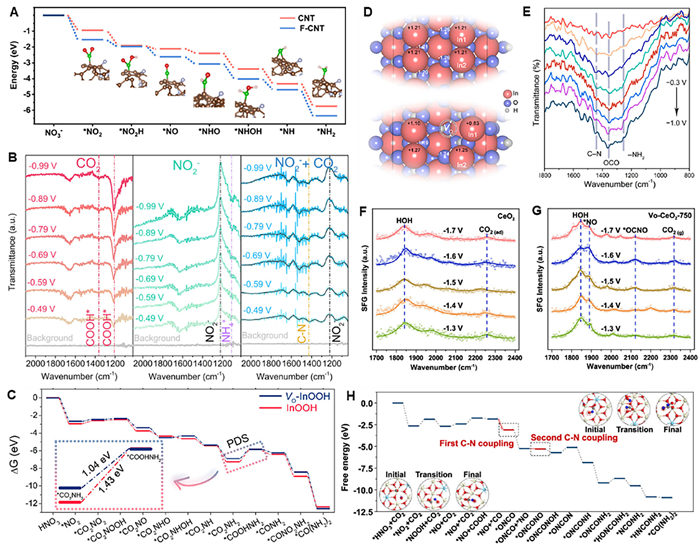
The mechanism of electrocatalytic urea synthesis from NO2–/NO3–-integrated CO2 reduction is still controversial, there are mainly four possible explanations in current reported works (Table 2). The *CO + *NH2 coupling mechanism observed in NO2–/NO3–-integrated electrocatalytic CO2 reduction for urea synthesis was firstly proposed by Shibata and Furuya et al. [33,71,72]. Recently, Liu et al. [39] proved the feasibility of this mechanism through density functional theory (DFT) calculations in their recent F-CNT work, and the formation of *CO and *NH2 intermediates was more favorable on F-doped C active sites ("C–F2" moieties) than on C active sites (Fig. 3A). The experimental results also proved the F-CNT showed better performance toward urea synthesis than CNT, hence we deduced the introduction of VO caused by F doping contributed to urea production. Of note, this coupling mechanism has long been controversial, and some new ideas have also put forward. For instance, Meng et al. [36] proposed that the VO–ZnO porous nanosheets mediated electrocatalytic C–N formation came from the coupling of *NH2 and *COOH intermediates, which were generated from the reduction of NO2– and CO2, respectively. In situ infrared (IR) spectra measurements indicated the *COOH peaks detected at reductive potentials under the CO2 + NO2– conditions were weaker than that detected at the CO2-free conditions, and a peak attributed to C–N bond was observed, indicative of the consumption of *COOH and occurrence of C–N coupling (Fig. 3B). The experimental results indicated VO promoted urea electrosynthesis, we therefor deduced VO was conducive to the formation of *NH2 and *COOH intermediates. Yu’s group [23,37] proposed the *CO2 + *NO2 coupling mechanism using In-based materials as electrocatalysts. In their VO–InOOH work [51], DFT calculations indicated the urea synthesis was triggered by the thermodynamically spontaneous reduction of NO3– to the *NO2 and the *CO2NH2 protonation was the potential-limiting step (PLS) for the overall urea formation process (Fig. 3C). Significantly, the VO contributed to lower the energy barrier for the protonation of the *CO2NH2 intermediate to accelerate urea synthesis, and Bader charge analysis (Fig. 3D) and the SR-FTIR analysis (Fig. 3E) further proved this idea. In the VO-S-IO-6 work reported by Li et al. [40] proposed the same C–N coupling mechanism, they illustrated the introduction of VO significantly tuned the electronic structure around the catalytic sites to accelerate the cleavage of In–O bond in VO-S-IO-6 adsorbed *CO2NH2, thereby benefiting the protonation process and boosting the electrocatalytic performance of urea synthesis. Recently, Wang’s group proposed the urea formation via a two-step coupling process. In their VO–CeO2 work [38], in situ sum frequency generation (SFG) spectroscopy analysis indicated the core of reaction mechanism was that *NO intermediate species adsorbed to VO sites of the catalyst, and the subsequent coupling process with *CO to form *OCNO was more favorable than the hydrogenation reaction of *NO, whereas the (proton-coupling electron-transfer, PCET) process of *NO tended to occur on the VO-deficient electrocatalyst (Figs. 3F and G). DFT calculations indicate the C–N coupling was thermodynamically and kinetically favorable on Vo-enriched CeO2, and meanwhile, a novel two-step coupling mechanism, namely, *CO + *NO → *ONCO and *ONCO + *NO → *ONCONO, was proposed (Fig. 3H). In addition, another two-step coupling mechanism of *CO + *NO → *ONCO; (2) *ONCO + *NO → *ONCONO was also proposed [59,60], but the related works did not involve defective electrocatalysts.
Table 2
Table 2.
Proposed mechanism of electrocatalytic urea synthesis from NO2–/NO3–-integrated CO2.
Figure 3.In situ characterization and proposed coupling mechanism for electrocatalytic urea synthesis from NO2–/NO3–-integrated CO2 reduction. (A) Potential energy diagrams of NO3– reduction to *NH2, undoped CNT and F-doped C active sites (F-CNT) are marked. C, O, H, F and N atoms are represented as brown, red, pinkish white, blue and green spheres, respectively. Reproduced with permission [39]. Copyright 2022, Elsevier Inc. (B) In situ ART-FTIR spectra of ZnO-V under CO2, NaNO2, and both. Reproduced with permission [36]. Copyright 2021, Cell Press. (C) Free-energy diagrams for urea production on the facets of VO-InOOH and pristine InOOH at 0 V. (D) Bader charge analysis on VO-InOOH and pristine InOOH. (E) Infrared signals in the range of 800−1800 cm−1 obtained from operando SR–FTIR spectroscopy measurements under various potentials (−0.3 V to −1.0 V) for VO-InOOH during the electrocatalytic coupling of NO3– and CO2. (C–E) Reproduced with permission [37]. Copyright 2021, American Chemical Society. SFG signals of intermediate species on (F) pristine CeO2 and (G) Vo–CeO2-750. (H) Free energy diagram of urea production on Vo-enriched CeO2. The insets are the initial, transition, and final states of C–N coupling processes. The gray, blue, red, white, and cyan balls represent C, N, O, Ce(IV), and Ce(III) atoms, respectively; the dash circles represent oxygen vacancies. (F–H) Reproduced with permission [38]. Copyright 2022, American Chemical Society.
N2-integrated electrocatalytic CO2 reduction reaction involves multistep electrochemical processes and nonelectrochemical processes, and probably share the similar C–N coupling mechanism as the co-reduction of CO2 and NO2–/NO3–. Generally, the coupling reaction occurs through the formation of C–N bonds via the thermodynamically spontaneous reaction between *N=N* and CO. In the Pd1Cu1/TiO2-VO-400 work [48], DFT calculations illustrated that the key urea precursor of tower-like *NCON*, was formed via the reaction between *N=N* and CO in a thermodynamically and kinetically feasible way, then *NCON* was hydrogenated following a distal pathway to form urea, and the third proton-coupled electron transfer process was energy demanding and was the PLS of urea (Fig. 4A). The formation of C–N bond was proved by SR-FTIR analysis (Figs. 2E and F), but the structure of *NCON* intermediates and the role of VO were not well identified. The same C–N coupling process was proposed by Zhang’s group [64] in the Bi–BiVO4 work, but *NCON* was hydrogenated in an alternating pathway rather than in a distal pathway, and the PLS was the generation of *NHCONH intermediate (Figs. 4B and C). This hypothetical *NCON* pathway for urea formation involved two N atoms bond with one C atom simultaneously, which was obviously different from the pathways in electrocatalytic co-reduction of CO2 and NO2–/NO3–. Of note, such a dual C–N bond-forming step circumvented the formation of highly unstable formamide or carbamic acid intermediates. In the defective Cu–Bi catalyst work reported by Wu et al. [50], Raman signal of C–N intermediates on defective Cu–Bi immersed in both N2 and CO2 atmosphere was observed (Fig. 4D), and no evidence of co-reduction N2 and CO2 was found on contrast catalysts including Cu, Bi, mixed Cu–Bi, and intact Cu–Bi (Fig. 4E), indicating the defective Cu–Bi had the most abundant active sites to produce urea by coupling N2 and CO2. DFT calculations indicated the defective Cu–Bi Gibbs free energy decreased in the presence of N2 and CO2, while the intact Cu–Bi did not show a significant preference for *COOH (*N2) and *COOH, which suggested that the defective Cu–Bi could offer important advantages over that intact Cu–Bi on coupling C–N intermediates (Fig. 4F). In addition, rate-determining step (RDS) barriers were also used to evaluate the catalytic activity. As shown in Figs. 4G and H, the RDS for the distal and alternating styles of intact Cu–Bi was from *NN + *CO to *NCON* step, amounting to a barrier of 2.63 eV, and relative lower RDS barriers were posed by comparing the distal (from *NHCON to *NHHCON, 0.78 eV) and alternating (from *NN + *CO to *NCON*, 0.76 eV) styles in defective Cu–Bi. This work paved the way to further advances in designing other defective electrocatalysts for N2 and CO2 co-reduction operation. Subsequent works also proposed the same coupling steps as the above two works, but further investigation was still desired to verify the C–N coupling process, especially the formation of *NCON* intermediate. Recently, theoretical scientists have carried out some theoretical calculation works to help researchers to better understand the C–N coupling mechanism of N2-integrated electrocatalytic CO2 reduction reaction [73–76], but the role of defect was not well explored.
Figure 4
Figure 4.In situ characterization and proposed coupling mechanism for Pd1Cu1/TiO2-VO-400 mediated electrocatalytic urea synthesis from N2-integrated CO2 reduction. (A) Free energy diagram of urea production. Reproduced with permission [48]. Copyright 2020, Nature Publishing. (B) Electrolytic urea production via the alternating mechanism. (C) Mechanism of the electrocatalytic urea synthesis based on synergistic effects of the Bi–BiVO4 Mott–Schottky heterostructure. (B, C) Reproduced with permission [64]. Copyright 2021, Wiley-VCH. Comparison of Raman signals with different (D) feeding gases and (E) catalysts. (F) Comparison of free-energy diagram of the CO2 reduction with and without the assistance of N2. (G, H) Free-energy diagram of the electrolytic urea production via distal and alternating mechanisms. (D–H) Reproduced with permission [50]. Copyright 2022, Cell Press.
The mechanisms for urea synthesis from N-integrated O2 reduction are controversial, and more detailed in situ characterization should be identified, especially the formation of key intermediates. Anyway, the defects are certainly proved to act the role of extra active sites to modulate the nearby active sites’ electronic structure, facilitating the formation of vital intermediates and higher reaction efficiency. Further research is bound to develop more robust defect engineering strategy to improve the selectivity, FE and yield of urea synthesis.
4.
Conclusions and outlooks
This minireview summarized the recent advances of defect engineered electrocatalysts in urea electrosynthesis from CO2 and various N reactants (NO2−/NO3− and N2). Although the works about defective engineered C–N coupling reactions toward urea synthesis were scarce, the introduction of defect was proved to be able to significantly tune the electronic structure around the catalytic sites to accelerate the formation of key intermediates, thereby benefiting the protonation process and boosting the electrocatalytic performance of urea synthesis. However, the selectivity, yield and FE of defective catalysts mediated urea production are still limited. The biggest obstacle to limiting the development of this field is the key intermediates and specific reaction path for urea production are not very well identified, which hindered the design and improvement of defective electrocatalysts.
In the future, more efforts must made to promote the development of defective engineering strategy mediated electrocatalytic urea production. Firstly, rational methods should be explored to accurately control the types of defects in electrocatalysts. Secondly, the relationship between defect type and catalytic reaction activity, and the specific role of defects in the C–N coupling reaction process should be uncovered by more robust and reliable in situ characterization techniques. Lastly, efficient strategies should be developed to synthesize large-scale defective catalysts and design specific reactors to realize large-scale production of urea. We sincerely hope further investigations of defect engineered electrocatalysts for building C–N bonds can be motivated by this minireview, and further achieve sustainable electrosynthesis of different types of organonitrogen compounds including urea.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 22278094 and 22209029), Outstanding Youth Project of Guangdong Natural Science Foundation (No. 2020B1515020028), Guangdong Natural Science Foundation (No. 2022A1515011775), University Innovation Team Scientific Research Project of Guangzhou Education Bureau (No. 202235246) and China Postdoctoral Science Foundation (No. 2023M730760).
Z. Zhang, L. Guo, J. Du, et al., New J. Chem. 46 (2022) 5278–5287. doi: 10.1039/D2NJ00095D
[76]
C. Zhu, Y. Geng, X. Yao, et al., Small Methods 7 (2023) 2201331. doi: 10.1002/smtd.202201331
Figure 1
Defective electrocatalysts mediated coupling of CO2 and NO2–/NO3– for urea synthesis. (A) XRD patterns of Cu–TiO2 and TiO2; (B) XPS spectra of Ti 2p orbital on Cu–TiO2. (C) FE of the urea synthesis on Cu–TiO2. (A–C) Reproduced with permission [35]. Copyright 2020, Elsevier Inc. (D) FEurea and (E) rurea under the given potentials over ZnO-V. (D, E) Reproduced with permission [36]. Copyright 2021, Cell Press. (F) EPR spectra CeO2, VO-CeO2-500, VO-CeO2-750, and VO-CeO2-1000. (G) rurea of CeO2, VO-CeO2-500, VO-CeO2-750, and VO-CeO2-1000 at various applied potentials. (F, G) Reproduced with permission [38]. Copyright 2022, American Chemical Society. (H) Raman spectra of F-CNT-300 and CNT sample. (I) Urea yield rates and FE values at different potentials for F-CNT-300 in CO2 saturated 0.1 mol/L KNO3 electrolyte with CO2 flow. (H, I) Reproduced with permission [39]. Copyright 2022, Elsevier Inc. (J) In 3d XPS spectra of M-IO, S-IO, and VO-S-IO-6. (K) LSV curves recorded on the M-IO, S-IO, and VO-S-IO-6 during urea electrosynthesis and the electroreduction of VO-S-IO-6 in electrolyte without NO3– or CO2. (L) Yields and FEs of urea during urea electrosynthesis with VO-S-IO-6 catalyst at different potentials after collecting –7.65 C of charge. (J–L) Reproduced with permission [40]. Copyright 2023, Elsevier Inc.
Figure 2
Defective electrocatalysts mediated coupling of CO2 and N2 for urea synthesis. (A) Pd 3d XPS spectra and (B) Cu 2p XPS spectra of Cu/TiO2, Pd1Cu1/TiO2 and Pd1Cu1/TiO2-400. (C) Competitive chemisorption of N2 (red) and CO2 (blue) on TiO2-400 and Pd1Cu1/TiO2-400. (D) Infrared signal in the range of 1100–1800 cm–1 under various potentials for Pd1Cu1/TiO2-400 during the electrocoupling of N2 and CO2. (E) Isotope-labeling infrared signals in the range of 1100–1800 cm–1 at −0.40 V for Pd1Cu1/TiO2-400 during the electrocoupling of 15N2 and/or 13CO2 processes. (F) Urea generation with CO2 and N2 as feeding gases. (G) The FEurea and the total current densities for all products at various potentials. (A–G) Reproduced with permission [48]. Copyright 2020, Nature Publishing. (H) TEM image from a double–corrected spherical aberration electron microscope. (I) Comparison of EPR spectra for intact and defective Cu–Bi. (J) The concentrations and faradaic efficiencies of urea. (K) Comparison of the urea concentrations by defective Cu–Bi (blue), the mixture of Cu–Bi (green), Cu (red), intact Cu–Bi (yellow), and Bi (gray) catalysts. (H–K) Reproduced with permission [50]. Copyright 2022, Cell Press.
Figure 3In situ characterization and proposed coupling mechanism for electrocatalytic urea synthesis from NO2–/NO3–-integrated CO2 reduction. (A) Potential energy diagrams of NO3– reduction to *NH2, undoped CNT and F-doped C active sites (F-CNT) are marked. C, O, H, F and N atoms are represented as brown, red, pinkish white, blue and green spheres, respectively. Reproduced with permission [39]. Copyright 2022, Elsevier Inc. (B) In situ ART-FTIR spectra of ZnO-V under CO2, NaNO2, and both. Reproduced with permission [36]. Copyright 2021, Cell Press. (C) Free-energy diagrams for urea production on the facets of VO-InOOH and pristine InOOH at 0 V. (D) Bader charge analysis on VO-InOOH and pristine InOOH. (E) Infrared signals in the range of 800−1800 cm−1 obtained from operando SR–FTIR spectroscopy measurements under various potentials (−0.3 V to −1.0 V) for VO-InOOH during the electrocatalytic coupling of NO3– and CO2. (C–E) Reproduced with permission [37]. Copyright 2021, American Chemical Society. SFG signals of intermediate species on (F) pristine CeO2 and (G) Vo–CeO2-750. (H) Free energy diagram of urea production on Vo-enriched CeO2. The insets are the initial, transition, and final states of C–N coupling processes. The gray, blue, red, white, and cyan balls represent C, N, O, Ce(IV), and Ce(III) atoms, respectively; the dash circles represent oxygen vacancies. (F–H) Reproduced with permission [38]. Copyright 2022, American Chemical Society.
Figure 4In situ characterization and proposed coupling mechanism for Pd1Cu1/TiO2-VO-400 mediated electrocatalytic urea synthesis from N2-integrated CO2 reduction. (A) Free energy diagram of urea production. Reproduced with permission [48]. Copyright 2020, Nature Publishing. (B) Electrolytic urea production via the alternating mechanism. (C) Mechanism of the electrocatalytic urea synthesis based on synergistic effects of the Bi–BiVO4 Mott–Schottky heterostructure. (B, C) Reproduced with permission [64]. Copyright 2021, Wiley-VCH. Comparison of Raman signals with different (D) feeding gases and (E) catalysts. (F) Comparison of free-energy diagram of the CO2 reduction with and without the assistance of N2. (G, H) Free-energy diagram of the electrolytic urea production via distal and alternating mechanisms. (D–H) Reproduced with permission [50]. Copyright 2022, Cell Press.

 DownLoad:
DownLoad:




 下载:
下载:





 下载:
下载:
