Figure 1.
Illustration of nanozyme-activating prodrug therapy, in which nanozymes catalyze the conversion of nontoxic prodrugs into biologically active drugs for cell death.

Enzyme prodrug therapies (EPTs) have been studied as very attractive strategies to locally activate systemically administered benign prodrugs by the exogenous enzymes or enzymes expressed at the targeted site, achieving localized, site-specific therapeutic effect [1,2]. Compared with conventional drug administration techniques, EPT strategies are capable of selective activation of systemically administered benign prodrugs, thus results in a higher local concentration of the therapeutic as well as a lower side effect [3]. The therapeutic effect of an EPT strategy depends on its two most critical components, enzymes and prodrugs. Prodrugs used in an EPT strategy should have no or only little pharmacological activity in the human body, which can be activated via an enzymatic transformation to release active drugs; while enzymes used in an EPT strategy should have no human analogues (e.g., bacterial lactamase) or only exhibit a localized distribution (e.g., glucuronidase) to avoid off-target toxicity [4,5]. And of course, it is also very essential to deliver drug-activating enzyme gene or functional protein to the targeted site for selective activation of benign prodrugs. In the past few decades, many effective deliver strategies have been developed, including antibody-, viral-, gene-, bacterial-, and polymer-directed EPT [6-8]. Nevertheless, the phenomenological limitation of EPT techniques as described above is the uncertainty in the delivery of the enzyme in vivo. The practical application of the EPT strategies is still a task that presents itself as highly challenging due to the complex intracellular environment and catalyst instability [9].
Recently, nanomaterials with intrinsic enzyme-like activity, referred to as nanozymes, have drawn substantial research interest [10-12]. Since the intrinsic peroxidase-like activity of Fe3O4 magnetic nanoparticles was reported by Yan's group in 2007, numerous advanced nanomaterials with intrinsic enzyme-like catalytic activity are mushrooming [13-15]. Due to the advantages of robust catalytic activity, high stability, low cost, as well as facile synthesis, nanozyme has emerging as a promising alternative to natural enzyme [16-18]. In the past years, nanozymes have found very wide applications in the field of biomedicine, biosensing and bioimaging [19-23]. With the continuous advances in nanotechnology, the biomedical application of nanozymes is still in the ascendant. In particular, the development of nanozymes that possess high stability and good biocompatibility in vivo brings a new opportunity for the theranostic applications [24,25]. It is not difficult to imagine that prodrug activation mediated by nanozyme also can be employed for the site-specific drug synthesis, thus achieving localized therapeutic effect. Indeed, many nanozyme-activated prodrug strategies have been developed for targeted therapy, referred to as nanozyme-activating prodrug therapies [26-32]. As shown in Fig. 1, nontoxic prodrugs can be effectively converted into toxic drugs under the activation of nanozymes, thus leading to cell death. These nanozyme-activated prodrug strategies combine the advantages of EPT (enhanced therapeutic efficacies as well as low off-target effects) and nanozyme (robust catalytic activity and good stability), which showed great potential in the field of targeted therapy [26]. Currently, researches on nanozyme-activating prodrug therapy are still in its initial stage [33]. The combinations of enzyme/prodrug used in the EPT strategies should be good references for developing novel nanozyme-activated prodrug strategies [5,34]. It is undeniable that the development of nanozymes has made very significant progress, however, nanozymes that used in the nanozyme-activating prodrug therapy are mostly focused on mimicking oxidoreductases (e.g., peroxidase, oxidase, and nitroreductase) and hydrolases (e.g., phosphatase, glucuronidase, and glycosidases), this range is only humble at the very best [27,28]. Thus, more advanced nanozymes with diverse catalytic activities, high stability, good biocompatibility, as well as excellent capability of navigating them to the desired action site should be developed. What is more, it is universally acknowledged that the long-term biological safety should be the first consideration for the use of nanomaterials in vivo [35,36]. At present, most nanozymes used in the nanozyme-activating prodrug therapy are metal-containing. However, most of metal- or metal oxide-based nanozymes are very susceptible to destabilizing oxidation under acidic conditions, whose in vivo applications are often limited by the potential metal toxicity [37-39]. In the future, nanozymes with relatively excellent biological safety should be developed, which may be more desirable in the development of nanozyme-activated prodrug strategies. In addition, identification of substrate scopes of the corresponding nanozyme is also of great importance in designing novel benign prodrugs for successful "nanozyme prodrug therapies" [33].
To date, there has not been a review that systematically summarizes nanozyme-activated prodrug strategies. Thus, we present an authoritative review on the nanozyme-activated prodrug strategies to summarize and comment on their development and advances. In this review, almost all the nanozyme-activated prodrug strategies reported in recent years are elaborated in detail. Furthermore, the combinations of enzyme/prodrug in EPT strategies are also briefly summarized to provide an inspiration for designing novel nanozyme-activated prodrug strategies. We firmly believed that this work will provide important enlightenment in the nanozyme-mediated activation of prodrug for the site-specific drug synthesis.
As described above, enzymes and prodrugs are the two most critical components in an EPT strategy. Summarizing enzyme/prodrug combinations in EPT strategies is of great significance in developing novel nanozyme-activated prodrug strategy. Next, commonly used enzyme/prodrug combinations in the field of EPT will be briefly summarized.
Cytosine deaminase is a non-mammalian protein derived from bacteria or yeast, which can catalyze conversion of nontoxic prodrug 5-fluorocytosine (5-FC) into chemotherapeutic agent 5-fluorouracil (5-FU). In the past decades, the combination of 5-FC/cytosine deaminase has been widely used in the field of targeted cancer therapy, especially for the development of antibody- and gene-directed EPT strategies [40,41]. Unfortunately, 5-FC/cytosine deaminase is suffered from its weak substrate flexibility, and 5-FC is the only prodrug at present that used for pair with cytosine deaminase [42]. Nevertheless, 5-FC/cytosine deaminase is still one of the most successful prodrug/enzyme pairs [43].
As another class of non-mammalian enzymes, bacterial lactamases have a high historical value in the context of EPT [44]. In the past few years, the integration of parent drugs (e.g., cephalosporins) and a prodrug strategy is now pushing the development of beta-lactamase-based EPT strategies [45]. As for the synthesis of beta-lactamase specific prodrugs, the introduction of either a carbonate or a carbamate into a cephalosporin is beneficial for the release kinetics [46,47]. In the beta-lactamase-based EPT, the efficient release of drugs is closely related to the leaving group potential of the drug, as well as the accessibility of the hydrolysis site for the enzyme [5]. Unfortunately, the toxicity ratio between the beta-lactamase-activated prodrug and parent drug is relatively low. It had been reported that the toxicity ratio could be improved through the introduction of a polar side group, such as a carboxylic acid or sulfate- group [48,49]. The overall broad substrate acceptance of beta-lactamase allowed the development of various different prodrugs, including but not limited to a cephalosporin-fluoroquinolone prodrug [50], a prodrug of pyrithione [51], and a 5-FU-cephalosporin prodrug [52].
The combination of glucuronide prodrugs/beta-glucuronidase is another popular enzyme/prodrug pair for the use in the field of EPT [53]. Beta-glucuronidase exists intracellularly within lysosomes at extremely low concentrations, of which is far less abundant in the human body when compared to phosphatases [54]. The attractive feature of glucuronide prodrugs in gene-directed EPT is their low toxicity, large range of possible glucuronide drug targets, as well as the bystander effect in the interstitial tumor space [55]. Nevertheless, the synthesis of glucuronide prodrugs is challenging and rather individual without universal reaction conditions. What is more, the very fast renal clearance of glucuronide prodrugs is extremely unfriendly for their clinical use [5]. Thus, a special attention should be paid to the development of novel glucuronide prodrugs with slower clearance, which is very beneficial for prolonging its exposure to beta-glucuronidase at the site of the tumor and thus reducing the administered dose [56]. In addition, the development of fluorescence probes for in vivo imaging of beta-glucuronidase activity is also of significance [57-59]. In the past few years, a large number of glucuronide prodrugs have been designed and synthesized, such as beta glucuronidase responsive albumin-binding prodrugs [60], and a self-immolative doxorubicin prodrug [61].
Recently, the combinations of beta-glucosidase/beta-galactosidase and glycoside prodrugs also have been intensively explored in gene-directed EPT and antibody-directed EPT [62,63]. Similarly, beta-glucosidase/beta-galactosidase exists exclusively within the cells, especially abundant in intestinal, spleen, liver cells, and lymphocytes. It is worth noting that most of the glucose- and galactose-based glycosidic prodrugs show a significantly higher capability to enter mammalian cells than those of glucuronide prodrugs [5,64]. Benefiting from these advantages, Tietze et al. once did very inspiring work in the field of EPT with their work on glycosidase-activated prodrugs [65]. In recent years, substantial advances have been made in medicinal chemistry, leading to the rapid development of glycosylated prodrugs for the glycosidases-based EPT, such as glycosyloxymethyl-prodrugs [66], β-Gal-NONOate (a prodrug that releases nitric oxide (NO) following enzymatic bioconversion) [67], a prodrug from the chemotherapeutic monomethyl auristatin E [68], and a β-galactosidase-activated gemcitabine-based theranostic, Gal-CGem [69].
Alkaline phosphatase (ALP), a member of phosphoesterases family found in all tissues throughout the entire body, including kidneys, liver, and apical membrane of enterocytes, has studied as another commonly used activating enzyme in the field of EPT [70]. Early studies highlighted that prodrug should have high stability under physiological conditions to avoid instability-induced undesired side effects, as well as a high value of half maximal inhibitory concentration ratio (QIC50) (i.e., toxicity ratio between the prodrug and parent drug). From this perspective, phosphate prodrugs are quite attractive due to the simplicity of their synthesis and their inability to cross cell membrane. Thus, the phosphate prodrug approach has emerged as an attractive strategy for increasing bioavailability of a drug candidate with poor hydrophilicity [71]. The major drawback of this enzyme/prodrug system is the undesired premature drug release and toxicity induced by the high endogenous concentration of phosphatase in healthy tissue and plasma [5]. Recently, a large number of phosphate prodrugs have been designed and synthesized, which can be activated by ALP into corresponding therapeutics, including but not limited to fosamprenavir [72], a phosphate prodrug of chalcone OC26 (BOC26P) [73], a water-soluble phosphate salt prodrug of triclabendazole [74], and phosphorylated calcipotriol [75].
As an activating enzyme for nitroaromatic prodrugs, nitroreductase is far less active in the healthy mammalian cells [76]. Expressing or delivering nitroreductase to the desired site of action for the site-specific prodrug activation makes it a prime candidate in the field of EPT. The therapeutic and catalytic potential of nitroaromatic prodrugs in partnership with nitroreductase enzymes were first demonstrated by the development of enzyme/prodrug pairing of Escherichia coli NfsB and CB1954 ([5-(aziridin-1-yl)−2, 4-dinitrobenzamide]) [77]. Despite promising results, the intrinsic limitations, such as the lowest Km for E. coli NfsB far exceeds the maximum dosage of CB1954, have seriously impeded their clinical progression [78]. In recent years, the biodiscovery and engineering of nitroreductase variants (e.g., the YfkO nitroreductase from Bacillus licheniformis) with desirable activities are now being investigated for their potential use in EPT, meanwhile, substantial advances have been made in medicinal chemistry, leading to the development of second- and third-generation nitroaromatic prodrugs with a universal masking group (i.e., nitrobenzyl alcohol) [79]. The substrate scope of nitroreductase has been expanded to paclitaxel and camptothecin prodrugs on the basis of 2‑nitroimidazole [80], nitroreductase-activatable morpholino oligonucleotides [81], nitroreductase-activated NO prodrugs [82,83], as well as nitroreductase-activated fluorescence probes [84].
Natural peroxidases (e.g., horseradish peroxidase (HRP)) also find many interesting applications in the field of EPT. For example, indole-3-acetic acid (IAA)/peroxidase has been explored as another promising enzyme/prodrug combination for tumor oxidation therapy [85]. As one of metabolites of tryptophan, IAA naturally exists in human plasma at micromoles level, which can be catalytically activated by exogenous peroxidase to release toxic oxidative species. Due to the intrinsic limitations of exogenous enzyme (e.g., easy biodegradability), the encapsulation of activating enzymes (e.g., HRP) in micro-/nano-carriers that is capable of retaining their catalytic activity has become a major development trend to construct novel advanced peroxidase-based EPT systems [86-89]. Besides that, the combination of natural peroxidases with many other prodrugs, such as IAA derivatives (e.g., 5-fluoroindole-3-acetic acid) [90], acetylacetone [91], and ethyl 3-indoleacetate [89], also have been demonstrated.
Recently, the increasing demand of more efficient enzyme-activating prodrug therapeutic schedules is pushing the investigation of a large variety of enzyme/prodrug combinations. And there are many other natural enzymes have been described as potential EPT candidates to catalyze the activation of theranostic prodrugs, mainly including various hydrolases [92-112], oxidoreductases [113-120], and transferases [121-126]. The detailed descriptions are shown in Supporting information.
As described above, the combinations of enzyme/prodrug used in EPT strategies can provide a very good reference for the development of nanozyme-activated prodrug strategies. The commonly used enzyme/prodrug combinations in the field of EPT are summarized and listed in Table S1 (Supporting information). In the future, the search for more efficient combinations of activating enzyme/prodrug in the development of advanced EPT strategies will never cease.
As described above, EPT strategies have made great progress in the field of targeted therapy. With the continuous advances in nanotechnology, numerous advanced nanomaterials with robust enzyme-like catalytic activity have been well-designed and synthesized [127]. Inspired by these, many nanozyme-activating prodrug therapy strategies have been developed in recent years. At present, nanozymes used in the nanozyme-activating prodrug therapy are mostly focused on mimicking oxidoreductases (e.g., peroxidase, oxidase, and nitroreductase) and hydrolases (e.g., phosphatase, glucuronidase, and glycosidases).
Significant progress has been made in the development of nanozymes since their discovery in 2007, nevertheless, current nanozymes offer mimicry to a very narrow range of mammalian enzymes. In 2019, Zelikin et al. performed a study on the identification of nanozymes with apparent mimicry of diverse mammalian enzymes, including non-proteinaceous pan-glycosidases, for efficient activation of therapeutic prodrugs, which was accomplished through screening of approximately 100 enzyme-mimic candidate materials (Fig. 2A) [28]. Moreover, the substrate scope for the lead candidates was identified, especially in the context of activation of glucuronides (human metabolites that hold a privileged position in the field of EPT). They found that poly(vinyl pyrrolidone)-stabilized copper nanocubes exhibited strong glycosidases-like activity even in biological media. The substrate scope of copper nanoparticles with regards to their glucuronidase mimicry was defined by the quality of the "leaving group" in the substrate (Fig. 2B). Finally, nanozymes with glucuronidase-like activity were employed for the activation of glucuronide prodrugs into marketed antibacterial and anti-inflammatory agents, as well as "nanozyme prodrug therapy" to mediate antibacterial measures (Fig. 2C). This work presents quite unique nanozymes, as well as a guideline for designing associated substrates, showing great potential for nanozyme engineering and applications. In 2020, they performed another study on the evaluation of nanozymes as glucuronidase mimics for the activation of glucuronide prodrugs, investigated the mechanism of prodrug activation [27]. They found that glucuronide prodrugs were activated by copper nanozymes via at least three different reactions: group transfer in acyl glucuronides, ester bond scission (and transesterification) in acyl glucuronides, as well as glucuronide glycosidic bond scission in activated aryl glucuronides. That is to say, copper nanozymes exhibited glucuronidase-like activity, but also catalyzed the activation of prodrugs via esterase-like mechanism and facilitating group transfer reactions. This study has great value in further designing glucuronidase-activated prodrugs for successful "nanozyme prodrug therapies". Besides that, they once demonstrated that zinc oxide particles with innate glutathione peroxidase and glycosidase activities could catalyze the decomposition of endogenous (S-nitrosoglutathione) and exogenous (β-gal-NONOate) prodrugs to generate NO at physiological conditions [128].

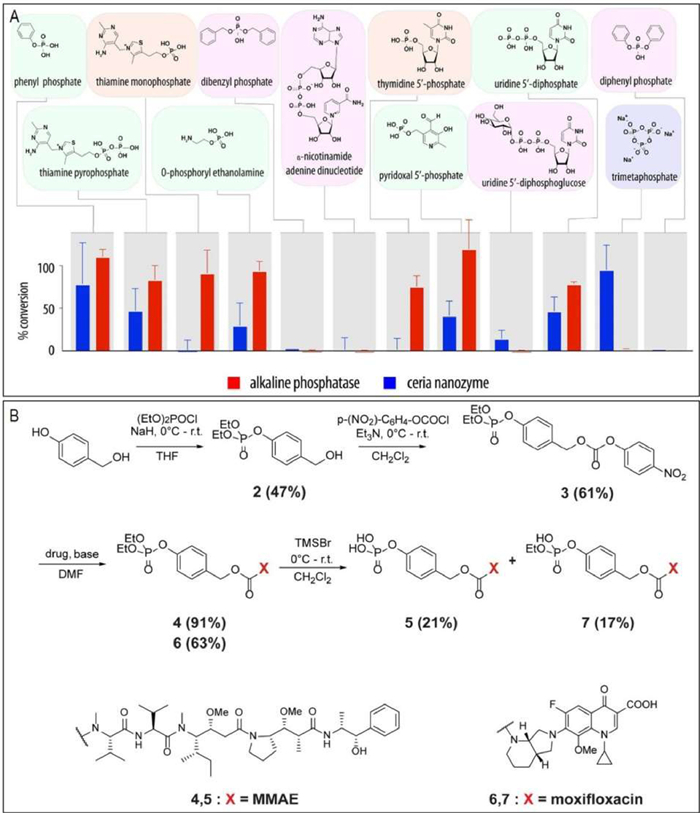
Ceria (ceriumⅢ/Ⅳ oxide) nanoparticles are well known for their multiple enzyme-like activities (e.g., peroxidase, catalase, oxidase, superoxide dismutase, photolyase, phospholipoperoxidase, nuclease, lipoperoxidase, phospholipase, and phosphatase mimetic activities) [129,130]. Most of previous studies mainly focused on exploiting the redox activity of ceria nanoparticles. Interestingly, recent studies indicated that ceria nanoparticles also exhibited phosphatase-like catalytic behavior [131]. Inspired by this, Zelikin et al. further demonstrated ceria nanoparticles as efficient phosphatase mimics to catalyze activation of phosphate prodrugs into corresponding therapeutics [33]. As shown in Fig. 3A, the substrate scope of ceria as a phosphatase mimic was investigated by using a broad range of natural pyrophosphates and phosphor(di)esters, thus providing a guide for the selection of existing phosphate prodrugs that can be activated by ceria into corresponding therapeutics. Moreover, the substrate scope was extended to a broad range of therapeutically active compounds via engineering in their structures a self-immolative linker (Fig. 3B). It is reported that ceria as a phosphatase mimic has been successfully used even in highly complex environment, including in vivo settings. Based on the defined nanozyme substrate scope, the rational pairing of ceria with the existing and the de novo designed "extended scaffold" phosphate prodrugs should be highly favorably for "nanozyme prodrug therapies". In another ceria nanoparticles-based study, Chandrawati et al. found that ceria nanoparticles showed an unexpected capability to controlled release NO from S-nitrosoglutathione [132]. The NO generation mechanism was strongly influenced by the oxidation of Ce3+ to Ce4+ on the surface.
Unfortunately, it has been reported that many metal complexes-based nanozymes (e.g., Ru, Pd) exhibit poor biocompatibility [133]. Thus, the development of bioinspired nanomaterials brings a new opportunity to overcome this challenge [134]. Inspired by the structures of zinc finger hydrolase, Yan et al. designed and synthesized minimal metallo-nanozymes via an amino acid coordinated self-assembly strategy by using zinc(Ⅱ) ions and amino acid derivatives as the building blocks (Fig. 4) [31]. The resulting metallo-nanozymes exhibit robust catalytic activity comparable to that of natural lipase in catalyzing the hydrolysis of p-nitrophenyl acetate. Moreover, the as-synthesized metallo-nanozymes was capable of catalyzing the activation of a hydrophobic prodrug (i.e., benorilate) to produce acetylsalicylic acid, opening up a new way for the development of efficient nanozymes and their application in the field of catalytic prodrug conversion.

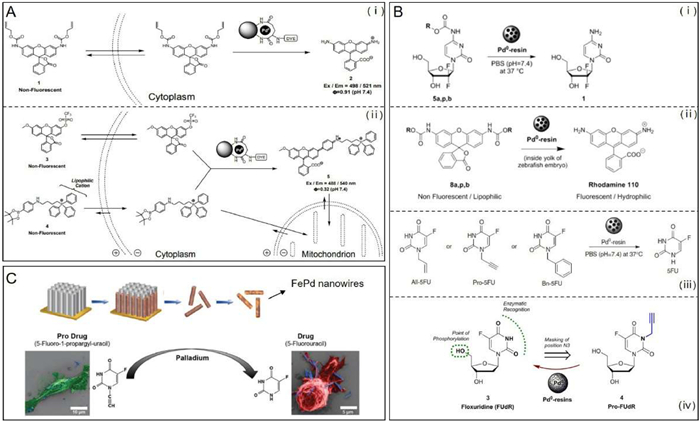
Recently, the application of bioorthogonal nanozymes in the in-situ synthesis of drug derivatives or deprotection of caged organic imaging and therapeutic agents has made great progress [135,136]. In 2015, Rotello et al. designed and synthesized protein-sized bioorthogonal nanozymes, NP_Pd or NP_Ru, via the encapsulation of hydrophobic transition metal catalysts (TMCs) into the monolayer of water-soluble gold nanoparticles (AuNPs) [26]. Then, a supramolecular cucurbit[7]uril (CB[7]) was bound onto the monolayer surface of the resulting nanozymes using host-guest chemistry to provide NP_Pd_CB[7] or NP_Ru_CB[7]. As shown in Fig. 5A, the complexation of CB[7] with NP_Pd or NP_Ru blocks access to the catalytic site, thus resulting in essentially complete inhibition of catalytic activity. The catalytic activity can be restored by the addition of 1-adamantylamine, a competitive guest molecule for CB[7] binding. The potential imaging and therapeutic applications of gated nanozymes were demonstrated by cleaving the allylcarbamate in an allylcarbamate caged fluorophore (i.e., bis-N, N′-allyloxycarbonyl rhodamine 110), as well as the propargyl groups in an inactive prodrug (i.e., pro-5-FU) inside living cell. This gated control of catalysis integrates biomimetic and bioorthogonal design elements, making them very attractive components for both in vitro and in vivo applications. As known to all, the spatial localization of imaging and therapeutic agents is central to their utility in biomedicine [137,138]. It has been reported that surface engineering contributes to the effective localization of nanomaterials [139]. Recently, they further performed a study to demonstrate the use of surface-engineered nanozymes to predictably localize bioorthogonal catalysis to either intra- or extracellular space (Fig. 5B) [140]. In this method, TMCs were encapsulated into hydrophobic core of AuNP (2 nm core) scaffolds, which were functionalized with ligands featuring three components, a hydrophobic interior segment, a tetraethylene glycol spacer, as well as a terminal interacting unit, using place-exchange reaction to provide surface-engineered nanozymes. A cell-penetrating cationic scaffold and a "stealth" zwitterionic scaffold were used to generate cationic nanozymes (Pos-NZ) for intracellular catalysis, as well as zwitterionic nanozymes (Zw-NZ) to limit catalysis to extracellular space, respectively. The specific location of catalytic activity of Pos-NZ and Zw-NZ was demonstrated by monitoring the deallylation-mediated fluorogenesis of two pro-fluorophores, alloc-protected resorufin and alloc-protected rhodamine 110, while the therapeutic applications of nanozymes Pos-NZ and Zw-NZ was also demonstrated through extra- and intracellular activation of a well-designed prodrug, allylcarbamate-protected doxorubicin. Taken together, these studies provide a promising strategy for bioorthogonal transformation of pro-fluorophores and prodrugs by spatially controlling TMC-mediated bioorthogonal catalysis. In 2022, they synthesized degradable ZnS-supported bioorthogonal nanozymes with an enhanced catalytic activity for intracellular activation of therapeutics [141]. The maximum rate of reaction (Vmax) increases ~2.5-fold as compared to the non-degradable AuNP analogue. In 2023, they fabricated bioorthogonal nanozymes through the encapsulation of TMCs into the monolayer of a cationic-functionalized AuNP, thus providing protection of the catalyst and adherence to tissue at the tumor site [142]. The resulting cationic bioorthogonal nanozymes maintained high catalytic activity at least seven days after injection. These nanozymes localized at the injection site in the tumor, activating systemically injected propargyl-protected imaging pro-fluorophores and prodrugs. Significantly, this nanozyme-activating prodrug strategy produced substantially less liver damage than the free therapeutic. Recently, they designed and synthesized novel biorthogonal nanocatalysts based on encapsulating TMCs into polymeric scaffolds, known as "polyzymes", for intracellular activation of anticancer therapeutics [143-145]. The as-synthesized "polyzymes" showed both controlled size and increased bioorthogonal catalytic efficiency. The biological applicability of polyzymes was demonstrated by efficiently transforming nontoxic prodrugs into the active drugs in cancer cells.

As a very promising transition metal candidate for bioorthogonal reactions, palladium is well known for its strong catalytic activity and good biocompatibility [138,146]. In recent years, palladium-mediated intracellular chemistry had attracted considerable attention, especially in the field of site-specific drug synthesis [147]. In particular, palladium nanoparticles (PdNPs) have proved to be very efficient for the deprotection of caged organic imaging and therapeutic agents [148,149]. Encouraged by vast possibilities and applications of palladium chemistry, very great efforts had been made for the development of bio-friendly Pd0-based heterogeneous catalysts. For example, Bradley and co-workers synthesized biocompatible cell-penetrating heterogeneous catalysts via the self-assembly of PdNPs on the surface of polystyrene microspheres [150,151]. PdNPs trapped within polystyrene microspheres was capable of mediating efficient bioorthogonal organometallic chemistries (e.g., allylcarbamate cleavage and Suzuki-Miyaura cross-coupling) (Fig. 6A). Once upon incubation with the as-synthesized Pd0 microspheres, alloc-Rh110 and alloc-amsacrine could be activated. Besides that, Unciti-Broceta and co-workers once designed and synthesized Pd0-labile gemcitabine prodrugs, N-propargyloxycarbonyl gemcitabine, which could be activated in cell culture by Pd0 nanoparticles trapped in an amino-functionalized polystyrene matrix, Pd0-resins [152]. The implanted Pd0-resins in the yolk sac of zebrafish embryos exhibited excellent biocompatibility and local catalytic activity. It had been demonstrated that the mixture of Pd0-resins and 5-fluoro-1-propargyluracil (Pro-5-FU) also could produce the comparable cytotoxicity with its active therapeutic counterpart 5-FU [153]. Similarly, another propargyl-protected prodrug, pro-floxuridine (FUdR), could be activated by Pd0-resins to produce FUdR, a clinically-used anticancer drug (Fig. 6B) [154]. However, further efforts should be made to promote in vivo demonstrations, particularly in mammalian model, which is very crucial to assess the actual therapeutic potential with regards to safety and pharmacological profiles. Recently, the utilize of magnetic nanorobots to activate therapeutic prodrugs represents a new trend in the development of chemotherapeutic treatments [155]. Inspired by this, Pané et al. synthesized a hybrid nanowire, which was composed of palladium and magnetic iron, for site-specific activation of the latent chemotherapeutic prodrug Pro-5-FU (Fig. 6C) [156]. The synthesized FePd nanowires induced no significant cytotoxic effect, while a significant reduction of cell viability was observed once upon incubation of the FePd nanowires with Pro-5-FU. The targeted therapeutic abilities of the FePd nanowires were demonstrated with the aid of magnetic fields to attract the FePd nanowires to a predefined area within a cultured cancer cell population, triggering the local activation of Pro-5-FU and thus inducing cell death exclusively in this region. Finally, the FePd nanowires were injected in MDA-MB-231 human breast cancer xenografts in nude mice, which significantly inhibited tumor growth without apparent side effects after intraperitoneal administration of Pro-5-FU over a period of two weeks.
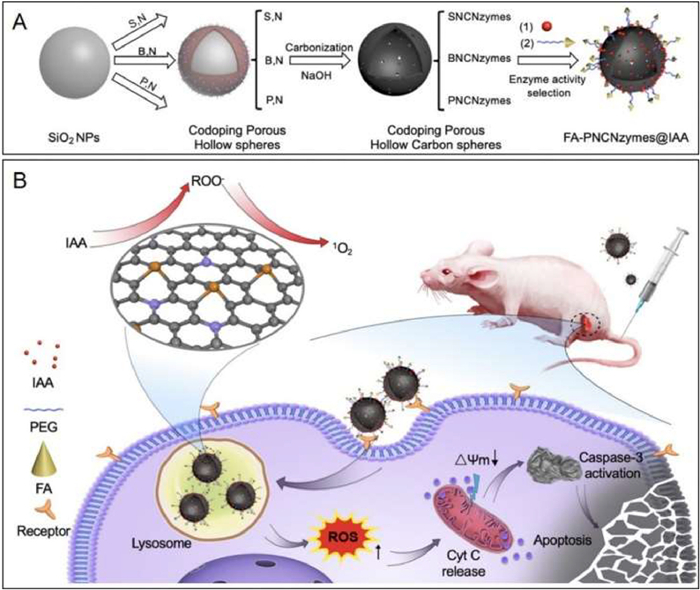
As described above, the combination of IAA and natural peroxidase is commonly used in enzyme prodrug therapy [86]. As one of the metabolites of tryptophan that naturally exists in human plasma, IAA exhibits negligible toxic effects under the dose of 100 mg/kg. Upon incubation with peroxidase, IAA can be effectively activated to produce free radical intermediates, and thus inducing the apoptosis of treated cells. In 2020, Yan's group demonstrated a nanozyme-activated prodrug strategy for targeted cancer therapy using a metal-free phosphorous and nitrogen dual-doped porous hollow carbon sphere nanozyme (PNCNzyme) with peroxidase-like activity in acidic environment [30]. As shown in Fig. 7A, IAA and folate (FA) were loaded on the surface of PNCNzymes to provide FA-PNCNzymes@IAA. IAA molecules released from the resulting FA-PNCNzymes@IAA only in an acidic environment, which can be activated by the PNCNzyme. To evaluate the antitumor activity and biological safety, FA-PNCNzymes@IAA nanoparticles were administrated to mice (Fig. 7B). The results indicated that this novel nanozyme-activated prodrug strategy exhibited very good antitumor activity and biological safety, which was capable of efficient activation of prodrug at the tumor site. In 2023, Duo and co-workers demonstrated a strategy for cancer therapy by innovatively combining gas therapy with nanocatalytic tumor therapy based on the use of a biomimetic single-atom nanozyme with peroxidase-like activity, an SO2 prodrug (benzothiazole sulfinate, BTS), as well as platelet membrane vesicles [157]. Under the acidic conditions of the tumor environment, the SO2 prodrug could release SO2 that subsequently induced the production of •SO3− to cause cell apoptosis. Meanwhile, the catalytic activity of single-atom nanozymes was improved with down-regulating glutathione (GSH) and up-regulating H2O2 in tumor cells, thus achieving self-enhanced nanocatalytic tumor therapy. As a result, more than 90% of the tumor could be suppressed.
Banoxantrone dihydrochloride (AQ4N) is a hypoxia-activated prodrug that has entered clinical trials, which can be activated to produce a cytotoxic DNA intercalator (AQ4) under the hypoxia condition [158]. However, the degree of tumor hypoxia is far from sufficient to maximize therapeutic potential of AQ4N. In 2022, Dong et al. synthesized bimetallic alloy nanoparticles (Cu-Ag NPs) with robust cytochrome c oxidase-like (CcO) catalytic activity via a two-step seeded growth procedure [29]. This bimetallic alloy nanozyme was capable of catalyzing the reduction of oxygen to hydroxyl radicals (·OH) and cytotoxic superoxide (·O2−) with the aid of cytochrome c overexpressed in cancer cells, whereby significantly aggravating tumor hypoxia. After being modified with the hydrophilic NH2-PEG2000 to enhance dispersibility, hypoxia-activated AQ4N prodrug molecules were loaded on the surface of the as-synthesized Cu-Ag NPs to prepare AQ4N@Cu-Ag NP, which unprecedentedly enabled simultaneous starvation, ferroptosis, and chemical therapy with very high efficiency and specificity, as well as low side effects (Fig. 8). Furthermore, the as-prepared AQ4N@Cu-Ag NP was also capable of totally eliminating tumor and prolonging the survival rate for 4T1-tumor-bearing mice. This study has great guiding significance for the site-specific activation of other hypoxia-activated prodrugs and brings new insights into nanocatalytic cancer therapy.

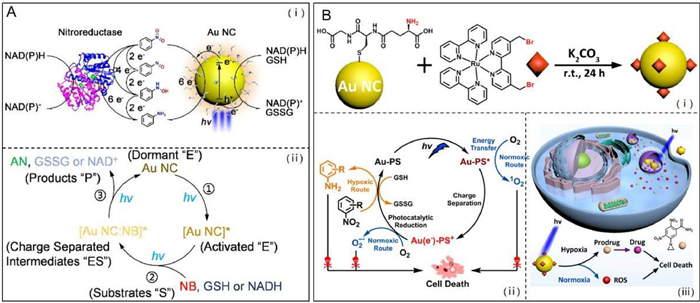
Photonanozymes refer to a class of nanomaterials that exhibit intrinsic enzyme-like catalytic activity activating with visible photons [159]. Gold nanoclusters (Au NCs) with a diameter less than 2 nm can be used as biocompatible photonanozymes, which hold great potential for the photocreation of biologically active molecules [160]. In 2021, Hu et al. synthesized GSH-modified Au NCs with nitroreductase-like catalytic behavior, which were capable of photoinduced catalytic the reduction of a prodrug moiety nitrobenzene to biologically active aniline (Fig. 9A) [32]. And they found that the photocatalytic reaction at variable light fluences was shown to follow the classical Michaelis-Menten enzyme kinetics. The viability of GSH-stabilized Au NCs as photonanozymes performing catalysis in a mammalian cell environment was demonstrated by intracellular reduction of a nitro-group-containing fluorescent probe. The successful implementation of this study will open a new avenue to utilize Au NCs as biocompatible photonanozymes that mimic natural nitroreductase for potential prodrug activation. Shortly after, they further performed a study on photocatalytic nitroaromatic prodrug activation by photosensitizer functionalized AuNCs, in which a Ru(Ⅱ) tris-bipyridine (bpy)-type coordination compound as the photosensitizer was covalently linked to the surface of glutathione-ligated Au NCs (Fig. 9B) [161]. Owning to the specific catalytic performance resulting from photoinduced charge separation within photosensitizer functionalized AuNCs, visible light excitation of the photocatalyst allows the reduction of nitrobenzene to aniline with 100% selectivity. Under light irradiation, the nitro-benzene moiety containing prodrug CB1954 could be efficiently activated by the photosensitizer functionalized AuNCs to produce significant cytotoxicity even in hypoxic cancer cells.
It had been reported that certain flavins could perform as potent redox photocatalysts, which were capable of prompting the conversion of transition metal substrates (e.g., PtⅣ anticancer prodrug complexes) into their biologically active counterparts in the biological environment (Fig. 10A) [162]. Salassa and co-workers once investigated the mechanism of these artificial photocatalytic reactions by employing four PtⅣ prodrug complexes and five flavins, namely, lumiflavin, riboflavin, tetra-O-acetyl riboflavin (TARF), riboflavin-5′-phosphate (FMN), flavoprotein miniSOG (mini Singlet Oxygen Generator) [163]. With the aid of nicotinamide adenine dinucleotide (NADH) as the electron donor, flavins could be transformed into the reduced FLH− species as the active photocatalyst to catalyze the PtⅣ to PtⅡ conversion under light irradiation. And they found that the catalytic efficiency was determined by both the ribityl and isoalloxazine moieties of the flavins, and may be influenced by H-bond interactions between the PtⅣ precursors and flavin catalysts. Overall, FMN-containing miniSOG is a less efficient catalyst, due to the reduced substrate accessibility to the flavin catalytic core when embedded in an amino acid scaffold. Thus, structure modification the isoalloxazine and ribityl groups in free flavins, as well as of site mutagenesis in miniSOG may be beneficial for boosting their catalytic activity. This study expands the catalyst and substrate scope for the activation of metal-based prodrugs and versatility of bioorthogonal catalytic reactions currently available in drug synthesis. In 2020, they further fabricated a supramolecular nanozyme via the encapsulation of FMN into 1.9 nm AuNPs decorated with a C11-thiol bearing a 1, 4, 7-triazacyclononane headgroup (Fig. 10B) [164]. With the aid of electron donors (e.g., zwitterionic buffers or NADH), the supramolecular nanozyme catalyzed activation of a Pt(Ⅳ) anticancer prodrug, cis, cis, trans-[Pt(NH3)2(Cl2)(O2CCH2CH2COOH)2], into clinically approved anticancer drug cisplatin in biological media (Fig. 10C). This novel activation strategy holds promise in the photocatalytic activation of Pt(Ⅳ) anticancer prodrugs (Fig. 10D).

There are still many other interesting "nanozyme prodrug therapies" strategies have been developed. In 2020, Li and co-workers demonstrated a novel therapeutic platform for multisynergistic cancer therapy based on taking full advantages of the glucose oxidase (GOD)-based β-D-glucose oxidation and palladium mediated in situ activation of doxorubicin prodrugs [165]. As shown in Fig. 11A, a GOD-modified nanoreactor was fabricated based on co-loading of prodrugs and nanocatalysts, in which the Pd0 nanoparticles were synthesized in situ within the pores of mesoporous silica nanoparticles and the doxorubicin prodrugs were encapsulated in β-cyclodextrins to reduce toxic side effects. To evaluate the therapeutic effect of this novel therapeutic platform in vivo, tumor-bearing nude mice were treated with the as-fabricated nanoreactor. The injected nanomaterials were significantly accumulated in the tumors, subsequently, a cascade chemical reaction was triggered: GOD efficiently and specifically catalyzed β-D-glucose oxidation to produce gluconic acid and H2O2; the as-produced gluconic acid decreased the local pH, which induced the disassembly of the doxorubicin prodrug-β-cyclodextrins complexes to release free doxorubicin prodrugs; the free doxorubicin prodrugs were catalytically activated by Pd0 nanoparticles to produce active anticancer drugs. Taken together, a multisynergistic cancer therapy was achieved, which involved GOD-based cancer starvation therapy, H2O2-mediated orchestrated oxidation therapy, as well as doxorubicin-induced chemotherapy. In 2023, Qu and his colleagues developed a bioorthogonal-activated in situ cancer vaccine based on the synthesis of a series of biocompatible covalent organic framework (COF)-based Fe(Ⅱ) catalysts, CFe-X (X = H, OH, OCH3, Br) [166]. Once upon incubation with pro-rhodamine 110, CFe-OH exhibited strongest catalytic activity, which was further modified with folic acid for targeting tumor cells (denoted as CFe-FA). Particularly, pro-DOX could be efficiently and bioorthogonally activated in situ by the as-synthesized CFe-FA, which could evoke immunogenic cancer cell death and thus releasing tumor-associated antigens to stimulate innate immune cells. What is more, the COF-based Fe(Ⅱ) catalysts were also capable of activating pro-imiquimod (pro-IMQ, a TLR7/8 immune agonist) to produce IMQ, which could be served as an adjuvant to amplify the antitumor immunity (Fig. 11B). As a result, this bioorthogonal-activated in situ cancer vaccine performed satisfying antitumor efficacy and prevented dose-dependent side effects in normal organs or tissues. More importantly, the encouraging results presented herein have paved way for the development of in situ cancer vaccines with safety and efficiency, and providing a paradigm for potent cancer immunotherapy. Recently, they demonstrated a spatially controlled macroautophagy degradation targeting chimeras strategy for tissue-specific mitochondrial depletion in tumors based on the use of a well-designed aptamer-based Cu nanocatalyst (Apt-Cu30) [167]. The bioorthogonal reaction between the alkynyl tagged LC3 recruiting warhead (alk-DP) and azido tagged ligand of mitochondria (azi-TPP) could be triggered by Apt-Cu30 to produce the lethal mitophagy inducer bio-autophagy-tethering compounds (ATTECs) at tumor sites, thus leading to mitochondrial depletion and autophagic cell death in malignant cells. The proposed bioorthogonal activation strategy can efficiently induce autophagic tumor cell death, providing new insights into the development of cancer therapy strategies to realize precision therapeutics by avoiding undesired side effects.

Besides that, some well-designed prodrug-based cancer therapeutic strategies may not fall under the category of "nanozyme prodrug therapy", but they still have very important implications in the field of targeted cancer therapy, which will be briefly discussed and summarized. Recently, fluoride (F−)-mediated desilylation has emerged as a burgeoning bioorthogonal system for prodrug activation [168]. Compared to F− for desilylation, metal fluoride nanocrystals are more effective for tumor targeting benefiting from the enhanced permeability and retention (EPR) effect. Due to solubility equilibrium, release of fluorides and metal ions from nanocrystals is intrinsic, which has always been considered as a drawback. Making good use of this so-called "drawback", Liu et al. unveiled that metal fluoride nanocrystals could induce rapid and efficient desilylation reactions for controlled release of active drugs and fluorophores in test tubes, living cells, and tumor-bearing mice [169]. They synthesized six model metal fluoride nanocrystals (LaF3, BaF2, SrF2, CaF2, NaYF4, and NaYF4@CaF2 nanocrystals) via a seed-mediated shell growth strategy. Due to the sufficient desilylation rate and low toxicity of metal ions, CaF2 nanocrystals were chosen for the subsequent biological assays. For more better biocompatibility and colloidal stability, CaF2 nanocrystals were decorated with a polyethylene glycol (PEG) coating. The as-synthesized biocompatible PEG-coated CaF2 nanocrystals was capable of releasing adequate F− to the desilylation of caged TBS-MeRho and monomethyl auristatin E both in vitro and in vivo. Inspired by the fluoride-mediated desilylation, Qu et al. also synthesized a pH-responsive, cancer cell-selective biomimetic nanocatalyst MIL-53@F−@M by coating cancer cell membranes on a F− absorbed metal-organic framework (MOF) for prodrug activation and enhanced synergistic chemodynamic therapy [170]. The synthesized pH-responsive biomimetic nanocatalyst prefers to be internalized by homotypic cancer cells with high efficiency, releasing F− and Fe3+ after entering cells. Then, the released F− was capable of initiating a desilylation reaction for site-specific activation of self-designed prodrug tert‑butyldimethyl silyl-hydroxycamptothecin (TBSO—CPT) into the anticancer drug OH—CPT (10-hydroxycamptothecin), which could kill cancer cells without apparent side effects. Simultaneously, the bioorthogonal-synthesized OH—CPT induced the upregulation of intracellular H2O2 by activating nicotinamide adenine dinucleotide phosphate oxidase, which greatly boosted formation of hydroxyl radical (•OH) from the Fe3+-induced Fenton reaction, thus resulting in augmented synergistic anticancer therapy. Both in vitro and in vivo studies demonstrated the good biocompatibility and excellent antitumor effect of this cancer cell selective fluoride-activated biomimetic platform. Finally, recently reported nanozyme-activating prodrug therapies are summarized and listed in Table 1.
EPT strategies have been proved to be attractive tools for site-specific drug synthesis thus achieving localized therapeutic effect. Recently, it has been demonstrated that the systemically administered benign prodrugs also can be activated in vivo by well-designed nanozymes, known as nanozyme-activating prodrug therapies. In this review, the development and recent advances of nanozyme-activated prodrug strategies are summarized and discussed. The combinations of enzyme/prodrug in EPT strategies are also briefly summarized, aiming to provide an inspiration for developing novel nanozyme-activated prodrug strategies.
The nanozyme-activated prodrug strategies combine the advantages of EPT (e.g., low side effect and high local concentration of the therapeutic) and nanozyme (e.g., robust catalytic activity and good stability), which show great potential in the field of targeted therapy. Although some progress has been made in this field, there are still many urgent issues that need to be addressed. (1) Currently, researches on nanozyme-activating prodrug therapy are still in its initial stage, there are relatively few reports on nanozyme-activating prodrug therapy; (2) Nanozymes that used in the nanozyme-activated prodrug strategies are mostly focused on mimicking oxidoreductases (e.g., peroxidase, oxidase, and nitroreductase) and hydrolases (e.g., phosphatase, glucuronidase, and glycosidases), this range is only humble at the very best; (3) Most currently developed nanozyme-activated prodrug strategies are demonstrated based on utilizing metal-containing nanozymes. However, metal- or metal oxide-based nanozymes are susceptible to destabilizing oxidation under acidic conditions, whose in vivo applications are often limited by the potential metal toxicity.
Predictably, the development of more advanced nanozymes with diverse catalytic activities, high stability, good biocompatibility, and excellent capability of navigating them to the desired action site remains the major trend in the future. Furthermore, the following aspects may also be taken into consideration. Firstly, the combinations of enzyme/prodrug used in EPT strategies should be good references for developing nanozyme-activated prodrug strategies; secondly, metal-free nanozymes, which exhibit relatively excellent biological safety, may be more desirable in the development of nanozyme-activated prodrug strategies; additionally, the identification of substrate scopes of the corresponding nanozyme may be also of great importance in designing novel benign prodrugs for successful "nanozyme prodrug therapies". With the continuous advances in nanotechnology, the application of nanozymes in the field of EPT is still in the ascendant. We firmly believed that this work will provide important enlightenment in the nanozyme-mediated activation of prodrug for the site-specific drug synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the Shandong Provincial Natural Science Foundation of China (No. ZR2021QC088).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Xu, H.L. McLeod, Clin. Cancer Res. 7 (2001) 3314–3324.
Q.X. Yao, F. Lin, X.Y. Fan, et al., Nat. Commun. 9 (2018) 5032. doi: 10.1038/s41467-018-07490-6
T.J. Gardner, J.P. Lee, C.M. Bourne, et al., Nat. Chem. Biol. 18 (2022) 216–225. doi: 10.1038/s41589-021-00932-1
B. Stadler, A.N. Zelikin, Adv. Drug Deliv. Rev. 118 (2017) 1. doi: 10.1016/j.addr.2017.10.006
R. Walther, J. Rautio, A.N. Zelikin, Adv. Drug. Deliv. Rev. 118 (2017) 65–77. doi: 10.1016/j.addr.2017.06.013
S.K. Sharma, K.D. Bagshawe, Adv. Drug. Deliv. Rev. 118 (2017) 2–7. doi: 10.1016/j.addr.2017.09.009
X. Zhang, Y.Q. Yang, T.Y. Kang, et al., Small 17 (2021) 2100501. doi: 10.1002/smll.202100501
G. Chung-Faye, D. Palmer, D. Anderson, et al., Clin. Cancer Res. 7 (2001) 2662–2668.
C. Li, M.F. Penet, P. Winnard, et al., Clin. Cancer Res. 14 (2008) 515–522. doi: 10.1158/1078-0432.CCR-07-1837
H. Wang, K.W. Wan, X.H. Shi, Adv. Mater. 31 (2019) 1805368. doi: 10.1002/adma.201805368
A. Robert, B. Meunier, ACS Nano 16 (2022) 6956–6959. doi: 10.1021/acsnano.2c02966
X.F. Chen, Y. Wang, M. Feng, et al., Chin. Chem. Lett. 34 (2023) 107969. doi: 10.1016/j.cclet.2022.107969
L.Z. Gao, J. Zhuang, L. Nie, et al., Nat. Nanotechnol. 2 (2007) 577–583. doi: 10.1038/nnano.2007.260
M. Xu, C. Ren, Y. Zhou, et al., Chin. Chem. Lett. 34 (2023) 107585. doi: 10.1016/j.cclet.2022.06.008
J. Hao, C. Zhang, C. Feng, et al., Chin. Chem. Lett. 34 (2023) 107650. doi: 10.1016/j.cclet.2022.06.073
D.W. Jiang, D.L. Ni, Z.T. Rosenkrans, et al., Chem. Soc. Rev. 48 (2019) 3683–3704. doi: 10.1039/C8CS00718G
B. Li, Q. Xu, X. Shen, et al., Chin. Chem. Lett. 34 (2023) 108015. doi: 10.1016/j.cclet.2022.108015
L. Yang, H. Guo, T. Hou, et al., Chin. Chem. Lett. 34 (2023) 107607. doi: 10.1016/j.cclet.2022.06.030
S.F. Ji, B. Jiang, H.G. Hao, et al., Nat. Catal. 4 (2021) 407–417. doi: 10.1038/s41929-021-00609-x
C. Zhang, P. Ni, B. Wang, et al., Chin. Chem. Lett. 33 (2022) 757–761. doi: 10.1016/j.cclet.2021.08.017
X. Hai, Y. Li, K. Yu, et al., Chin. Chem. Lett. 32 (2021) 1215–1219. doi: 10.1016/j.cclet.2020.09.0136|2021|||
J. Ju, Y. Chen, Z. Liu, et al., Chin. Chem. Lett. 34 (2023) 107820. doi: 10.1016/j.cclet.2022.107820
Y. Xia, K. Sun, Y. Zuo, et al., Chin. Chem. Lett. 33 (2022) 2081–2085. doi: 10.1016/j.cclet.2021.08.083
Y. Wu, Q. Chen, S. Liu, et al., Chin. Chem. Lett. 30 (2019) 2186–2190. doi: 10.1016/j.cclet.2019.08.014
Y.W. Peng, M.C. Huang, L.J. Chen, et al., Nano Res. 15 (2022) 8783–8790. doi: 10.1007/s12274-022-4541-x
G.Y. Tonga, Y.D. Jeong, B. Duncan, et al., Nat. Chem. 7 (2015) 597–603. doi: 10.1038/nchem.2284
R. Walther, W. van den Akker, A.S. Fruergaard, A.N. Zelikin, Small 16 (2020) e2004280. doi: 10.1002/smll.202004280
R. Walther, A.K. Winther, A.S. Fruergaard, et al., Angew. Chem. Int. Ed. 58 (2019) 278–282. doi: 10.1002/anie.201812668
C. Cao, N. Yang, Y. Su, et al., Adv. Mater. 34 (2022) e2203236. doi: 10.1002/adma.202203236
Q. Liang, J.Q. Xi, X.J.J. Gao, et al., Nano Today 35 (2020) 100935. doi: 10.1016/j.nantod.2020.100935
J.J. Han, Q.L. Zou, W.W. Su, X.H. Yan, Chem. Eng. J. 394 (2020) 124987. doi: 10.1016/j.cej.2020.124987
R. Liu, D.J. Cheng, Q. Zhou, et al., ACS Appl. Nano Mater. 4 (2021) 990–994. doi: 10.1021/acsanm.1c00014
R. Walther, T.H. Huynh, P. Monge, et al., ACS Appl. Mater. Interfaces 13 (2021) 25685–25693. doi: 10.1021/acsami.1c03890
A.C. Mendes, A.N. Zelikin, Adv. Funct. Mater. 24 (2014) 5202–5210. doi: 10.1002/adfm.201304312
M.J. Cao, R. Cai, L.N. Zhao, et al., Nat. Nanotechnol. 16 (2021) 708–716. doi: 10.1038/s41565-021-00856-w
X. Chang, L. Chen, B. Liu, et al., Chin. Chem. Lett. 33 (2022) 3303–3314. doi: 10.1016/j.cclet.2022.03.057
X.P. Liu, Z.Q. Yan, Y. Zhang, et al., ACS Nano 13 (2019) 5222–5230. doi: 10.1021/acsnano.8b09501
Y. Dai, Y. Ding, L. Li, Chin. Chem. Lett. 32 (2021) 2715–2728. doi: 10.1016/j.cclet.2021.03.036
D. He, M. Yan, P. Sun, et al., Chin. Chem. Lett. 32 (2021) 2994–3006. doi: 10.1016/j.cclet.2021.03.078
Z.H. Chen, M.F. Penet, B. Krishnamachary, et al., Biomaterials 80 (2016) 57–67. doi: 10.1016/j.biomaterials.2015.11.048
W.H. Fan, M.Y. Shao, J.W. Zhang, et al., Adv. Funct. Mater. 29 (2019) 1807104. doi: 10.1002/adfm.201807104
P.C. Despres, A.F. Cisneros, E.M. Alexander, et al., Nat. Ecol. Evol. 6 (2022) 1501–1515. doi: 10.1038/s41559-022-01846-4
W.P. Accomando, A.R. Rao, D.J. Hogan, et al., Clin. Cancer Res. 26 (2020) 6176–6186. doi: 10.1158/1078-0432.CCR-20-0536
K. Bush, P.A. Bradford, Nat. Rev. Microbiol. 17 (2019) 295–306. doi: 10.1038/s41579-019-0159-8
N. Barraud, B.G. Kardak, N.R. Yepuri, et al., Angew. Chem. Int. Ed. 51 (2012) 9057–9060. doi: 10.1002/anie.201202414
D.E. Kerr, Z. Li, N.O. Siemers, et al., Bioconjug. Chem. 9 (1998) 255–259. doi: 10.1021/bc970163l
L.N. Jungheim, T.A. Shepherd, J.K. Kling, Heterocycles 35 (1993) 339–348. doi: 10.3987/COM-92-S22
K.R. Reddy, J. Parkinson, M. Sabet, et al., J. Med. Chem. 64 (2021) 17523–17529. doi: 10.1021/acs.jmedchem.1c01722
A.B. Shapiro, N. Gao, ACS Infect. Dis. 7 (2021) 114–122. doi: 10.1021/acsinfecdis.0c00656
L.E. Evans, A. Krishna, Y. Ma, et al., J. Med. Chem. 62 (2019) 4411–4425. doi: 10.1021/acs.jmedchem.8b01923
N. Wiebelhaus, J.M. Zaengle-Barone, K.K. Hwang, et al., ACS Chem. Biol. 16 (2021) 214–224. doi: 10.1021/acschembio.0c00900
R.M. Phelan, M. Ostermeier, C.A. Townsend, Bioorg. Med. Chem. Lett. 19 (2009) 1261–1263. doi: 10.1016/j.bmcl.2008.12.057
M.T. Jarlstad Olesen, R. Walther, P.P. Poier, et al., Angew. Chem. Int. Ed. 59 (2020) 7390–7396. doi: 10.1002/anie.201916124
P. Awolade, N. Cele, N. Kerru, et al., Eur. J. Med. Chem. 187 (2020) 111921. doi: 10.1016/j.ejmech.2019.111921
I. Tranoy-Opalinski, T. Legigan, R. Barat, et al., Eur. J. Med. Chem. 74 (2014) 302–313. doi: 10.1016/j.ejmech.2013.12.045
R. Walther, S.M. Nielsen, R. Christiansen, et al., J. Control. Release 287 (2018) 94–102. doi: 10.1016/j.jconrel.2018.08.025
P.W. Gong, L. Sun, F. Wang, et al., Chem. Eng. J. 356 (2019) 994–1002. doi: 10.1016/j.cej.2018.09.100
J.M. Wang, L. Zhang, Y.L. Su, et al., Anal. Chem. 94 (2022) 7012–7020. doi: 10.1021/acs.analchem.1c05635
X.F. Lou, T.B. Ren, H.M. Chen, et al., Biomaterials 287 (2022) 121657. doi: 10.1016/j.biomaterials.2022.121657
G. Compain, N. Oumata, J. Clarhaut, et al., Eur. J. Med. Chem. 158 (2018) 1–6. doi: 10.1016/j.ejmech.2018.08.100
K. Roemhild, H.C. Besse, B. Wang, et al., Theranostics 12 (2022) 4791–4801. doi: 10.7150/thno.69168
H. Martin, L.R. Lazaro, T. Gunnlaugsson, E.M. Scanlan, Chem. Soc. Rev. 51 (2022) 9694–9716. doi: 10.1039/D2CS00379A
Y.H. Song, X.M. Li, D.L. Shi, et al., Chem. Sci. 13 (2022) 11738–11745. doi: 10.1039/D2SC03525A
N. Yu, T. Liu, X. Zhang, et al., Nano Lett. 20 (2020) 5465–5472. doi: 10.1021/acs.nanolett.0c01973
L.F. Tietze, F. Major, I. Schuberth, Angew. Chem. Int. Ed. 45 (2006) 6574–6577. doi: 10.1002/anie.200600936
H. Elferink, W.H.C. Titulaer, M.G.N. Derks, et al., Chem. Eur. J. 28 (2022) e202103910. doi: 10.1002/chem.202103910
A.K. Winther, B. Fejerskov, M. ter Meer, et al., ACS Appl. Mater. Interfaces 10 (2018) 10741–10751. doi: 10.1021/acsami.8b01658
S. Gnaim, A. Scomparin, S. Das, et al., Angew. Chem. Int. Ed. 57 (2018) 9033–9037. doi: 10.1002/anie.201804816
M. Maiti, K. Kikuchi, K.K. Athul, et al., Chem. Commun. 58 (2022) 6413–6416.
J. Zhang, Y. Fu, R.T. Zhou, et al., Adv. Healthc. Mater. (2023) 2202421.
T. Tantra, Y. Singh, R. Patekar, et al., Curr. Med. Chem. 31 (2024) 336–357. doi: 10.2174/0929867330666230209094738
J.B. Eriksen, J.J. Christiansen, A. Bauer-Brandl, et al., Eur. J. Pharm. Sci. 181 (2023) 106366. doi: 10.1016/j.ejps.2022.106366
C.G. Zhu, R.M. Wang, W.C. Zheng, et al., Biomed. Pharmacother. 96 (2017) 551–562. doi: 10.1016/j.biopha.2017.10.006
M. Flores-Ramos, F. Ibarra-Velarde, H. Jung-Cook, et al., Bioorg. Med. Chem. Lett. 27 (2017) 616–619. doi: 10.1016/j.bmcl.2016.12.004
N.X. Duan, J.J. Li, S. Song, et al., Adv. Funct. Mater. 31 (2021) 2100605. doi: 10.1002/adfm.202100605
R. Dhankhar, A. Kawatra, A. Mohanty, P. Gulati, Curr. Protein Pept. Sci. 22 (2021) 514–525. doi: 10.2174/1389203721666201207231932
S.O. Vass, D. Jarrom, W.R. Wilson, et al., Brit. J. Cancer 100 (2009) 1903–1911. doi: 10.1038/sj.bjc.6605094
P. Ball, R. Hobbs, S. Anderson, et al., Pharmaceutics 13 (2021) 517. doi: 10.3390/pharmaceutics13040517
E.M. Williams, R.F. Little, A.M. Mowday, et al., Biochem. J. 471 (2015) 131–153. doi: 10.1042/BJ20150650
C. Jin, S.A. Wen, Q.M. Zhang, et al., ACS Med. Chem. Lett. 8 (2017) 762–765. doi: 10.1021/acsmedchemlett.7b00189
S. Yamazoe, L.E. McQuade, J.K. Chen, ACS Chem. Biol. 9 (2014) 1985–1990. doi: 10.1021/cb500429u
H.A.J. Hibbard, M.M. Reynolds, Bioorg. Chem. 93 (2019) 103318. doi: 10.1016/j.bioorg.2019.103318
K. Sharma, K. Sengupta, H. Chakrapani, Bioorg. Med. Chem. Lett. 23 (2013) 5964–5967. doi: 10.1016/j.bmcl.2013.08.066
X.F. Zhang, X.H. Li, W. Shi, H.M. Ma, Chem. Commun. 57 (2021) 8174–8177. doi: 10.1039/D1CC03232A
O. Greco, L.K. Folkes, P. Wardman, et al., Cancer Gene Ther. 7 (2000) 1414–1420. doi: 10.1038/sj.cgt.7700258
Y.H. Weng, H.H. Chen, X.Q. Chen, et al., Nat. Commun. 13 (2022) 4712. doi: 10.1038/s41467-022-32453-3
F. Wang, J. Yang, Y.S. Li, et al., J. Mater. Chem. B 8 (2020) 6139–6147. doi: 10.1039/D0TB01004A
X.Y. Wan, J.Q. Yin, Q.Q. Yan, et al., Chem. Commun. 58 (2022) 5877–5880. doi: 10.1039/D2CC00373B
H. Li, L. Chen, Y. Shi, et al., Chem. Asian J. 12 (2017) 176–180. doi: 10.1002/asia.201601198
L.K. Folkes, O. Greco, G.U. Dachs, et al., Biochem. Pharmacol. 63 (2002) 265–272. doi: 10.1016/S0006-2952(01)00868-1
A.P. Rodrigues, L.M. da Fonseca, O.M.D. Oliveira, et al., Biochim. Biophys. Acta 1760 (2006) 1755–1761. doi: 10.1016/j.bbagen.2006.09.008
W.B. Zhang, Y. Guo, J.L. Yang, et al., ACS Omega 8 (2023) 3484–3492. doi: 10.1021/acsomega.2c07511
S. Pereira, G.L. Ma, L. Na, et al., Acta Biomater. 140 (2022) 530–546. doi: 10.1016/j.actbio.2021.12.019
L.S. Zhang, J.R. Zhou, Y.C. Yan, et al., Cancer Lett. 465 (2019) 36–44. doi: 10.1016/j.canlet.2019.08.019
M. Ni, W.J. Zeng, X. Xie, et al., Chin. Chem. Lett. 28 (2017) 1345–1351. doi: 10.1016/j.cclet.2017.04.024
M. Markovic, S. Ben-Shabat, J.N. Manda, et al., Pharmaceutics 14 (2022) 675. doi: 10.3390/pharmaceutics14030675
X.R. Guan, Y. Chen, X. Wu, et al., Chem. Commun. 55 (2019) 953–956. doi: 10.1039/C8CC09047E
Q.F. Liu, X.C. Zhong, Y. Zhang, et al., Mol. Pharmaceutics 17 (2020) 1922–1932. doi: 10.1021/acs.molpharmaceut.0c00008
M. Xiao, W. Sun, J.L. Fan, et al., Adv. Funct. Mater. 28 (2018) 1805128. doi: 10.1002/adfm.201805128
F.Y. Wang, S.S. Hu, Q. Sun, et al., ACS Appl. Bio Mater. 2 (2019) 4904–4910. doi: 10.1021/acsabm.9b00655
A. Diez-Torrubia, S. Cabrera, S. de Castro, et al., Eur. J. Med. Chem. 70 (2013) 456–468. doi: 10.1016/j.ejmech.2013.10.001
M.Q. Sun, S.B. Yao, L.Y. Fan, et al., Small 18 (2022) 2106296. doi: 10.1002/smll.202106296abm.9b00655
E. Rango, L. D'Antona, G. Iovenitti, et al., Eur. J. Med. Chem. 223 (2021) 113653. doi: 10.1016/j.ejmech.2021.113653
J. Jiang, N. Shen, T.Y. Ci, et al., Adv. Mater. 31 (2019) 1904278. doi: 10.1002/adma.201904278
J. Wang, H. Wang, H. Cui, et al., Chin. Chem. Lett. 31 (2020) 3143–3148. doi: 10.1016/j.cclet.2020.07.027
N. Shim, S.I. Jeon, S.H. Yang, et al., Biomaterials 289 (2022) 121806. doi: 10.1016/j.biomaterials.2022.121806
Y. Li, T. Mei, S. Han, et al., Chin. Chem. Lett. 31 (2020) 3027–3040. doi: 10.1016/j.cclet.2020.05.027
P. Wang, H. Yang, C. Liu, et al., Chin. Chem. Lett. 32 (2021) 168–178. doi: 10.1016/j.cclet.2020.11.056
H. Kim, Y.S. Cho, S.W. Chung, et al., J. Control. Release 346 (2022) 136–147. doi: 10.1016/j.jconrel.2022.04.014
Y. Liu, W. Dong, Y.C. Ma, et al., Biomaterials 294 (2023) 122023. doi: 10.1016/j.biomaterials.2023.122023
F. Liu, X. Ding, X.B. Xu, et al., Angew. Chem. Int. Ed. 61 (2022) e202203243. doi: 10.1002/anie.202203243
M. Richter, M.M. Leuthold, D. Graf, et al., ACS Med. Chem. Lett. 10 (2019) 1115–1121. doi: 10.1021/acsmedchemlett.9b00058
D.D. Liang, L.H. Li, C. Lynch, et al., Eur. J. Med. Chem. 179 (2019) 84–99. doi: 10.1016/j.ejmech.2019.06.031
Q. Gong, X. Li, T. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202210001. doi: 10.1002/anie.202210001
Y.Y. Pu, B.G. Zhou, H.J. Xiang, et al., Biomaterials 259 (2020) 120329. doi: 10.1016/j.biomaterials.2020.120329
C.W. Li, D.C. Quenelle, M.N. Prichard, et al., Bioorg. Med. Chem. 20 (2012) 2669–2674. doi: 10.1016/j.bmc.2012.02.031
D.C. Yang, X.Z. Yang, C.M. Luo, et al., Eur. J. Med. Chem. 243 (2022) 114749. doi: 10.1016/j.ejmech.2022.114749
X.B. Zhao, W. Ha, K. Gao, Y.P. Shi, Anal. Chem. 92 (2020) 9039–9047. doi: 10.1021/acs.analchem.0c01220
X.M. Li, Y.A. Hou, X.K. Meng, et al., Angew. Chem. Int. Ed. 57 (2018) 6141–6145. doi: 10.1002/anie.201801058
Y. Zhou, C.Y. Zhang, Y.F. Wang, et al., Colloid Surf. B 220 (2022) 112853. doi: 10.1016/j.colsurfb.2022.112853
A.J. Sawdon, J. Zhang, S. Peng, et al., Molecules 26 (2021) 1759. doi: 10.3390/molecules26061759
S. Murty, L. Labanieh, T. Murty, et al., Cancer Res. 80 (2020) 4731–4740. doi: 10.1158/0008-5472.CAN-19-3579
Q. Li, P. Zou, J. Sun, L. Chen, Eur. J. Med. Chem. 143 (2018) 732–744. doi: 10.1016/j.ejmech.2017.11.081energy.2017.05.159
C. Trudeau, S. Yuan, J. Galipeau, et al., Hum. Gene Ther. 12 (2001) 1673–1680. doi: 10.1089/10430340152528165
G. Chen, B. Tang, B.Y. Yang, et al., Appl. Microbiol. Biot. 97 (2013) 4393–4401. doi: 10.1007/s00253-012-4321-8
Y.B. Sun, Y. Ke, C.S. Li, et al., J. Med. Chem. 63 (2020) 10816–10828. doi: 10.1021/acs.jmedchem.0c00149
M. Cong, G.L. Xu, S.Y. Yang, et al., Chin. Chem. Lett. 33 (2022) 2481–2485. doi: 10.1016/j.cclet.2021.11.083
T. Yang, A.S. Fruergaard, A.K. Winther, et al., Small 16 (2020) 1906744. doi: 10.1002/smll.201906744
H. Yu, F.Y. Jin, D. Liu, et al., Theranostics 10 (2020) 2342–2357. doi: 10.7150/thno.40395
L. Yu, W.L. Liu, Z.F. Zhang, Z.T. Song, Chin. Chem. Lett. 26 (2015) 700–704. doi: 10.1016/j.cclet.2015.01.039
L. Jiang, M. Tinoco, S. Fernandez-Garcia, et al., ACS Appl. Mater. Interfaces 13 (2021) 38061–38073. doi: 10.1021/acsami.1c09992
Z.J. Luo, Y.Z. Zhou, T. Yang, et al., Small 18 (2022) 2105762. doi: 10.1002/smll.202105762
L.M. Wang, J.L. Xue, J. Chang, et al., J. Mater. Sci. 56 (2021) 13579–13589. doi: 10.1007/s10853-021-06190-9
Y.W. Zhang, J. Mei, C. Yan, et al., Adv. Mater. 32 (2020) 1902806. doi: 10.1002/adma.201902806
Z.A. Zhang, K.L. Fan, Nanoscale 15 (2022) 41–62.
Z.Y. Sun, Q.Q. Liu, X.Y. Wang, et al., Theranostics 12 (2022) 1132–1147. doi: 10.7150/thno.66325
Y. Okamoto, R. Kojima, F. Schwizer, et al., Nat. Commun. 9 (2018) 1943. doi: 10.1038/s41467-018-04440-0
J. Clavadetscher, E. Indrigo, S.V. Chankeshwara, et al., Angew Chem. Int. Ed. 56 (2017) 6864–6868. doi: 10.1002/anie.201702404
W. Zhu, Z. Chen, Y. Pan, et al., Adv. Mater. 31 (2019) 1800426. doi: 10.1002/adma.201800426
R. Das, R.F. Landis, G.Y. Tonga, et al., ACS Nano 13 (2019) 229–235. doi: 10.1021/acsnano.8b05370
X.Z. Zhang, S.C. Lin, R. Huang, et al., J. Am. Chem. Soc. 144 (2022) 12893–12900. doi: 10.1021/jacs.2c04571
X.Z. Zhang, Y.C. Liu, J. Doungchawee, et al., J. Control. Release 357 (2023) 31–39. doi: 10.1016/j.jconrel.2023.03.032
R. Huang, C.M. Hirschbiegel, X.Z. Zhang, et al., ACS Appl. Mater. Interfaces 14 (2022) 31594–31600. doi: 10.1021/acsami.2c04496
X.Z. Zhang, R.F. Landis, P. Keshri, et al., Adv. Healthc. Mater. 10 (2021) 2001627. doi: 10.1002/adhm.202001627
S. Fedeli, R. Huang, Y. Oz, et al., ACS Appl. Mater. Interfaces 15 (2023) 15260–15268. doi: 10.1021/acsami.3c02640
M. Sancho-Albero, B. Rubio-Ruiz, A.M. Perez-Lopez, et al., Nat. Catal. 2 (2019) 864–872. doi: 10.1038/s41929-019-0333-4
G. Lu, R. Franzen, X.J. Yu, Y.J. Xu, Chin. Chem. Lett. 17 (2006) 461–464.
J. Li, J.T. Yu, J.Y. Zhao, et al., Nat. Chem. 6 (2014) 352–361. doi: 10.1038/nchem.1887
K. Zhan, P. Lu, J. Dong, X. Hou, Chin. Chem. Lett. 31 (2020) 1630–1634. doi: 10.1016/j.cclet.2019.09.006
R.M. Yusop, A. Unciti-Broceta, E.M.V. Johansson, et al., Nat. Chem. 3 (2011) 239–243. doi: 10.1038/nchem.981
A. Unciti-Broceta, E.M. Johansson, R.M. Yusop, et al., Nat. Protoc. 7 (2012) 1207–1218. doi: 10.1038/nprot.2012.052
J.T. Weiss, J.C. Dawson, C. Fraser, et al., J. Med. Chem. 57 (2014) 5395–5404. doi: 10.1021/jm500531z
J.T. Weiss, J.C. Dawson, K.G. Macleod, et al., Nat. Commun. 5 (2014) 3277. doi: 10.1038/ncomms4277
J.T. Weiss, N.O. Carragher, A. Unciti-Broceta, Sci. Rep. 5 (2015) 9329. doi: 10.1038/srep09329
H.J. Zhou, C.C. Mayorga-Martinez, S. Pane, et al., Chem. Rev. 121 (2021) 4999–5041. doi: 10.1021/acs.chemrev.0c01234386
M. Hoop, A.S. Ribeiro, D. Rosch, et al., Adv. Funct. Mater. 28 (2018) 1705920. doi: 10.1002/adfm.201705920
T. Chen, X. Luo, L.W. Zhu, et al., Chem. Eng. J. 467 (2023) 143386. doi: 10.1016/j.cej.2023.143386
L.Z. Feng, L. Cheng, Z.L. Dong, et al., ACS Nano 11 (2017) 927–937. doi: 10.1021/acsnano.6b07525
S.L. Lin, W.L. Tan, P.F. Han, et al., Nano Res. 15 (2022) 9073–9081.
K.G. Stamplecoskie, P.V. Kamat, J. Am. Chem. Soc. 136 (2014) 11093–11099. doi: 10.1021/ja505361n
D.J. Cheng, R. Liu, L.M. Tian, et al., ACS Appl. Nano Mater. 4 (2021) 13413–13424. doi: 10.1021/acsanm.1c02926
S. Alonso-de Castro, A.L. Cortajarena, F. Lopez-Gallego, L. Salassa, Angew. Chem. Int. Ed. 57 (2018) 3143–3147. doi: 10.1002/anie.201800288
J. Gurruchaga-Pereda, V. Martinez-Martinez, E. Rezabal, et al., ACS Catal. 10 (2020) 187–196. doi: 10.1021/acscatal.9b02863
L.F. Mazzei, L. Martinez, L. Trevisan, et al., Chem. Commun. 56 (2020) 10461–10464. doi: 10.1039/D0CC03450A
C. Ren, H.F. Liu, F.F. Lv, et al., ACS Appl. Mater. Interfaces 12 (2020) 34667–34677. doi: 10.1021/acsami.0c09489
M.Y. Sun, Z.W. Liu, L. Wu, et al., J. Am. Chem. Soc. 145 (2023) 5330–5341. doi: 10.1021/jacs.2c13010
M.M. Liu, Z.Q. Liu, G. Qin, et al., Nano Lett. 23 (2023) 4965–4973. doi: 10.1021/acs.nanolett.3c00798
J. Wu, R. Huang, T.L. Wang, et al., Org. Biomol. Chem. 11 (2013) 2365–2369. doi: 10.1039/c2ob27324a
D.B. Duan, H. Dong, Z.Y. Tu, et al., J. Am. Chem. Soc. 143 (2021) 2250–2255. doi: 10.1021/jacs.0c10399
W.T. Zhang, C. Liu, Z.W. Liu, et al., ACS Nano 16 (2022) 20975–20984. doi: 10.1021/acsnano.2c08604

Figure 1 Illustration of nanozyme-activating prodrug therapy, in which nanozymes catalyze the conversion of nontoxic prodrugs into biologically active drugs for cell death.

Figure 2 Identification and directed development of nanozymes with β-glucuronidase activity and pan-enzymatic activity for efficient prodrug conversion. (A) High-throughput screen of nanozymes identifying nanoparticles with β-glucuronidase activity based on metals, metal oxides and other nanomaterials using resorufin β-D-glucuronide as an enzyme-specific fluorogenic probe, which can be converted into the corresponding fluorescent product, resorufin. (B) Substrate scope of the copper nanozyme as a β-glucuronidase mimic is closely related to the quality of the leaving group in the substrate: high performance liquid chromatography (HPLC) characterization of conversion for glucuronide prodrugs with increasing quality of the leaving group from (ⅰ) to (ⅳ). (C) The "Zone of inhibition" experiment reveals that nanoengineered copper nanozymes and antibacterial prodrugs afford a strong antibacterial effect. Reproduced with permission [28]. Copyright 2019, Wiley-VCH.

Figure 3 Investigation of the ceria nanozyme as a phosphatase mimic for efficient conversion of phosphate prodrugs into corresponding therapeutics. (A) Comparative analysis of the substrate scope of alkaline phosphatase (2 mg/L, 2 h) and CeO2 nanozyme (0.125 g/L, 24 h) with regard to hydrolysis of organic phosphates. (B) Schematic illustration of synthesis of "extended scaffold phosphate" prodrugs for moxifloxacin and monomethyl auristatin E (MMAE). Reproduced with permission [33]. Copyright 2021, American Chemical Society.

Figure 4 The synthesis of novel minimal metallo-nanozymes with hydrolase-like catalytic behavior via an amino acid coordinated self-assembly strategy by using zinc(Ⅱ) ions and amino acid derivatives as the building blocks, which can catalyze the hydrolysis of p-nitrophenyl acetate and benorilate to produce p-nitrophenol and acetylsalicylic acid, respectively. Reproduced with permission [31]. Copyright 2020, Elsevier.

Figure 5 (A) Bioorthogonal nanozyme design and supramolecular regulation of intracellular catalysis. (ⅰ) AuNP, AuNP-encapsulated catalyst, and AuNP-encapsulated CB[7]-capped catalyst used in this study; (ⅱ) Endosomal uptake of AuNP-encapsulated CB[7]-capped catalysts; (ⅲ) Intracellular catalysis with AuNP-encapsulated catalysts allowing the conversion of substrate into product; (ⅳ) The complexation of CB[7] with the ligand headgroup induces complete inhibition of catalytic activity; (ⅴ) The catalytic activity is restored by addition of 1-adamantylamine; (ⅵ) Structures of AuNP with the surface ligand bearing a dimethylbenzylammonium headgroup, 1-adamantylamine, and CB[7]. (ⅶ) TMCs catalyzes allylcarbamate cleavage of pro-fluorescent substrate (rhodamine 110 derivative). Reproduced with permission [26]. Copyright 2015, Nature Publishing Group. (B) The synthesis of surface-engineered nanozymes (cationic nanozymes (Pos-NZ) and zwitterionic nanozymes (Zw-NZ), which can catalyze activation of alloc-protected rhodamine 110 and alloc-protected resorufin, respectively) that enable control of intra- versus extracellular bioorthogonal catalysis. Reproduced with permission [140]. Copyright 2018, American Chemical Society.

Figure 6 The development of bioorthogonally activated prodrug approaches based on the palladium chemistry. (A) The use of Pd0-microspheres that enable mediating efficient bioorthogonal organometallic chemistries. (ⅰ) The Pd0-microspheres enable catalyzing the allylcarbamate cleavage of pro-fluorescent substrate (rhodamine 110 derivative). (ⅱ) The Pd0-microspheres enable catalyzing the Suzuki-Miyaura cross-coupling reaction. Reproduced with permission [150]. Copyright 2011, Nature Publishing Group. (B) The use of Pd0-resins for catalyzing the activation of Pd0-labile gemcitabine prodrugs in cell culture (ⅰ), rhodamine precursors in zebrafish (ⅱ), Reproduced with permission [152]. Copyright 2022, American Chemical Society. Pro-5-FU (ⅲ), Reproduced with permission [153]. Copyright 2014, Nature Publishing Group. as well as another propargyl-protected prodrug, Pro-FUdR (ⅳ). Reproduced with permission [154]. Copyright 2015, Nature Publishing Group. (C) The synthesis of mobile magnetic nanocatalysts (FePd nanowires, which are capable of site-specific activation of the latent chemotherapeutic prodrug Pro-5-FU) for bioorthogonal targeted cancer therapy. Reproduced with permission [156]. Copyright 2018, Wiley-VCH.

Figure 7 The activation of IAA catalyzed by a metal-free nanozyme, PNCNzyme, for targeted tumor catalytic therapy. (A) Schematic illustration of synthesis of the therapeutic reagents, FA-PNCNzymes@IAA. (B) The prodrug activation strategy for tumor catalytic therapy of FA-PNCNzymes@IAA. Reproduced with permission [30]. Copyright 2020, Elsevier.

Figure 8 Starvation, ferroptosis, and prodrug therapy synergistically enabled by the use of a cytochrome c oxidase like nanozyme (Cu-Ag NP) and AQ4N (a prodrug that can be converted into a cytotoxic DNA intercalator (AQ4) by the hypoxia condition in tumor tissues). Reproduced with permission [29]. Copyright 2022, Wiley-VCH.

Figure 9 (A) GSH-modified Au NCs perform enzyme-like photocatalysis for catalyzing the reduction of a prodrug moiety nitrobenzene to biologically active aniline under light. (ⅰ) Comparative illustration of nitroreductase (left) along with Au NC photonanozymes (right) for the activation of nitrobenzene; (ⅱ) The catalytic mechanism of Au NCs-catalyzed the reduction of nitrobenzene. Reproduced with permission [32]. Copyright 2021, American Chemical Society. (B) Photocatalytic nitroaromatic prodrug activation by photosensitizer functionalized AuNCs. (ⅰ) Schematic illustration of the synthesis of the photosensitizer functionalized AuNCs. (ⅱ) The mechanism of the photosensitizer functionalized Au NCs for photocatalytic and photodynamic double killing of cancer cells. (ⅲ) Schematic illustration of the generation of reactive oxygen species (ROS) and the photocatalytic reduction of the prodrug moiety nitrobenzene triggered by the photosensitizer functionalized AuNCs. Reproduced with permission [161]. Copyright 2021, American Chemical Society.

Figure 10 Flavin bioorthogonal photocatalysis toward platinum substrates. (A) With the aid of NADH as the electron donor, flavins can be transformed into the reduced FLH− species under light irradiation, which serves as the active photocatalyst to catalyze the PtⅣ to PtⅡ conversion. Reproduced with permission [163]. Copyright 2019, American Chemical Society. (B) The mechanism of FMN for the activation of the cisplatin prodrug cis, cis, -trans-[Pt(NH3)2(Cl2)(O2CCH2CH2COOH)2]. (C) Structures of FMN and AuNPs decorated with a C11-thiol bearing a 1, 4, 7-triazacyclononane headgroup. (D) Schematic illustration of the supramolecular nanozyme developed in this work. Reproduced with permission [164]. Copyright 2020, Royal Society of Chemistry.

Figure 11 (A) A novel therapeutic platform for multisynergistic cancer therapy based on taking full advantages of the GOD-based β-D-glucose oxidation and palladium mediated in situ activation of doxorubicin prodrugs. Reproduced with permission [165]. Copyright 2020, American Chemical Society. (B) The design and synthesis of a COF-based Fe(Ⅱ) catalyst as a bioorthogonal catalyst for the bioorthogonal-activated in situ cancer vaccine. Reproduced with permission [166]. Copyright 2023, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: