Citation:
Min Song, Qian Zhang, Tao Shen, Guanyu Luo, Deli Wang. Surface reconstruction enabled o-PdTe@Pd core-shell electrocatalyst for efficient oxygen reduction reaction[J]. Chinese Chemical Letters,
2024, 35(8): 109083.
doi:
10.1016/j.cclet.2023.109083
Key Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology (HUST), Wuhan 430074, China
Received Date:
25 June 2023 Accepted Date:
11 September 2023 Revised Date:
23 July 2023 Available Online:
15 August 2024
Abstract:
Palladium-based alloy catalysts have been employed as one of the potential candidates for oxygen reduction reaction (ORR), but the dissolution of transition metal hinders their application. Herein, structure ordered PdTe intermetallic with Pd shell (o-PdTe@Pd) are synthesized via an electrochemical etching driven surface reconstruction strategy. The surface reconstruction could tune the electronic structure, weaken the adsorption energy of reaction intermediates on o-PdTe@Pd, resulting in enhanced electrocatalytic activity for ORR. The mass activity of o-PdTe@Pd is about 3.3 and 2.7 times higher than that of Pd/C in acid and alkaline, respectively. Besides, the half-potentials for ORR decay only about 44 mV and 12 mV after 30 k cycles accelerated durability test in acid and alkaline media, respectively. The enhanced durability originates from the resistance of Te atoms dissolve in the ordered PdTe intermetallic core and the core-shell structure. When assembled in a Zn-air battery, o-PdTe@Pd electrode delivers a higher specific capacity (794 mAh/g) and better cycling stability than Pt/C.
Developing green energy, such as hydrogen energy, is crucial to overcome the global energy crisis and climate change. The application of hydrogen energy in fuel cells is an important indicator for achieving hydrogen economy [1,2]. Oxygen reduction reaction (ORR) has been extensively investigated over the past decades as one of the most important reactions for fuel cells [3–6]. However, the sluggish kinetics of ORR greatly limit its commercial application. Highly active and durable electrocatalysts are required to compensate the high overpotential mainly caused by the sluggish kinetics of ORR [7,8]. Pt-based catalysts are currently the first choice for commercial application due to their excellent activity and durability. Unfortunately, the reserves and prices of Pt limit its commercialization [9–13]. In recent years, numerous studies have attempted to develop low-platinum or non-platinum catalysts to address this challenge.
Pd-based catalysts have great potential for ORR due to their similar electronic properties to Pt and better methanol tolerance than Pt [14–16]. However, the strong binding energy of oxygen on the surface of pure Pd catalyst hinders the desorption of the reaction intermediates [17–19], thus it is difficult to achieve similar ORR performance as Pt. Alloying Pd with 3d-transition metals (M) to form PdM alloy is a feasible strategy to enhance the ORR activity by modulating the electronic structure [20,21], but the dissolution of transition metals results in poor durability. Constructing core-shell structure with Pd shell could enhance the durability by protecting the inner M atoms from dissolution during electrochemical test [22–24]. On the other hand, ordered Pd-based intermetallic compounds have attracted attention because it is generally accepted that intermetallic compounds exhibit improved durability compared to disordered alloy due to the strong interaction between Pd and M in intermetallic. In addition, intermetallic compounds have definite composition and atomic arrangement that can accurately regulate the activity [25–29]. Inspired by this, it is desirable to construct ordered Pd-based intermetallic with a core-shell structure.
Tellurium (Te), a metalloid element with high electronegativity, can easily modulate the electronic structure of active sites, resulting in stronger tunability for Pd [30,31]. However, the research on Pd-Te electrocatalysts for ORR is rare. Herein, an ordered PdTe intermetallic was synthesized by a simple hydrothermal method, and obtained a o-PdTe@Pd core-shell structure via an electrochemical etching strategy. The leaching of Te results in the rearrangement of the surface atoms, which enhances the ORR activity. In addition, the durability of o-PdTe@Pd electrocatalyst is enhanced by the formation of core-shell structure and the strong interaction between Pd and Te in ordered PdTe intermetallic core. More importantly, o-PdTe@Pd shows better cycling stability as a cathode catalyst for Zn-air batteries.
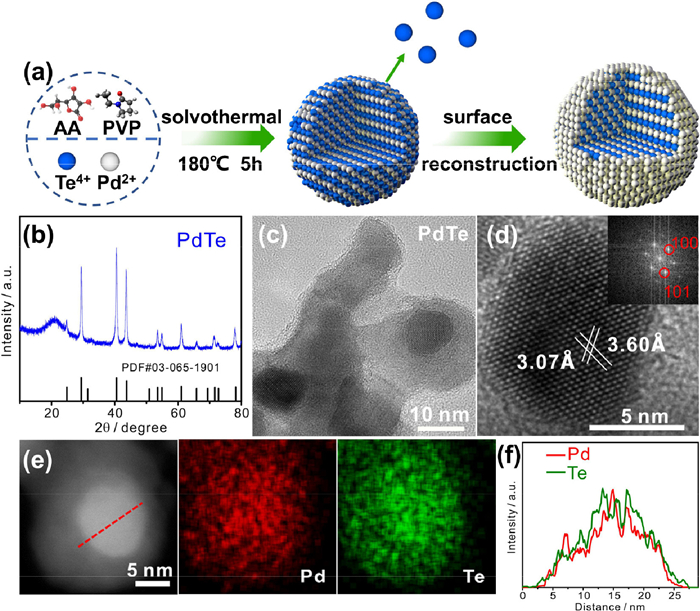
The ordered PdTe intermetallic electrocatalysts were prepared via a solvothermal method using palladium chloride and potassium tellurite as raw materials, polyvinyl pyrrolidone (PVP) as surfactant and ascorbic acid as reducing agent (Fig. 1a). The resulting product was harvested through centrifugation utilizing a mixture of ethanol and acetone. X-ray diffraction (XRD) was conducted to characterize the crystal structure of the synthesized catalyst. It can be observed in Fig. 1b that all the peaks match well with the hexagonal phase PdTe (hcp-PdTe, PDF#03–065–1901) [32], indicating that the prepared PdTe has ordered structure. The transmission electron microscope (TEM) was utilized to characterize the morphology and structure of the prepared catalysts. As shown in Fig. S1 (Supporting information) and Fig. 1c, the particles are dispersed on the carbon support with an average size of approximately 6 nm. The high resolution TEM (HRTEM) image in Fig. 1d shows that the calculated lattice spacing of 3.07 Å and 3.60 Å correspond to the (101) and (100) facets of hcp-PdTe, which matches well with the lattice spacing of (101) and (100) in XRD. Moreover, we also analysed the distribution of Pd and Te elements by energy dispersive X-ray spectroscopy (EDX) mapping and line scan techniques. The EDX mapping in Fig. 1e indicates that Pd and Te are uniformly dispersed on the surface of the catalysts, and the line-scan result in Fig. 1f confirms that the atomic ratio of Pd and Te is close to 1:1.
Figure 1
Figure 1.
(a) Schematic diagram of the synthesis for o-PdTe@Pd. (b) XRD pattern, (c) TEM image and (d) HRTEM image of PdTe. Inset shows the corresponding FFT image. (e) STEM and corresponding elemental mappings of PdTe. (f) EDX line scan across the PdTe as indicated by the dashed line in (e).
The surface reconstruction was conducted through electrochemical etching by cyclic voltammetry (CV) in the potential range between 0.05 and 1.2 V. The Te elements were precipitated from the surface of PdTe, resulting in composition change and the structural evolution, as verified by the CV curves in Fig. S2 (Supporting information). At the initial state, the hydrogen adsorption/desorption peaks in the lower potential region (0.05–0.4 V) are not observed, indicating that there are few continues Pd active sites on the surface of PdTe. However, a pair of strong redox peaks between 0.8 V and 1.2 V is obviously observed that are assigned to the redox of Te [33]. In contrast, two distinct peaks appeared in the low potential region after 100 potential cycles, which is related to the adsorption/desorption of H on the continuous Pd active sites. Moreover, the current response of Te oxidation is weakened. The peaks of the H adsorption/desorption are gradually obvious, while the redox peaks of Te gradually decrease during the etching process. This change reveals that most of the surface of PdTe is occupied by Pd atoms, in other words, o-PdTe@Pd core-shell structure with Pd-rich surface are formed during the etching process.
The HRTEM was further conducted to perform the structural evolution of the catalysts. As shown in Figs. S3a and b (Supporting information), the lattice distance at the inner region is 3.03 Å, corresponding to the (101) facet of hcp-PdTe. However, the lattice distance at the edge is 2.00 Å, which matches with the (200) facet of Pd. The elemental mapping and line-scan in Figs. S3c and d (Supporting information) further verified the core-shell structure (o-PdTe@Pd). The atomic ratio of Pd and Te increased to 64.3:36.7, higher than that of initial PdTe intermetallic (Fig. S3e in Supporting information).
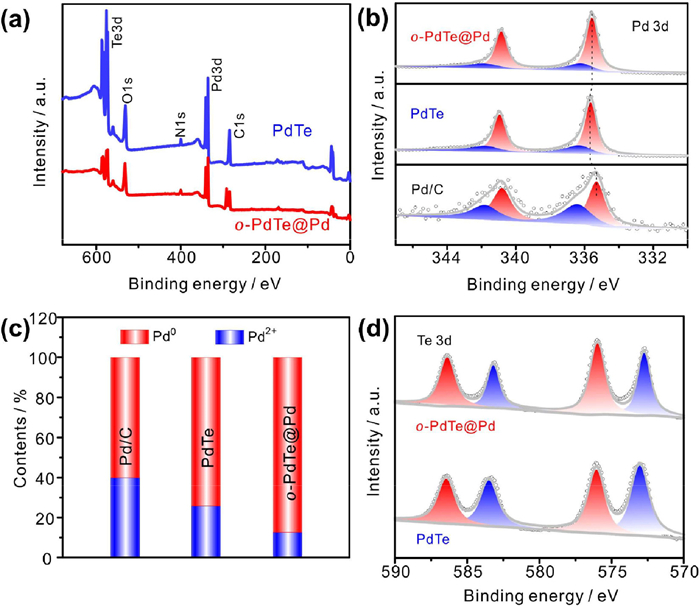
X-ray photoelectron spectroscopy (XPS) was performed to analyze the surface structure and chemical state of catalysts. It demonstrates that the content of Te in o-PdTe@Pd is greatly reduced relative to that in PdTe (Fig. 2a). The Pd 3d XPS spectrum was fitted into metallic state Pd0 and oxidation state Pd2+. As shown in Fig. 2b, the peaks at 335.7 eV and 341.0 eV of PdTe are assigned to Pd0. The binding energy of Pd0 in PdTe is positively shifted compared to that in Pd/C, indicating the electronic structure of Pd was tuned by Te element. The peaks at 336.2 eV and 341.9 eV correspond to Pd2+ arising from surface oxidation. It can be observed from Fig. 2c that the oxidation of Pd on the surface of PdTe is reduced than Pd/C, which is attributed to the strong interaction between Pd and Te in PdTe intermetallic. Meanwhile, the binding energy of Pd0 in o-PdTe@Pd is negatively shifted relative to that in PdTe due to the formation of metallic Pd shell on the surface. The change in the binding energy of Pd0 in all samples indicates that structural evolution leads to different electronic structure. In addition, the formation of Pd shell further prevents the oxidation of Pd in o-PdTe@Pd (Fig. 2c). The red and blue filled peaks in Fig. 2d are attributed to Te4+ and Te0. The strong covalent nature of Pd-Te bond causes Te exhibiting an oxidation state of 0 to +4 instead of −2 in Fig. 2d. The change of electronic structure will enhance the ORR activity caused by the weakened adsorption energy of the reaction intermediates on the active sites. In addition, the strong interaction between Pd and Te in PdTe intermetallic and the core-shell structure may enhance the structure stability of the catalyst.
Figure 2
Figure 2.
(a) XPS survey spectra of PdTe and o-PdTe@Pd. (b) Pd 3d of Pd/C, PdTe and o-PdTe@Pd. (c) The contents of Pd species in Pd/C, PdTe and o-PdTe@Pd. (d) Te 3d of PdTe and o-PdTe@Pd.
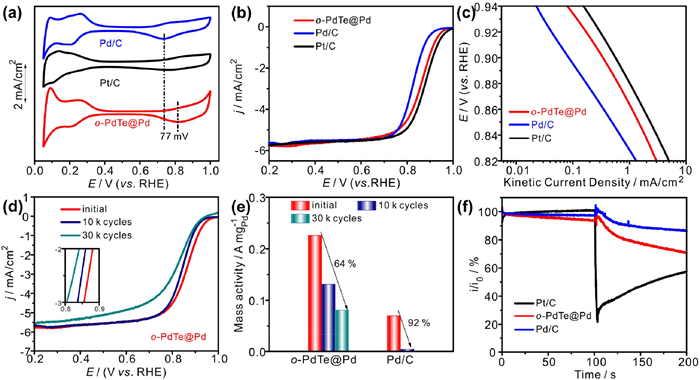
o-PdTe@Pd was evaluated in 0.1 mol/L HClO4 to investigate the effect of the structural change on the electrochemical performance, Pt/C and Pd/C were also evaluated under the same condition as benchmarks. CV curves were measured to reveal the surface properties of the prepared catalysts. As shown in Fig. 3a, o-PdTe@Pd exhibits obvious hydrogen adsorption/desorption peaks similar to those in Pd/C, hinting the continuous Pd is exposed on the surface of o-PdTe@Pd. According to the literature [34,35], the potential of the adsorbed hydroxyl oxidation peaks reflects the binding strength of oxygenated species on the surface of the catalyst, and the more positive potential contributed to the weaker binding strength of oxygenated species. The onset potential for the oxidation peak of adsorbed hydroxyl on o-PdTe@Pd positively shifts relative to Pd/C, revealing a weaker oxygen binding energy on the o-PdTe@Pd surface, which is benefit to enhance the ORR activity.
Figure 3
Figure 3.
(a) CV curves, (b) LSV curves and (c) Tafel plots of Pd/C, Pt/C and o-PdTe@Pd. (d) LSV curves of o-PdTe@Pd after ADT. (e) Comparison of MA at 0.85 V of o-PdTe@Pd and Pd/C before and after ADT. (f) Methanol tolerance of o-PdTe@Pd, Pd/C and Pt/C.
The ORR polarization curves were measured under O2-saturated 0.1 mol/L HClO4 (Fig. S4 in Supporting information and Fig. 3b). It can be seen that the ORR performance increases with electrochemical etching. The o-PdTe@Pd was formed after 300 potential cycles, which displays a higher onset potential than Pd/C, and the half-potential of o-PdTe@Pd positively shifts 35 mV relative to Pd/C (Fig. 3b and Fig. S5 in Supporting information). The Tafel plots (potential versus kinetic current density) displayed in Fig. 3c show that o-PdTe@Pd has higher kinetic current densities than Pd/C, indicating faster reaction kinetics. To evaluate the intrinsic activity of o-PdTe@Pd, the catalytic activity was further normalized by the mass of noble metal and ECSA, respectively. The loading of Pd was determined by Inductively coupled plasma atomic emission spectrometer (ICP-AES) and determined to be 26%. The mass activity and specific activity of o-PdTe@Pd is 0.23 A/mgPd and 0.37 mA/cm2 at 0.85 V, superior to that of Pd/C (0.07 A/mgPd and 0.15 mA/cm2) as shown in Figs. S5 and S6 (Supporting information). Table S1 (Supporting information) lists the E1/2 and mass activity of the prepared catalyst and state-of-art catalysts in acid medium, o-PdTe exhibits better ORR performance than most reported catalysts. The increased ORR performance of o-PdTe@Pd is probably attributed to the surface reconstruction, which can efficiently tune the electronic structure of Pd and then weaken the adsorption energy of the reaction intermediates, thereby modulate the ORR activity.
In order to further investigate the reaction kinetics of o-PdTe@Pd, the ORR polarization curves were measured under different rotation rates. As shown in Fig. S7d–f (Supporting information), the Koutecky-Levich plots at 0.75, 0.775 and 0.8 V exhibit good linearity. The slope of these plots remains approximately constant, indicating the electron transfer numbers for ORR are similar at the overall potential range, and the average electron transfer number (n) of o-PdTe@Pd is close to theoretical value (4.0). Moreover, the electrochemical durability of Pd/C and o-PdTe@Pd was evaluated by accelerated durability tests (ADT) between 0.6 V and 1.0 V. It can be seen in Fig. 3d and Figs. S8 and S9 (Supporting information) that o-PdTe@Pd shows smaller attenuation than Pd/C after 10 k cycles, and the half-potential of o-PdTe@Pd decays 44 mV after 30 k cycles. Moreover, o-PdTe@Pd still exhibits higher MA with a smaller decay rate of 64% after 30 k cycles in Fig. 3e, while Pd/C shows a decay rate of 92% after just 10 k cycles. The ADT results reveal that o-PdTe@Pd exhibits better stability than Pd/C, both ordered intermetallic structure and the core-shell structure may contribute to the enhanced stability by inhibiting the dissolution of Te elements. Furthermore, methanol tolerance was studied by monitoring the current change after adding methanol in the chronoamperometric test. As displayed in Fig. 3f, o-PdTe@Pd can maintain a higher current than Pt/C catalysts, indicating o-PdTe@Pd owns better methanol tolerance capability.
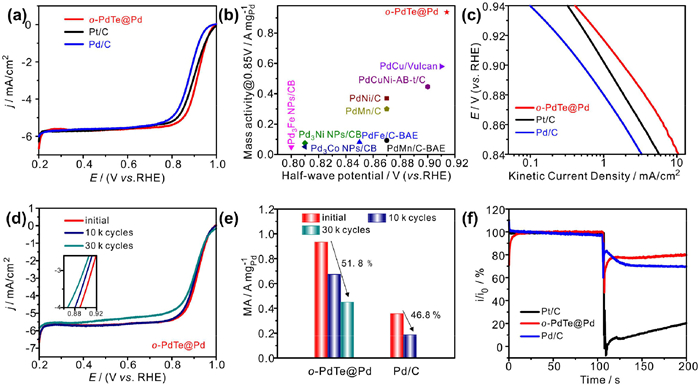
Except for acid electrolyte, the ORR activity and durability of o-PdTe@Pd were also evaluated in alkaline electrolyte. As shown in Fig. 4a, o-PdTe@Pd exhibits high activity with a positively shift half-potential to 0.914 V, which is higher than that of Pt/C (0.890 V) and Pd/C (0.873 V). The mass activity of o-PdTe@Pd is 0.934 A/mgPd at 0.85 V, superior to that of Pt/C (0.626 A/mgPt) and Pd/C (0.345 A/mgPd) in Fig. S10 (Supporting information), and also takes the leading position among the report catalysts in Fig. 4b and Table S2 (Supporting information). The Tafel plots are calculated from the polarization curves to reveal the ORR kinetics. As displayed in Fig. 4c, o-PdTe@Pd exhibits higher kinetic current density than others, hinting the fast reaction kinetics on o-PdTe@Pd. The LSV curves of o-PdTe@Pd at different rotating speeds were performed to determine the electron transfer number. It can be seen that the average electron transfer number of o-PdTe@Pd is ~4.0 according to the Koutecky-Levich plots in Fig. S11 (Supporting information), indicating that oxygen was completely reduced through the four-electron pathway. And the good linearity of the Koutecky-Levich plots further revealed the first-order reaction kinetics [36]. ADT was conducted for 30 k cycles to assess the long-term stability of o-PdTe@Pd in alkaline medium. It can be seen that the half-potential only drops 8 mV and 12 mV after 10 k cycles and 30 k cycles in Fig. 4d and Fig. S12b (Supporting information). The mass activity of o-PdTe@Pd only decays 28%, lower than Pd/C (46.8%) after 10 k cycles in Fig. 4e. In addition, the mass activity only decays 51.8% after 30 k cycles. The methanol tolerance of o-PdTe@Pd was also tested in Fig. 4f, and it was found that o-PdTe@Pd can retain 80% current density after adding methanol. These results manifest that o-PdTe@Pd shows superior activity, durability and methanol tolerance in alkaline.
Figure 4
Figure 4.
(a) LSV curves of o-PdTe@Pd, Pt/C and Pd/C catalysts. (b) Comparison of MA for o-PdTe@Pd and the reported catalysts. (c) The tafel slops of o-PdTe@Pd, Pd/C and Pt/C. (d) LSV curves of o-PdTe@Pd before and after 10 k cycles. (e) The change of mass activity on o-PdTe@Pd and Pd/C after ADT. (f) Methanol tolerance of o-PdTe@Pd, Pd/C and Pt/C.
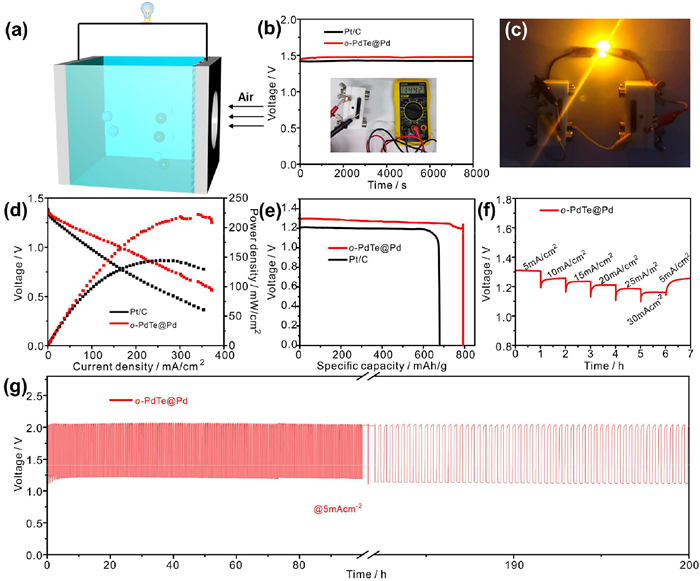
Benefiting from the excellent ORR performance of o-PdTe@Pd in alkaline electrolytes, a zinc-air battery was constructed utilizing o-PdTe@Pd/RuO2 as the cathode and polished zinc sheet as anode (Fig. 5a). The commercial Pt/C/RuO2 was also assembled for comparison. As shown in Fig. 5b, the zinc-air battery assembled with o-PdTe@Pd exhibits a higher open circuit potential (OCP) of 1.44 V for an extended period, surpassing that of Pt/C. The light-emitting diode (LED, with a rated voltage of 1.8-2.2 V) was powered by two connected Zn-air batteries (Fig. 5c). Additionally, the fan with a rated voltage of 1.2–1.5 can be powered by one o-PdTe@Pd Zn-air battery in Movie S1 (Supporting information). The discharge polarization curves and their corresponding power densities were examined to reveal the discharge performance of the o-PdTe@Pd. As shown in Fig. 5d, o-PdTe@Pd-based battery exhibits a peak power density of 222 mW/cm2, which is greater than that of the Pt/C-based battery (144 mW/cm2). After normalizing the mass of consumed Zn foil, the o-PdTe@Pd-based battery exhibits a specific capacity of 794 mAh/g in Fig. 5e, which is superior to that of Pt/C. Furthermore, the o-PdTe@Pd-based battery exhibits excellent rate capability at different discharge current densities in Fig. 5f, which further reveals the facilitated reaction kinetics. The operation durability of o-PdTe@Pd-based Zn-air battery was also investigated at 5 mA/cm2 (10 min for discharge and 10 min for charge). It can be seen in Fig. 5g that nearly no loss was observed after cycling for 200 h. Hence, the o-PdTe@Pd is an efficient ORR catalyst and can be used for Zn-air batteries.
Figure 5
Figure 5.
(a) Schematic diagram of Zn-air battery. (b) Long-term circle potential testing of o-PdTe@Pd-based and Pt/C-based batteries. The inset is the OCP recorded by a multimeter. (c) Optical images of an LED (1.8–2.2 V) driven by two Zn-air batteries. (d) LSV curves and their corresponding power density curves of Zn-air batteries. (e) Discharge curves of o-PdTe@Pd tested at 5 mA/cm2. (f) Rate capacity under different current density. (g) Long-term galvanostatic discharge and charge curves of o-PdTe@Pd at 5 mA/cm2.
In conclusion, the ordered o-PdTe@Pd intermetallic core-shell structure catalysts were successfully synthesized through surface reconstruction of PdTe. Due to the changes in surface atomic arrangement and electronic structure, o-PdTe@Pd exhibits enhanced ORR activity with 3.3 times and 2.7 times higher mass activity than Pd/C in both acidic and alkaline medium. In addition, the strong interaction between Pd and Te in ordered PdTe intermetallic and core-shell structure could alleviate the dissolution of Te element during ADT, thereby improving the durability. The better activity and durability enable o-PdTe@Pd significantly improved cyclability (500 cycles) and specific capacity (794 mAh/gZn) in Zn-air battery. This work provides a new way to design efficient and durable Pd-based catalysts for fuel cells.
Declaration of competing interest
The authors declare no competing financial interest.
Acknowledgments
This work was supported by the National Natural Science Foundation (No. 22279036) and the Innovation and Talent Recruitment Base of New Energy Chemistry and Device (No. B21003). We thank the Analytical and Testing Center of Huazhong University of Science and Technology for allowing us to use its facilities for XRD, XPS, and TEM measurements.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109083.
[1]
I. Staffell, D. Scamman, A. Velazquez Abad, et al., Energy Environ. Sci. 12 (2019) 463–491. doi: 10.1039/C8EE01157E
[2]
J. Cui, Q. Chen, X. Li, S. Zhang, Green Chem. 23 (2021) 6898–6925. doi: 10.1039/D1GC01040A
[3]
X. Huang, Z. Zhao, L. Cao, et al., Science 348 (2015) 1230–1234. doi: 10.1126/science.aaa8765
[4]
W. Xiong, F. Du, Y. Liu, et al., J. Am. Chem. Soc. 132 (2010) 15839–15841. doi: 10.1021/ja104425h
[5]
M. Shao, Q. Chang, J. Dodelet, R. Chenitz, Chem. Rev. 116 (2016) 3594–3657. doi: 10.1021/acs.chemrev.5b00462
[6]
Y. Gao, D. Zheng, Li Q, et al., Adv. Funct. Mater. 32 (2022) 2203206. doi: 10.1002/adfm.202203206
Y. Zhang, B. Huang, G. Luo, et al., Sci. Adv. 6 (2020) eaba9731. doi: 10.1126/sciadv.aba9731
[34]
J. Hu, K.A. Kuttiyiel, K. Sasaki, et al., Catalysts 5 (2015) 1321–1332. doi: 10.3390/catal5031321
[35]
Y. Xiong, Y. Yang, F.J. DiSalvo, H.C.D. Abruña, J. Am. Chem. Soc. 140 (2018) 7248–7255. doi: 10.1021/jacs.8b03365
[36]
R. Liu, D. Wu, X. Feng, K. Müllen, Angew. Chem. Int. Ed. 122 (2010) 2619–2623. doi: 10.1002/ange.200907289
Figure 1
(a) Schematic diagram of the synthesis for o-PdTe@Pd. (b) XRD pattern, (c) TEM image and (d) HRTEM image of PdTe. Inset shows the corresponding FFT image. (e) STEM and corresponding elemental mappings of PdTe. (f) EDX line scan across the PdTe as indicated by the dashed line in (e).
Figure 2
(a) XPS survey spectra of PdTe and o-PdTe@Pd. (b) Pd 3d of Pd/C, PdTe and o-PdTe@Pd. (c) The contents of Pd species in Pd/C, PdTe and o-PdTe@Pd. (d) Te 3d of PdTe and o-PdTe@Pd.
Figure 3
(a) CV curves, (b) LSV curves and (c) Tafel plots of Pd/C, Pt/C and o-PdTe@Pd. (d) LSV curves of o-PdTe@Pd after ADT. (e) Comparison of MA at 0.85 V of o-PdTe@Pd and Pd/C before and after ADT. (f) Methanol tolerance of o-PdTe@Pd, Pd/C and Pt/C.
Figure 4
(a) LSV curves of o-PdTe@Pd, Pt/C and Pd/C catalysts. (b) Comparison of MA for o-PdTe@Pd and the reported catalysts. (c) The tafel slops of o-PdTe@Pd, Pd/C and Pt/C. (d) LSV curves of o-PdTe@Pd before and after 10 k cycles. (e) The change of mass activity on o-PdTe@Pd and Pd/C after ADT. (f) Methanol tolerance of o-PdTe@Pd, Pd/C and Pt/C.
Figure 5
(a) Schematic diagram of Zn-air battery. (b) Long-term circle potential testing of o-PdTe@Pd-based and Pt/C-based batteries. The inset is the OCP recorded by a multimeter. (c) Optical images of an LED (1.8–2.2 V) driven by two Zn-air batteries. (d) LSV curves and their corresponding power density curves of Zn-air batteries. (e) Discharge curves of o-PdTe@Pd tested at 5 mA/cm2. (f) Rate capacity under different current density. (g) Long-term galvanostatic discharge and charge curves of o-PdTe@Pd at 5 mA/cm2.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
