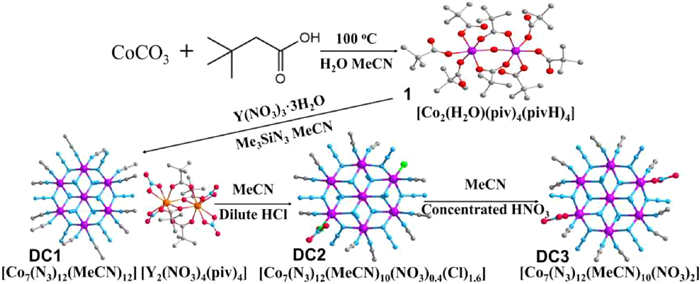
Scheme 1.
Synthetic route of complexes DC1-DC3.
Structural evolution and zero-field SMM behaviour in ferromagnetically-coupled disk-type Co7 clusters bearing exclusively end-on azido bridges
Yijia Jiao , Yuzhu Li , Yuting Zhou , Peipei Cen , Yi Ding , Yan Guo , Xiangyu Liu

As a kind of basically structural units existing in discrete or extended systems, paramagnetic multi-metal cluster with the nuclear number over five is usually an intriguing research frontier due to their attractive motifs [1–3], prospective applications in diverse research fields, such as magnetic materials [4], optical materials [5], and catalyst [6]. Particularly, magnetic molecule, high-spin molecule [7] and single-molecule magnet (SMM) [8–11] are doubtlessly two nontrivial research fields where multinuclear 3d transition metal complexes are regarded as an indispensable constituent. One of the challenges in this field is that the currently reported 3d-metal clusters are more complex in the self-assembly process, with variable structures and poor controllability. The general synthetic approach usually offered plenty of structurally unpredictable metal clusters, such that the cognitions of the assembly process and mechanism are still ambiguous [12–16].
It is well-known that multi-spin CoⅡ-clusters have becoming appealing candidate for molecular magnetism especially for SMM, due to its significant magnetic anisotropy and large spin-orbit coupling [17–22]. However, among the reported polynuclear CoⅡ-clusters [23–25], SMM behaviour without an extra magnetic field was still difficult to be observed above 2 K [26,27]. An important reason is that the competent ferromagnetic (F) interaction is absent to give a large spin ground state, as oxygen-containing bridges usually tend to transfer antiferromagnetic (AF) coupling. It has been reported that azido (N3−) ligand is one of the versatile, flexible moieties in the fabrication of coordination polymers, qualified for connecting diverse metal ions and giving rise to attractive networks with remarkable magnetisms [28–31]. In principle, end-on (EO) azido bridge is capable of promoting F coupling between metal centers, thus causing SMM with spin-canting behaviour or long-range ordering [32–34].
In most known 3d-metal clusters, azido ligand has been predominantly employed in combination with other organic bridges/chelates [35,36]. Although essential for the crystallinity and thermodynamic stability of the preparing complexes, the attendance of other bridging/chelating ligands often impacts the dynamic magnetic relaxations of the resulting complexes, producing competing AF coupling, among others [37–39]. A potential strategy to overcome the problem and fabricate strong ferromagnetic system is the development of promising synthetic methods for the construction of polynuclear azido-metal clusters without requiring the coexistence of other bridging/chelating ligands [40]. Krause et al. suggested an exercisable solution for the assembly of structurally and magnetically impressive multinuclear NiⅡ complexes by using Me3SiN3 as the azido-bridge pioneer in the absence of any bridging/chelating ligands [41–44]. Inspired by this case, we demonstrate an effective route to obtain such a unique class of ferromagnetic species, N-rich and O-free disk-like CoⅡ-clusters with the attendance of SMM behaviour in zero dc-field. More importantly, we also show that access to the structural manipulation of polynuclear CoⅡ-clusters becomes feasible through the exploration of the modulating mechanism on coordination microenvironment around terminal metal ions.
Through carefully adjusting the reaction conditions, a known complex [Co2(H2O)(piv)4(pivH)4] (1) [45] prepared by CoCO3 and pivalic acid was used to mix with Y(NO3)3·3H2O in a molar ratio of 2:1 in MeCN, in excess of Me3SiN3, leading to a deep purple solution, which upon diffusion with Et2O, yielded the purple crystals of a new complex [Co7(N3)12(CH3CN)12] [Y2(NO3)4(piv)4]·2CH3CN (DC1) (pivH = pivalic acid), of which the crystal samples react with dilute hydrochloric acid in MeCN giving complex [Co7(N3)12(CH3CN)10(NO3)0.4(Cl)1.6]·4CH3CN (DC2). Furthermore, complex [Co7(N3)12(CH3CN)10(NO3)2]·4CH3CN (DC3) could be yielded by dissolving the DC2 crystals in MeCN with the addition of concentrated nitric acid (Scheme 1).
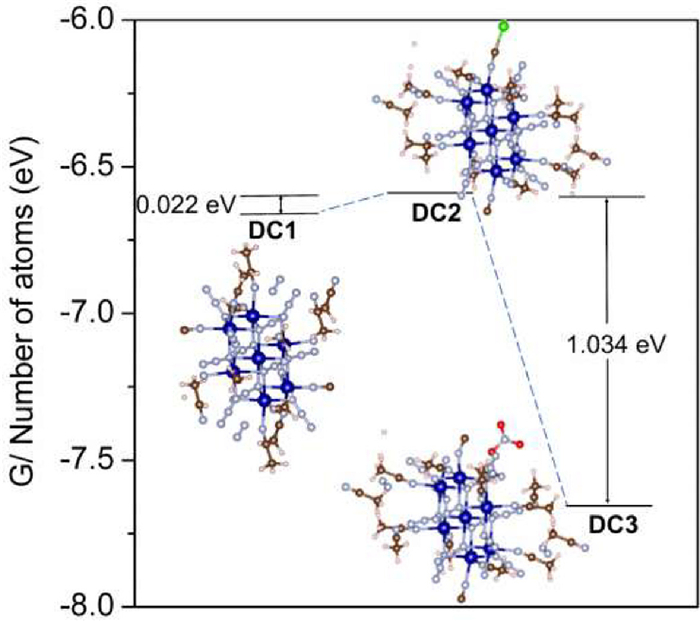
In view of such an interesting chemical process as above, it is significant to understand the formation of these complexes with the aid of theoretical calculation which can be used to explore the thermodynamic stability for DC1-DC3. The calculations were performed in the Vienna ab-initio simulation package (VASP), using DFT to exchange the correlation potential energy in the generalized gradient approximation (GGA), and the projector-augmented wave (PAW) method. To begin with, the crystallographic segment of CoN6 for the complex, whose symmetry and structure remained unchanged, were selected as substrates, and all possible structures were further optimized with the addition and removal of small ligand molecules (Fig. S1 in Supporting information) in an attempt to find the respective optimal binding sites. The adsorption structures of three different ligand molecules CH3CN, NO3, and Cl species at four different positions of CoN6 were obtained by systematically investigating various binding sites and Gibbs free energies (Figs. S2–S4 in Supporting information). Notably, the substrate energies were negative after small molecule linking, and the results all converged as expected. By comparing the Gibbs free energies of small ligand molecules at different binding sites (Table S1 in Supporting information), it is found that CH3CN is located directly above C, whereas NO3 and Cl were located on the C-N vertical substrate with more negative Gibbs free energies and more stable bonding. The steady state energies of DC1-DC3 are calculated as -683.485, -646.968 and -774.086 eV, respectively. The corrected Gibbs free energy values for individual atoms are -6.624, -6.602 and -7.636 eV for DC1-DC3, respectively (Table S2 in Supporting information), yielding the differences of 0.022 eV between DC1 and DC2, and 1.034 eV between DC2 and DC3 (Fig. 1). Thermodynamically, it is found that DC3 is of the most steady state, DC1 comes second place, while complex DC2 can be considered as a thermodynamically metastable state, which corresponds to the single-crystal incubation method during the synthetic process.
Single-crystal structure analysis reveals that DC1-DC3 are indicative of Co7 disk-like unit with a highly structural similarity. All CoⅡ centers are hexa-coordinated with quasi-octahedral geometry. The centrosymmetric heptanuclear motif presents an ideal hexagonal plane (Fig. 2a) in which a central CoⅡ atom is surrounded by six alternating CoⅡ atoms (Fig. 2b and Fig. S5 in Supporting information). The Co2 pairs of the hexagon are linked by six azido bridges with μ-1,1 EO mode, while the linkages between the external Co6 hexagon and the centric Co atom are provided by six μ3-1,1,1 EO azides. All complexes display a layer-like configuration, with N atoms from azido ligands and C atoms from acetonitrile above and below the Co7 plane (Fig. 2c). Then, the Co7 fragment provides a virtual C3 symmetry. Structurally, DC1-DC3 are comparable to a few of previously reported disc-like Co7 clusters [18,19,46–52].
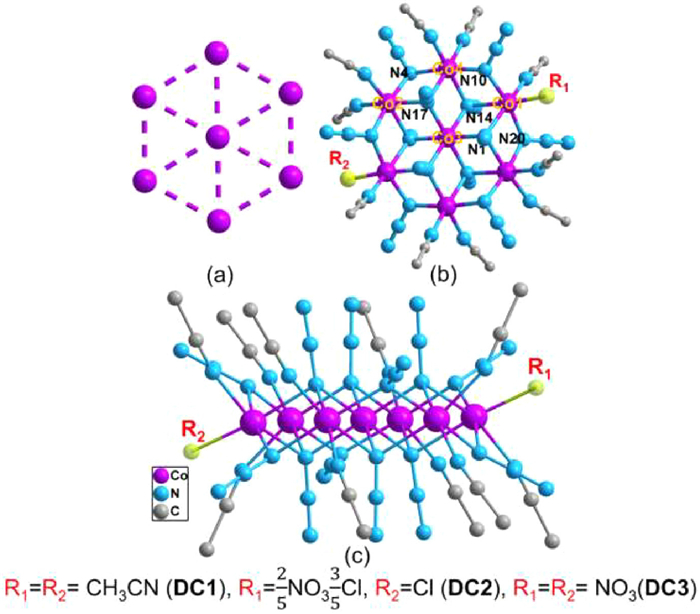
In addition to similarities, however, there are subtle differences among these three complexes. Complex DC1 is composed of one heptanuclear cluster [Co7(N3)12(MeCN)12]2+, and one [Y2(NO3)4 (piv)4]2− counter anions (Fig. S6 in Supporting information). Inorganic core of DC1 could alternatively be regarded as composing of six {Co3(N3)4} partial cubane groups, each biface sharing, and all six vertexes sharing at the core CoⅡ ion. All YⅢ atoms in [Y2(NO3)4(piv)4]2− are non-coordinated and surrounded by four O atoms from two bidentate chelating NO3− groups, and five O atoms from four bidentate chelating piv groups. The shortest distance between the YⅢ ion in the [Y2(NO3)4(piv)4]2− counter ion and the {Co7}2+ moiety is 6.942 Å, while the intramolecular Y···Y distance is 3.818 Å. The coordination geometric spheres of all YⅢ atoms were determined by the SHAPE 2.1 software [53] according to the crystallographic data, and the typical coordination geometries are listed in (Table S4 in Supporting information). The calculated continuous shape measures (CShMs) values suggest that the YⅢ ions in DC1 indicate the unified Muffin polyhedron with the identical Cs symmetry (Fig. S5). The peripheral ligation of Co6 hexagon in DC1 is filled with 12 terminal acetonitrile molecules, two on each of the external CoⅡ ions. For DC2, noteworthily, one of the peripheral Co atoms (Co1) is linked to one MeCN molecule and one chloride ion, whereas another Co1A atom is coordinated with mixed moieties of NO3−/Cl− (proportion 2/3) (Fig. S7 in Supporting information). Different from DC1 and DC2, external six Co atoms in DC3 bond with ten terminal MeCN molecules and two NO3− groups, in which Co1 and Co1A atoms are surrounded by one MeCN molecule and one NO3− group, respectively (Fig. S8 in Supporting information). The peripheral Co···Co separations in DC1-DC3 span the range 3.251–3.265 Å, 3.271–3.316 Å and 3.265–3.271 Å, and the Co-N-Co angles are in the ranges of 95.45°–103.80°, 117.597°–121.57° and 95.65°–103.72°, respectively (Tables S5–S7 in Supporting information). The stacking of the Co7 units in DC1-DC3 show long intermetallic distances between CoⅡ ions of adjacent molecules, with the shortest intermolecular Co···Co distances being 9.581, 8.275 and 8.294 Å, respectively (Figs. S9-S11 in Supporting information).
Direct-current (dc) magnetic data for DC1-DC3 were collected in the 2–300 K range under 0.1 T applied field. The χMT values at room temperature are 23.01, 24.58 and 25.03 cm3 K/mol for DC1-DC3, respectively, which are larger than the spin-only (g = 2) values of 13.13 cm3 K/mol expected for non-interacting Co7 species, implying significant orbital contribution of CoⅡ cations in an octahedral symmetry [54]. Upon decreasing the temperature, the χMT values increase continuously, reaching the maximums of 88.43 (DC1) cm3 K/mol at 18 K, 89.88 (DC2) and 74.80 (DC3) cm3 K/mol at 7 K, before reducing to 36.35, 43.76 and 60.55 cm3 K/mol for DC1-DC3 at 2 K, respectively (Figs. 3a, c and e, Fig. S12 in Supporting information). This rising behaviour indicates overall F coupling between the CoⅡ ions, while the rapid decline might depend on the onset of ZFS of CoⅡ, and/or the AF intermolecular interactions, as well as the more general integrations due to spin orbital coupling (SOC), which is a common source of difficulty in the cognition of magnetism for CoⅡ species [19,55]. The temperature dependent χM−1 curves of three complexes above 25 K keep to the Curie–Weiss law, giving C of 21.9 cm3 K/mol and θ of 18.8 K for DC1, C of 24 cm3 K/mol and θ of 10.8 K for DC2 and C of 24.1 cm3 K/mol and θ of 13.4 K for DC3. The positive θ values support a predominantly intermetallic F coupling in the Co7 cluster (Figs. 3a, c and e). In view of the coordination patterns in the heptanuclear Co cluster, the magnetic susceptibilities were tentatively simulated by a fitting model that was previously proposed by Gao for a structurally similar CoⅡ cluster with a quasi S6 symmetric geometry [46]. All magnetic data was fitted by using PHI program [55], and all J constants are referred to the -J Hamiltonian. In the high-T (>150K) range, the fitting gives an average Jav = +12.1 cm−1 (DC1), +8.4 cm−1 (DC2) and +10.5 cm−1 (DC3) with each SCo = 3/2 and g = 2.50; fitting with 2J parameters is inclined to obtain approximate J values. Then, an effective spin S'Co = 1/2 system was considered in the low-T (2–30 K) area, obtaining an excellent fit with with Jav = +33.9 cm−1, zJ' = -0.23 cm−1, and g = 6.62 for DC1, Jav = +21.3 cm−1, zJ' = -0.73 cm−1, and g = 6.58 for DC2, Jav = +27.2 cm−1, zJ' = -0.48 cm−1, and g = 6.69 for DC3. The results further confirm the significant intramolecular F interactions in the Co7 cluster of three complexes, which motivate an effective ground state of S = 7/2.
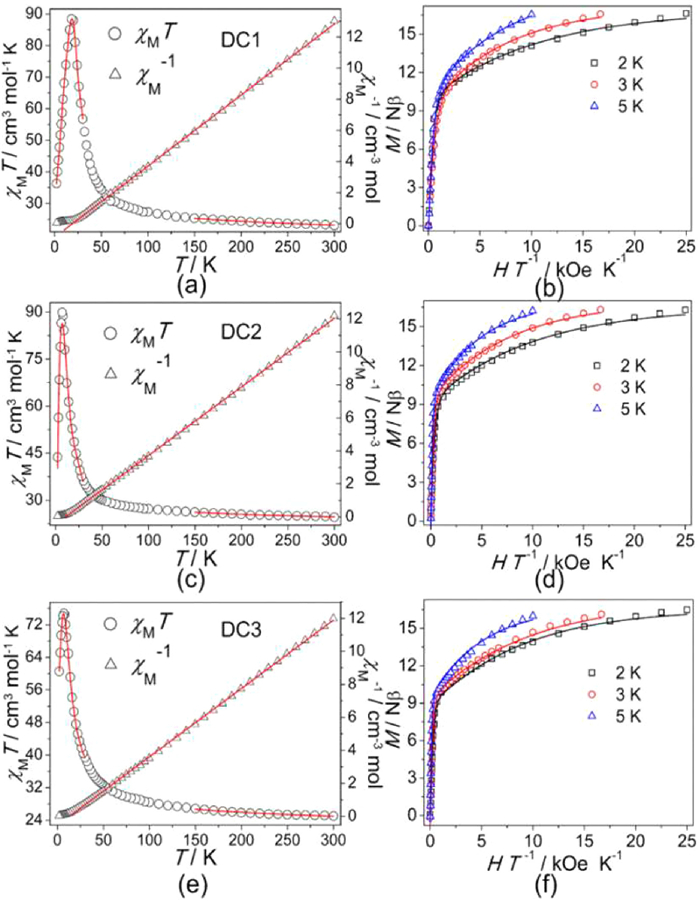
The M vs. H data for DC1-DC3 were collected up to 5 T at low temperatures (Fig. S13 in Supporting information). The sharp rise of M at low fields supports the existence of intracluster F couplings, then slowly reach the maximal values of 16.63 μB (DC1), 16.28 μB (DC2) and 16.49 μB (DC3) with a lack of saturation even at 2.0 K. Moreover, the non-saturated M values together with the non-superimposition of M vs. H/T plots (Figs. 3b, d and f) imply the occurrence of strong single-ion anisotropy, which is depending on the well-known significant SOC of the CoⅡ cation [56–59]. Fitting of the decreased M data is performed assuming a well-isolated, obtaining S = 7/2 ground state with D = -4.52, -5.08, -4.72 cm−1 and g = 6.56, 6.63, 6.59 for DC1-DC3, respectively.
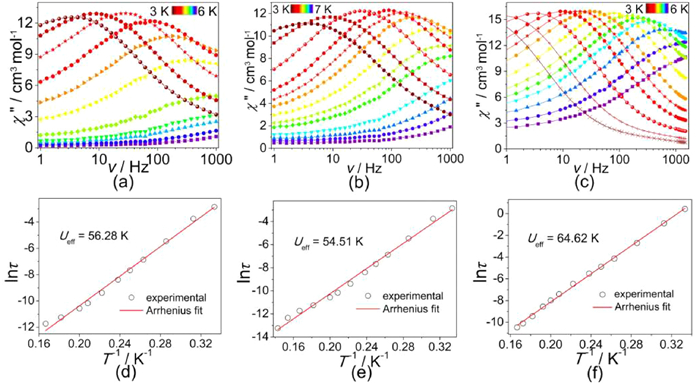
Alternating current (ac) magnetic data were measured in the absence of dc field to unravel the dynamic magnetisms of DC1-DC3. All complexes show explicit temperature and frequency-dependent signals for in-phase (χ') and out-of-phase (χ″) products (Figs. S14–S17 in Supporting information). Significant temperature-dependent peaks appear in the low temperature range, illustrating that the relaxation process via the quantum channel was clearly weaken or repressed. For frequency-dependent ac susceptibility with increased frequency, the maxima in χ″ plots move to high-temperature region, characteristic of a superparamagnetic behaviour (Figs. 4a–c). When DC1-DC3 are subjected to an ac magnetic field at temperatures between 3 K and 6 K, a peak in the χ″ component and a concurrent decrease in the χ' component (Fig. S17) are observed, confirming magnetic blocking on a millisecond to second time scale. An Arrhenius fitting of ln(τ) vs. T−1 for DC1 (Fig. 4d) obtains Ueff = 56.28 K and τ0 = 4.08 × 10−10 s. Similar analyses yield Ueff = 54.51 K and τ0 = 6.55 × 10−10 s for DC2, Ueff = 64.62 K, τ0 = 7.58 × 10−10 s for DC3 (Figs. 4e and f). This places DC1-DC3 among the small collection of Co7-based single molecule magnets [17–22]. The Cole-Cole curves for DC1-DC3 are also studied at variable temperatures (Fig. S18 in Supporting information). These curves are based on a generalised Debye function, with α parameters in the ranges of 0.324–0.458 for DC1, 0.262–0.509 for DC2 and 0.291–0.331 for DC3 (Tables S8–S10 in Supporting information), signifying a wide distribution of relaxation time for the magnetic relaxations of DC1-DC3.
To gain further insight into the SMM behaviour, hysteresis loops were performed at different temperatures. The fields were swept from 0 T to +1.5 T and then to -1.5 T and back. The results indicate that the tiny hysteresis openings are observed at 5 K for DC1, 4 K for DC2 and 2 K for DC3 (Fig. S19 in Supporting information). Moreover, Gatteschi et al. emphasized that for SMM a second temperature, TIRREV, should also be noticed, which is the point where the FC and ZFC curves diverge, as this is the temperature below which the magnetic observables are out-of-equilibrium and show history-dependent behaviour [60]. In this case, TIRREV are observed at 5, 4 and 2 K for DC1-DC3, respectively, and the ZFC plots cross the FC plots (Fig. S20 in Supporting information); conventionally convergence of the two curves should appear at the maxima of the ZFC data, and the FC data should usually be larger than the ZFC one. Compared with the rarely existing disk-like Co7 complexes, even the previously reported multinuclear Co clusters, the present three cases for the first time exhibit the slow relaxation of the magnetization at zero dc field, which is a significant enhancement of the magnetic properties for polynuclear Co-containing clusters (Table S11 in Supporting information) [18–20,46,61].
In summary, we have herein exemplified an elaborate synthetic route and structural transformation of three new disc-type heptanuclear CoⅡ clusters that are prepared by only using the flexible and versatile azido without requiring the simultaneous presence of any bridging/chelating organic liangs. Thermodynamically, DC3 stands in the most stable state, followed by DC1, and DC2 keeps in a metastable state. The overall intracluster ferromagnetic interaction contributes to a large ground state and dynamic magnetic relaxation under zero static field for three complexes. Besides the fascinating magnetic properties, investigations on the other application fields, for instance, high-energy materials due to the nitrogen-rich content, are in progress.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (NSFC, Nos. 21863009 and 22063008), the Natural Science Foundation of Ningxia Province (Nos. 2023AAC03014, 2023AAC03227, 2021AAC03136 and 2021BEB04062), the Young Top-notch Talent Cultivation Program of Ningxia Province, the Discipline Project of Ningxia (No. NXYLXK2017A04) and the China Postdoctoral Science Foundation (No. 2022M723148).
Supplementary material associated with this article can be found, in the online version, at doi:
M. Nakano, H. Oshio, Chem. Soc. Rev. 40 (2011) 3239-3248. doi: 10.1039/c0cs00223b
A. Landart-Gereka, M.M. Quesada-Moreno, I.F. Díaz-Ortega, et al., Inorg. Chem. Front. 9 (2022) 2810-2831. doi: 10.1039/d2qi00275b
P. Kumar Sahu, R. Kharel, S. Shome, S. Goswami, S. Konar, Coordin. Chem. Rev. 475 (2023) 214871. doi: 10.1016/j.ccr.2022.214871
E. Arias-Egido, M.A. Laguna-Marco, C. Piquer, et al., Mater. Des. 196 (2020) 109083. doi: 10.1016/j.matdes.2020.109083
Z. Wang, J.J. Liu, M.Y. Li, G. Chen, Chem. Eng. J. 462 (2023) 142154. doi: 10.1016/j.cej.2023.142154
B.D. Nath, K. Takaishi, T. Ema, Catal. Sci. Technol. 10 (2020) 12-34. doi: 10.1039/c9cy01894h
S. Schmitz, X. Qiu, M. Gloss, et al., Front. Chem. 7 (2019) 681. doi: 10.3389/fchem.2019.00681
G.P. Guedes, S. Soriano, N.M. Comerlato, et al., Inorg. Chem. Commun. 37 (2013) 101-105. doi: 10.1016/j.inoche.2013.09.034
M. Wang, Y. Guo, Z. Han, et al., Inorg. Chem. 61 (2022) 9785-9791. doi: 10.1021/acs.inorgchem.2c01299
Q.C. Luo, N. Ge, Y.Q. Zhai, et al., Chin. Chem. Lett. 34 (2023) 107547. doi: 10.1016/j.cclet.2022.05.061
M. Wang, X. Meng, N. Liu, et al., Chin. Chem. Lett. 34 (2023) 107995. doi: 10.1016/j.cclet.2022.107995
B.H. Wilson, J.S. Ward, D.C. Young, et al., Angew. Chem. Int. Ed. 61 (2022) e202113837. doi: 10.1002/anie.202113837
S. Mondal, A. Lunghi, J. Am. Chem. Soc. 144 (2022) 22965-22975. doi: 10.1021/jacs.2c08876
C.M. Liu, D.Q. Zhang, X. Hao, D.B. Zhu, Inorg. Chem. Front. 7 (2020) 3340-3351. doi: 10.1039/d0qi00632g
S. Li, Z. Weng, L. Jiang, et al., Chin. Chem. Lett. 34 (2023) 107251. doi: 10.1016/j.cclet.2022.02.056
X.L. Li, L. Zhao, J. Wu, et al., Chem. Sci. 13 (2022) 10048-10056. doi: 10.1039/d2sc03156f
Q. Chen, M.H. Zeng, Y.L. Zhou, H.H. Zou, M. Kurmoo, Chem. Mater. 22 (2010) 2114-2119. doi: 10.1021/cm903651e
D.I. Alexandropoulos, L. Cunha-Silva, A. Escuer, T.C. Stamatatos, Chem 20 (2014) 13860-13864. doi: 10.1002/chem.201403815
X.T. Wang, B.W. Wang, Z.M. Wang, W. Zhang, S. Gao, Inorg. Chim. Acta 361 (2008) 3895-3902. doi: 10.1016/j.ica.2008.03.020
D.M. Chen, X.Z. Ma, X.J. Zhang, N. Xu, P. Cheng, Inorg. Chem. 54 (2015) 2976-2982. doi: 10.1021/acs.inorgchem.5b00074
L.Q. Wei, B.W. Li, S. Hu, M.H. Zeng, CrystEngComm 13 (2011) 510-516. doi: 10.1039/C0CE00085J
E.C. Yang, Z.Y. Liu, L. Zhang, N. Yang, X.J. Zhao, Dalton Trans. 45 (2016) 8134-8141. doi: 10.1039/C6DT00010J
I. Radu, V.C. Kravtsov, S.M. Ostrovsky, et al., Inorg. Chem. 56 (2017) 2662-2676. doi: 10.1021/acs.inorgchem.6b02827
D. Shao, F.X. Xu, L. Yin, et al., Chin. J. Chem. 40 (2022) 2193-2202. doi: 10.1002/cjoc.202200284
A. Świtlicka, B. Machura, M. Penkala, et al., Inorg. Chem. Front. 7 (2020) 2637-2650. doi: 10.1039/d0qi00257g
A. Castro-Alvarez, Y. Gil, L. Llanos, D. Aravena, Inorg. Chem. Front. 7 (2020) 2478-2486. doi: 10.1039/d0qi00487a
W. Wang, H. Hai, S.H. Zhang, et al., J. Clust. Sci. 25 (2013) 357-365. doi: 10.1504/IJCEELL.2013.055406
A. Escuer, J. Esteban, S.P. Perlepes, T.C. Stamatatos, Coordin. Chem. Rev. 275 (2014) 87-129. doi: 10.1016/j.ccr.2014.04.001
S. Li, Z. Weng, L. Jiang, et al., Chin. Chem. Lett. 34 (2023) 107489. doi: 10.1016/j.cclet.2022.05.003
Y.F. Zeng, X. Hu, F.C. Liu, X.H. Bu, Chem. Soc. Rev. 38 (2009) 469-480. doi: 10.1039/B718581M
J. Netz, A.O. Mitrushchenkov, A. Kohn, J. Chem. Theory Comput. 17 (2021) 5530-5537. doi: 10.1021/acs.jctc.1c00294
S.F.M. Schmidt, M.P. Merkel, G.E. Kostakis, et al., Dalton Trans. 46 (2017) 15661-15665. doi: 10.1039/C7DT03149A
J.K. Staab, N.F. Chilton, J. Chem. Theory Comput. 18 (2022) 6588-6599. doi: 10.1021/acs.jctc.2c00611
J. Han, R. Cheng, L. Liu, H. Ohno, S. Fukami, Nat. Mater. 22 (2023) 684-695. doi: 10.1038/s41563-023-01492-6
T.C. Stamatatos, E. Rentschler, Chem. Commun. 55 (2018) 11-26.
K.X. Yu, J.G.C. Kragskow, Y.S. Ding, et al., Chem. Eur. J. 6 (2020) 1777-1793.
X.M. Zhang, K. Wang, Y.Q. Wang, E.Q. Gao, Dalton Trans. 40 (2011) 12742-12749. doi: 10.1039/c1dt11068c
Y.S. Ding, K.X. Yu, D. Reta, et al., Nat. Commun. 9 (2018) 3134. doi: 10.1038/s41467-018-05587-6
A. Lunghi, Sci. Adv. 8 (2022) eabn7880. doi: 10.1126/sciadv.abn7880
D. Krylov, G. Velkos, C.H. Chen, et al., Inorg. Chem. Front. 7 (2020) 3521-3532. doi: 10.1039/d0qi00771d
J. Krause, D.I. Alexandropoulos, L.M. Carrella, E. Rentschler, T.C. Stamatatos, Chem. Commun. 54 (2018) 12499-12502. doi: 10.1039/c8cc07722c
A.A. Athanasopoulou, M. Pilkington, C.P. Raptopoulou, A. Escuer, T.C. Stamatatos, Chem. Commun. 50 (2014) 14942-14945. doi: 10.1039/C4CC07192A
P.S. Perlepe, A.A. Athanasopoulou, K.I. Alexopoulou, et al., Dalton Trans. 43 (2014) 16605-16609. doi: 10.1039/C4DT02434F
A.A. Athanasopoulou, C.P. Raptopoulou, A. Escuer, T.C. Stamatatos, RSC Adv. 4 (2014) 12680-12684. doi: 10.1039/C4RA00738G
V.I. Ovcharenko, O.V. Kuznetsova, Russ. Chem. Rev. 89 (2020) 1261-1273. doi: 10.1070/rcr4981
Y.Z. Zhang, W. Wernsdorfer, F. Pan, Z.M. Wang, S. Gao, Chem. Commun. 31 (2006) 3302-3304. doi: 10.1039/b605459e
Y.L. Zhou, M.H. Zeng, L.Q. Wei, B.W. Li, M. Kurmoo, Chem. Mater. 22 (2010) 4295-4303. doi: 10.1021/cm1011229
R.X. Zhao, Q.P. Huang, G. Li, et al., J. Clust. Sci. 25 (2014) 1099-1108. doi: 10.1007/s10876-014-0692-6
S.H. Zhang, Y. Song, H. Liang, M.H. Zeng, CrystEngComm 11 (2009) 865-872. doi: 10.1039/b815675a
M.E. Slater-Parry, J.P. Durrant, J.M. Howells, et al., Dalton Trans. 48 (2019) 1477-1488. doi: 10.1039/c8dt04229b
H.L. Zheng, X.L. Chen, T. Li, et al., Chem 24 (2018) 7906-7912. doi: 10.1002/chem.201800516
S.T. Meally, C. McDonald, P. Kealy, et al., Dalton Trans. 41 (2012) 5610-5616. doi: 10.1039/c2dt12229d
M. Llunell, D. Casanova, J. Cirera, P. Alemany, S. Alvarez SHAPE Version 2.1, Shape Software, Barcelona, 2013.
M. Kurmoo, Chem. Soc. Rev. 38 (2009) 1353-1379. doi: 10.1039/b804757j
N.F. Chilton, R.P. Anderson, L.D. Turner, A. Soncini, K.S. Murray, J. Comput. Chem. 34 (2013) 1164-1175. doi: 10.1002/jcc.23234
Y. Wu, J. Xi, T. Xiao, et al., Inorg. Chem. Front. 8 (2021) 5158-5168. doi: 10.1039/d1qi01208h
M.R. Saber, M.K. Singh, K.R. Dunbar, Chem. Commun. 56 (2020) 8492-8495. doi: 10.1039/d0cc03238g
C.E. Patrick, S. Kumar, G. Balakrishnan, et al., Phys. Rev. Lett. 120 (2018) 097202. doi: 10.1103/PhysRevLett.120.097202
J. Mohapatra, M. Xing, J. Elkins, J.P. Liu, J. Alloys Compd. 824 (2020) 153874. doi: 10.1016/j.jallcom.2020.153874
D. Gatteschi, A. Cornia, M. Mannini, R. Sessoli, Inorg. Chem. 48 (2009) 3408-3419. doi: 10.1021/ic8013283
T.S. Mahapatra, D. Basak, S. Chand, et al., Dalton Trans. 45 (2016) 13576-13589. doi: 10.1039/C6DT02494G

Figure 2 (a) Representation of the Co7 disc core. (b) Partially labeled representation of the dications and (c) a side view of complexes DC1-DC3. Color scheme: CoⅡ purple, N pale blue, C light gray. All hydrogen atoms were omitted for clarity.

Figure 3 Plots of χMT vs. T and 1/χM vs. T for (a) DC1, (c) DC2 and (e) DC3 at H = 1 kOe. Plots of M vs. H/T for (b) DC1, (d) DC2 and (f) DC3 at different temperatures. The solid lines are the best fitting.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: