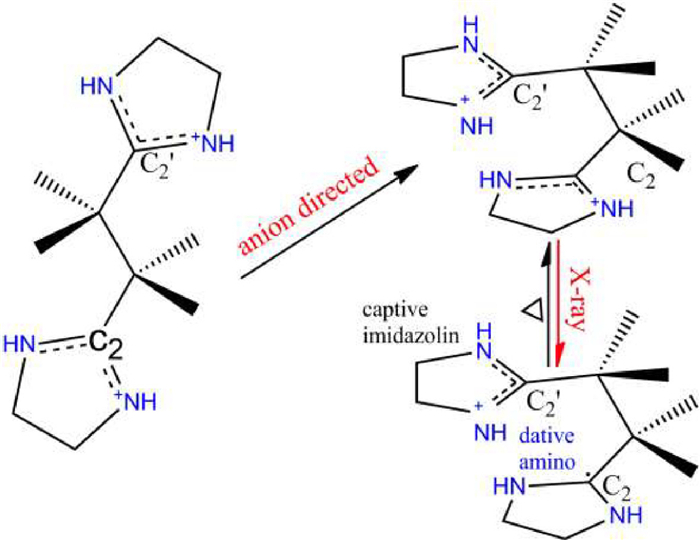
Scheme 1.
The concept of anion directed folding (H2imb)2+ leading to the postulated captodative effect for stabilizing the X-ray induced (H2imb) +• radical.
A new class of crystalline X-ray induced photochromic materials assembled from anion-directed folding of a flexible cation
Hong-Jin Liao , Zhu Zhuo , Qing Li , Yoshihito Shiota , Jonathan P. Hill , Katsuhiko Ariga , Zi-Xiu Lu , Lu-Yao Liu , Zi-Ang Nan , Wei Wang , You-Gui Huang

X-ray detection has received considerable attention due to the widespread use of X-ray in medical services and industrial crack detection applications [1–4]. Traditional X-ray detection methods rely on complicated instruments, which greatly limit their accessibility and practicality. Therefore, it is on demand to develop an economical and convenient X-ray detection technique. The emergence of X-ray induced photochromic complexes has opened up an avenue toward this important technique, providing direct visual detection of X-ray [5–9].
In recent years, the progress on photochromic complexes is mainly on metal-organic frameworks (MOFs). These MOFs, utilizing viologens as ligands or structure-directing agents (SDAs), have exhibited promising photochromic behaviors [10–13]. However, the structure change in these MOFs before and after X-ray induced photochromism is usually too subtle to be detected [14,15] because the photochromic reaction can occur only near the surface of the materials except an exceptional framework [13] reported by Zhang et al., and understanding their structure-related chromic behavior by theoretical calculation is also hindered for structural complexities.
In this context, X-ray induced photochromic organic molecules or salts may outperform MOFs because their relatively simple structures may make it feasible to investigate their photochromic processes by theoretical calculation [16]. This provides an opportunity to gain insights into the correlation between structure and photochromism. Therefore, it is highly desirable to develop rational strategies for X-ray induced photochromic organic molecules or salts yet remains a great challenge.
To address this issue, we aimed to develop a supramolecular approach. Herein we demonstrate a rational strategy of precise conformational control of an oxidative cation directed by anions. Photo-induced charge transfer has been demonstrated to be an effective approach for photochromism, in this regard, the key issue is to design appropriate electron [donor−acceptor] system and stabilize the yielded radicals [10–20]. (H2imb)2+ which can be readily synthesized from (H2azoimp)2+ through a radical-radical cross-coupling [21,22] attracted our attention, because it is oxidative so that cyclic alkyl amino carbene (CAAC) radical (H2imb)+• might be obtained by one electron reduction, as the synthesis of stable N-Heterocyclic carbene (NHC) radicals reported recently [23]. On the other hand, the great flexibility of (H2imb)2+ allows it to respond to the special geometry demands of counter anions [24,25]. We envisaged that X-ray induced metastable (H2imb)+• could be achieved by using various electron-donating anions with different geometries to modulate the conformation of (H2imb)2+ (Scheme 1).
We were able to synthesize single crystals of (H2imb)(NO3)2 (1) (Table S1 in Supporting information) by refluxing the mixture of (H2azoimp)Cl2 and Co(NO3)2·6H2O in methanol followed by crystallization through slow solvent evaporation ((H2azoimp)Cl2 = (2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride). In complex 1, (H2imb)2+ crystallize in an anti-conformation with the C2‒C2ʹ distance (the distance between the central C atoms of imidazolinium moieties) of 3.833 Å and are connected into a one-dimensional ribbon by NO3− through [N−H···O] hydrogen bonds (Fig. 1a). The PXRD pattern and IR spectrum for complex 1 are shown in Figs. S1 and S2 (Supporting information), respectively. Complex 1 is X-ray silent (Fig. S3 in Supporting information) probably because of the poor electron donating performance of NO3− and/or poor stability of postulated anti-(H2imb)+•. This finding promoted us to modulate the conformation of (H2imb)2+ with Cl− or Cl− containing anions for their greater electron donating performance. The following couple of advantages could be benefited from this strategy: (1) Cl− has been demonstrated as an excellent X-ray induced electron donor while that of NO3− has rarely been found [10,26], thus [Cl−−(H2imb)2+] would be a better electron [donor−acceptor] system than [NO3−−(H2imb)2+]; (2) (H2imb)2+ might be folded to syn-(H2imb)2+ by Cl− or Cl− containing anions. Since both the captive imidazolinium moiety and the dative alkyl amino groups are close to the central C• atom in syn-(H2imb)+• (Scheme 1), the captodative effect resulted from the synergy of the electron drawing of the imidazolinium moiety and the electron pulling of the alkyl amino groups is beneficial for stabilizing syn-(H2imb)+• [27–29]. As if absorption bands of the X-ray induced charge separated species appear in visible region, X-ray induced photochromism could be achieved. To validate our postulation, we used Cl− and (ZnCl4)2− to direct folding of (H2imb)2+.
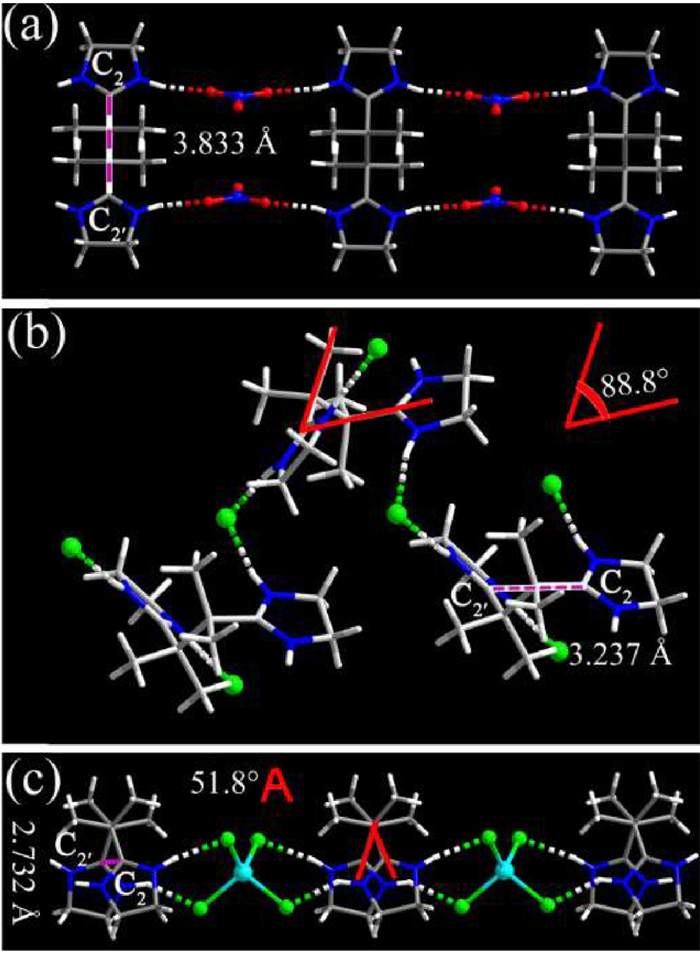
Crystals of (H2imb)Cl2·H2O (2) were synthesized by refluxing (H2azoimp)Cl2 in methanol followed by recrystallizing in H2O. In complex 2, (H2imb)2+ is folded into a gauche conformation (Fig. 1b) with the dihedral angle between imidazolinium moieties of 88.8° and the C2‒C2ʹ distance of 3.237 Å. (H2imb)2+ are assembled into one dimensional chains by Cl− through N−H···Cl hydrogen bonds which are further assembled into a three dimensional framework structure by H2O molecules through N−H···O and O−H···Cl hydrogen bonds (Fig. S4 in Supporting information).
To enhance the postulated captodative effect in (H2imb)+•, we then aimed to further shorten the C2‒C2ʹ distance by further folding the cation with Cl− containing anions. We envisaged that anions with tetrahedral geometries would direct (H2imb)2+ to crystallize in an approximately syn conformation for intermolecular hydrogen bonds, therefore (H2imb)ZnCl4 (3) was synthesized by refluxing the mixture of (H2azoimp)Cl2 and ZnCl2 in methanol (Fig. S5 in Supporting information). Compared with those in complex 2, (H2imb)2+ in complex 3 are further folded into a gauche conformation with the dihedral angle between imidazolinium moieties of 51.8° (Fig. 1c) and the C2‒C2ʹ distance of 2.732 Å. (H2imb)2+ are assembled into a one-dimensional supramolecular chain by (ZnCl4)2− anions through N−H···Cl hydrogen bonds. Remarkably, complex 1 can transit to complex 3 and complex 2 can transit to complexes 1 and 3 by anion exchange. The C2‒C2ʹ distance has been shorten from 3.883 Å in complex 1 to 2.732 Å in complex 3, thus great (H2imb)+• stability enhancement can be expected [27].
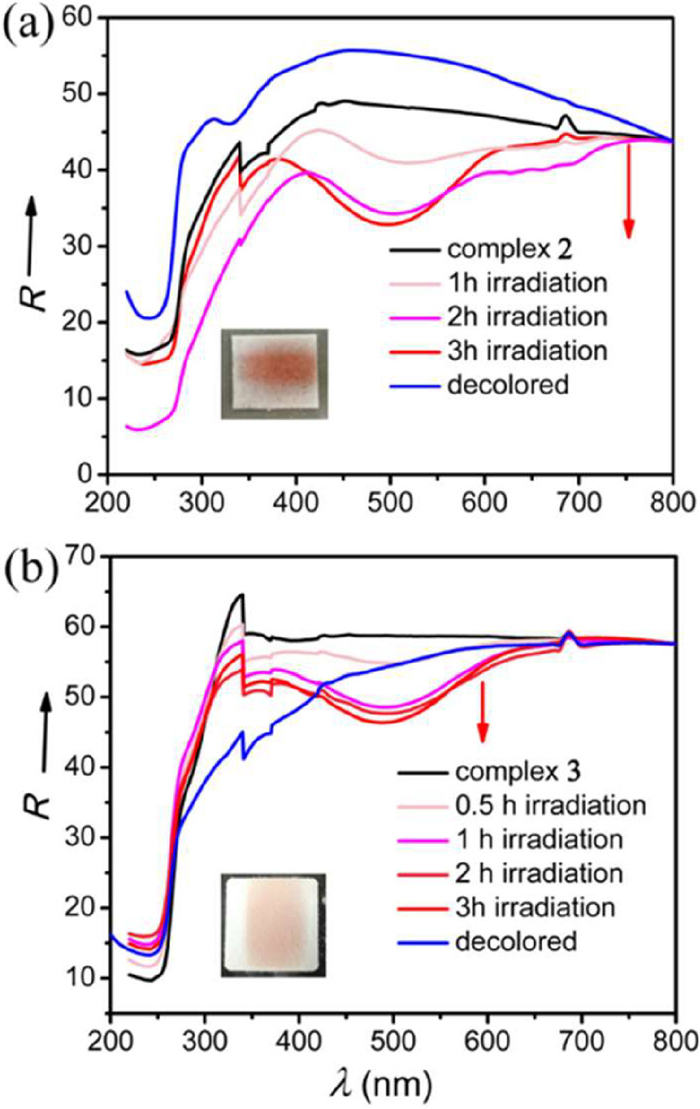
With the directing effect of different anions, the conformation of (H2imb)2+ has been successfully modulated. The main structural parameters related to the conformation of (H2imb)2+ are listed in Table 1. To examine whether our design strategy is effective for photochromic complexes, crystalline powder samples of complex 2 and 3 were irradiated with soft X-ray (Cu-K, λ = 1.54056 Å, 0.6 kW) on a powder X-ray diffractometer. Both complexes showed a noticeable color change from colorless to red in 0.5 h upon irradiation at room temperature and the color became deeper and deeper with increasing exposure time (Figs. S6 and S7 in Supporting information). The sensitivities (Obvious color change can be observed within 0.5 h with an X-ray source of 0.6 kW) of complex 2 and 3 to X-ray are comparable to other X-ray induced photochromic materials (Table S2 in Supporting information) [11–13]. The color of both the red photoproducts could slowly fade in approximate one month in atmosphere condition, and full decoloration could be completed in 2 h by annealing at 130 ℃. Decoloration of complex 2 at 130 ℃ for 2 h gives rise to pale yellow dehydrated 2 (H2imb)Cl2 as indicated by Thermogravimetric (TG) analysis (Fig. S8 in Supporting information). Although the crystals of dehydrated 2 were not suitable for single-crystal X-ray structural determination, PXRD measurement indicated that the one-dimensional hydrogen bonded chains assembled from Cl− bridged (H2imb)2+ remain after dehydration (Fig. S9 in Supporting information). indicating the photochromism is not related to the lattice H2O molecule (Figs. S10 and S11 in Supporting information). In comparison with the colorless samples of complexes 2 and 3, a new broad band centered at ~487 nm emerges in the time-dependent UV-vis spectra of both the red photoproducts which increases with prolonged irradiation time and disappears after decoloration (Fig. 2). This absorption band is characteristic of CAAC radicals [23,30,31]. Dehydrated 2 shows similar photochromic behavior. The reversible coloration-decoloration processes for both complexes can be repeated for at least three cycles (Figs. S12 and S13 in Supporting information) while the structures remain almost unchanged as revealed by SCXRD, PXRD (Figs. S5 and S14 in Supporting information) and IR analyses (Figs. S15 and S16 in Supporting information). Coloration of both complexes also can be induced by soft X-ray (Al-K, λ = 8.357 Å) (Fig. S17 in Supporting information), but no obvious color change can be observed upon UV irradiation.
 DownLoad:
CSV
DownLoad:
CSV

|
X-ray photoelectron spectroscopy (XPS) measurements were performed on complexes 2 and 3 before and after coloration to get insights into the mechanism of the phtochromic behavior. Early studies indicated that the photochromism of viologen halides is usually induced by charge-transfer from halide to viologen [10,13,26], therefore X-ray induced charge-transfer from Cl− to (H2imb)2+ and core-lever spectra variation of Cl 2p, C 1s, and N 1s can be expected before and after X-ray irradiation. Indeed, the core-level spectra of Zn 2p of complex 3 were almost the same before and after coloration, while the variation in the core-level spectra of Cl 2p, C 1s, and N 1s of both complexes is discernible (Figs. S18−S21 in Supporting information). For both complexes, the spectra of Cl 2p and N 1s slightly shift toward high binding energy region after coloration, while the spectra of C 1s slightly shift toward low binding energy region (Tables S3 and S4 in Supporting information). Cl− is electron-rich and (H2imb)2+ is electron-deficient, therefore the charge transferred from Cl− to (H2imb)2+ generating open-shelled Cl• and (H2imb)+• radicals upon X-ray irradiation.
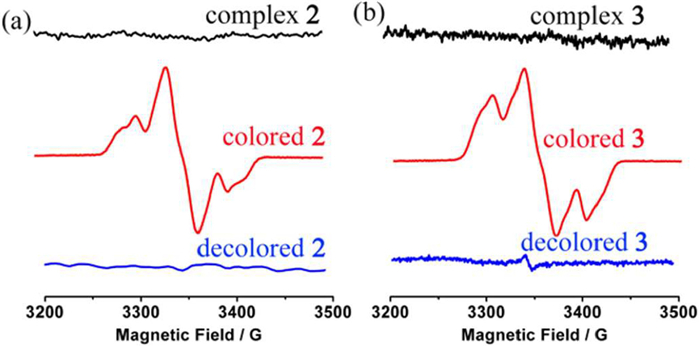
To characterize the feature of the yielded radical species, X-band electron spin resonance (ESR) spectroscopy and magnetic measurements were also performed on colorless and colored complexes 2 and 3. As shown in Fig. 3, colorless complex 2, complex 3 are ESR silent, but a typical broaden triplet-state signal [32–34] emerged with a zero-field parameter of D = 6.2 mT and E = 1.5 mT for colored complex 2 and D = 7.2 mT and E = 1.9 mT for colored complex 3, as determined by spectrum simulation (Fig. S22 in Supporting information). Both the g factors are anisotropic with g = 2.0026, g = 2.0082, g = 2.0045 for colored 2 and g = 2.0022, g = 2.0062, g = 2.0052 for colored 3, respectively. The average spin−spin distance was estimated from D to be 7.61 Å for colored 2 and 7.26 Å for colored 3, respectively. The magnetic susceptibilities of colored complexes 2 and 3 obtained through X-ray irradiation of original samples for 3 h were measured under an applied field H = 1 kOe, after the color completely faded the magnetic susceptibilities were measured again under the same condition. In such way, the data allow subtraction of the diamagnetic contribution from the sample holders and the intrinsic diamagnetism of the samples. The χ values for both colored 2 and 3 decrease linearly upon cooling indicating dominant antiferromagnetic interaction between spins (Fig. S23 in Supporting information) [35,36]. The field dependence of magnetization for colored 2 and 3 at 2.0 K saturated at high fields suggesting unidirectionally aligned spins were developed across the materials (Figs. S24 and S25 in Supporting information) [37].
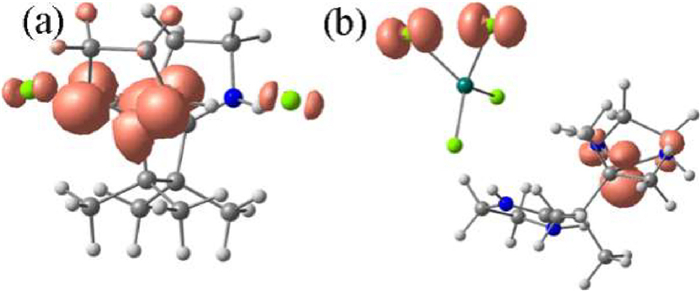
To clarify whether the folding of (H2imb)2+ in complex 2 and 3 is crucial for the X-ray induced photochromism, density functional theory (DFT) calculations were performed at the B3LYP/6-311+G** level using the Gaussian 09 program package [38] to shed light on the electronic structures of colored 2 and 3 (Fig. S26 in Supporting information). For both complexes, DFT calculations suggest the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are located on Cl− and the two π-systems of (H2imb)2+, respectively (Fig. S27 in Supporting information). The energy differences ΔEBT−CS between the biradical triplet state (BT) and closed-shell state (CS) were calculated to be 3.3 eV and 2.4 eV for complex 2 and 3, respectively. The ΔEBT−CS value for complex 1 was also calculated, giving a value of 4.36 eV which is higher than those of complex 2 and 3. This result implies the triplet state of complex 1 is not stable and probably too short-lived to be detected by naked eyes. The spin densities of triplet complex 2 and 3 are distributed on both the Cl and (H2imb)+• radicals, and the spin densities on (H2imb)+• are delocalized (Fig. 4). To give further insights into the photochromic behaviours, the electron transition configurations of complex 2 were calculated (Table S5 in Supporting information). Several significant HOMO→LUMO transition configurations (amplitude f larger than 0.04) can be identified, suggesting the observed X-ray induced photochromism originating from HOMO→LUMO charge transfer.
In summary, X-ray induced photochromism has been achieved on two organic salts designed by anion-directed folding a flexible cation. The photo responsive mechanism has been elucidated on the basis of UV–vis, ESR, XPS measurements, and DFT calculations. This work has developed a new class of X-ray induced photochromic materials, demonstrated that anion directed precise conformational control of cations is critical for the functionalities, therefore opened a new avenue for design of X-ray induced photochromic materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was supported by National Natural Science Foundation of China (No. 92261109), Natural Science Foundation of Fujian Province (No. 2020J05080), Project Funded by China Postdoctoral Science Foundation (No. 2023M733496), Natural Science Foundation of Xiamen (No. 3502Z20206080), Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (No. 2021ZR110), Recruitment Program of Global Youth Experts, Youth Innovation Promotion Association CAS (No. 2021302). A portion of this work was performed on the Steady High Magnetic Field Facilities, High Magnetic Field Laboratory, CAS.
Supplementary material associated with this article can be found, in the online version, at doi:
V. Kiryukhin, D. Casa, J.P. Hill, et al., Nature 386 (1997) 813–815. doi: 10.1038/386813a0
V. Kiryukhin, Y. Horibe, Y.S. Hor, et al., Phys. Rev. Lett. 97 (2006) 225503. doi: 10.1103/PhysRevLett.97.225503
G. Poneti, M. Mannini, L. Sorace, et al., Angew. Chem. Int. Ed. 49 (2010) 1954–1957. doi: 10.1002/anie.200906895
K.D. Irwin, G.C. Hilton, D.A. Wollman, et al., Appl. Phys. Lett. 69 (1996) 1945–1947. doi: 10.1063/1.117630
J.A. Sobrinho, J.H.K.S. Monteiro, M.R. Davolos, et al., ChemistrySelect 2 (2017) 3538–3548. doi: 10.1002/slct.201700287
K. Kinashi, Y. Miyamae, R. Nakamura, et al., Chem. Commun. 51 (2015) 11170–11173. doi: 10.1039/C5CC03977K
B.D. Ge, Y. Han, S.D. Han, et al., Inorg. Chem. Front. 6 (2019) 2435–2440. doi: 10.1039/c9qi00730j
P.F. Hao, X. Liu, C.Y. Guo, et al., Inorg. Chem. Front. 9 (2022) 879–888. doi: 10.1039/d1qi01242h
D. Gupta, A.K. Gaur, D. Chauhan, et al., Inorg. Chem. Front. 9 (2022) 2315–2327. doi: 10.1039/d2qi00325b
M.S. Wang, C. Yang, G.E. Wang, et al., Angew. Chem. Int. Ed. 51 (2012) 3432–3435. doi: 10.1002/anie.201108220
J.B. Wu, C.Y. Tao, Y. Li, et al., Chem. Sci. 5 (2014) 4237–4241. doi: 10.1039/C4SC01396D
H. Zhang, X.T. Wu, Adv. Sci. 3 (2016) 1500224. doi: 10.1002/advs.201500224
C. Chen, J.K. Sun, Y.J. Zhang, et al., Angew. Chem. Int. Ed. 56 (2017) 14458–14462. doi: 10.1002/anie.201707290
J.Z. Qiu, Y. Yu, Z.F. Chen, et al., Chin. Chem. Lett. 34 (2023) 107346. doi: 10.1016/j.cclet.2022.03.069
Q. Zhang, J.X. Hu, Q. Li, et al., Chin. Chem. Lett. 33 (2022) 1417‒1421. doi: 10.1016/j.cclet.2021.08.029
T. Yamaguchi, Y. Kobayashi, J. Abe, J. Am. Chem. Soc. 138 (2016) 906–913. doi: 10.1021/jacs.5b10924
J.Z. Liao, S.S. Wang, X.Y. Wu, et al., Dalton Trans. 47 (2018) 1027–1031. doi: 10.1039/c7dt04276k
M. Morimoto, S. Kobatake, M. Irie, Chem. Commun. 2006, 2656–2658. doi: 10.1039/b604388g
P.X. Li, M.S. Wang, M.J. Zhang, et al., Angew. Chem. Int. Ed. 53 (2014) 11529–11531. doi: 10.1002/anie.201406554
C. Sun, G. Xu, X.M. Jiang, et al., J. Am. Chem. Soc. 140 (2018) 2805–2811. doi: 10.1021/jacs.7b10101
Y.I. González, M. Stjerndahl, D. Danino, Langmuir 20 (2004) 7053–7063. doi: 10.1021/la0493464
D. Spaccini, N. Pastori, A. Clerici, et al., J. Am. Chem. Soc. 130 (2008) 18018–18024. doi: 10.1021/ja807613q
D. Rottschäfer, B. Neumann, H. Stammler, et al., Angew. Chem. Int. Ed. 57 (2018) 4765–4768. doi: 10.1002/anie.201801596
H. Maeda, Y. Bando, Chem. Commun. 49 (2013) 4100–4113. doi: 10.1039/c2cc35759c
Y. Haketa, Y. Bando, K. Takaishi, et al., Angew. Chem. Int. Ed. 51 (2012) 7967–7971. doi: 10.1002/anie.201202196
G. Xu, G.C. Guo, M.S. Wang, et al., Angew. Chem. Int. Ed. 46 (2007) 3249–3251. doi: 10.1002/anie.200700122
E.V. Anslyn, D.A. Dougherty, Modern Physical Organic Chemistry, Sausalito, CA, 2005.
H.G. Viehe, Z. Janousek, R. Merenyi, et al., Acc. Chem. Res. 18 (1985) 148–154. doi: 10.1021/ar00113a004
J.R. David, R. Brook, C. Haltiwanger, J. Am. Chem. Soc. 113 (1991) 5910–5913. doi: 10.1021/ja00015a082
M.M. Hansmann, M. Melaimi, G. Bertrand, J. Am. Chem. Soc. 140 (2018) 2206–2213. doi: 10.1021/jacs.7b11184
M.M. Hansmann, M. Melaimi, D. Munz, et al., J. Am. Chem. Soc. 140 (2018) 2546–2554. doi: 10.1021/jacs.7b11183
T. Nozawa, M. Nagata, M. Ichinohe, et al., J. Am. Chem. Soc. 133 (2011) 5773–5775. doi: 10.1021/ja2014746
Y.T. Su, X.Y. Wang, Y.T. Li, et al., Angew. Chem. Int. Ed. 54 (2015) 1634–1637. doi: 10.1002/anie.201410256
T. Li, G.W. Tan, D. Shao, et al., J. Am. Chem. Soc. 138 (2016) 10092–10095. doi: 10.1021/jacs.6b05863
C.P. Constantinides, A.A. Berezin, M. Manoli, et al., Chem. Eur. J. 20 (2014) 5388–5396. doi: 10.1002/chem.201304538
X.Y. Li, Y. Zou, S.D. Han, et al., Inorg. Chem. Front. 8 (2021) 4186–4191. doi: 10.1039/d1qi00750e
E.Q. Jin, M. Asada, Q. Xu, et al., Science 357 (2017) 673–676. doi: 10.1126/science.aan0202
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision E. 01, Gaussian, Inc., Wallingford CT, 2004.

Scheme 1 The concept of anion directed folding (H2imb)2+ leading to the postulated captodative effect for stabilizing the X-ray induced (H2imb) +• radical.

Figure 1 (a) The ribbon structure of complex 1. (b) The hydrogen bonded chain in complex 2. (c) The chain structure of complex 3 (Zn cyan, Cl green, O red, N blue, C gray, H white).

Figure 2 Time-dependent UV–vis diffuse reflectance spectra of complex (a) 2 and (b) 3, showing the color change of complex (a inset) 2 and (b inset) 3 upon X-ray irradiation for 3 h and 10 h, respectively.

Figure 3 ESR spectrum evolution of complex (a) 2 and (b) 3 upon coloration-decoloration processes.

Figure 4 Spin densities of (a) colored 2 and (b) 3 calculated at B3LYP/6-311+G** level.
Table 1. Comparison of C2‒C2ʹ distance and the dihedral angle between imidazolinium moieties of compounds 1, 2, and 3.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: