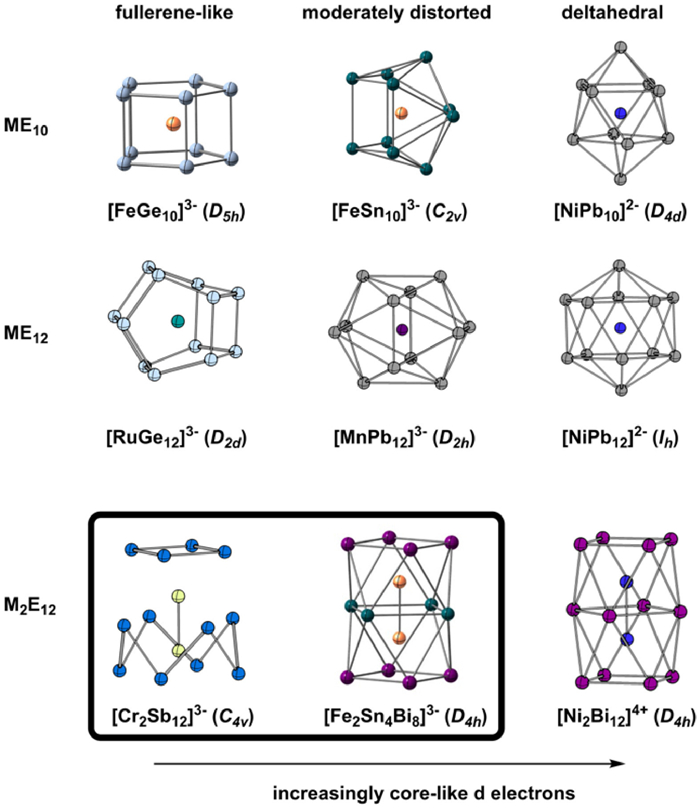
Figure 1.
Structural trends within the ME10, ME12, M2E16 and M2E12 families. The two new clusters reported in this paper are highlighted in the box.
Metal-metal bonds in Zintl clusters: Synthesis, structure and bonding in [Fe2Sn4Bi8]3– and [Cr2Sb12]3–
Ya-Nan Yang , Zi-Sheng Li , Sourav Mondal , Lei Qiao , Cui-Cui Wang , Wen-Juan Tian , Zhong-Ming Sun , John E. McGrady

The family of endohedral Zintl clusters, in which a d-block or f-block metal atom is enclosed by a cage of p-block (semi)metal atoms, is an increasingly diverse one that now includes examples with many of the d-block elements [1,2], the lanthanides and the actinides [3–6]. Applications of these clusters are beginning to emerge in materials science and catalysis [7], but there remains an enduring fascination with the nature of the chemical bond between the endohedral metal and the cage [8,9]. These clusters are perhaps unique in their electronic flexibility, in the sense that although strong bonding interactions between the cage and the endohedral atom are certainly possible, they are not essential to the integrity of the cluster. It is possible, therefore, to find a wide spectrum of bond types within a closely-related family of clusters, and numerous reviews have been devoted to their electronic structure using density functional theory as well as wavefunction-based methods [2,9–12].
Many of the earliest endohedral Zintl ions contained metal ions with a closed-shell d10 configuration, classic examples being the icosahedral triad [Ni/Pd/PtPb12]2– and bi-capped square antiprismatic [PtPb10]2– (Fig. 1, right column) [13]. These deltahedra are based on triangular faces and highly-connected vertices, and their geometries are straightforward to rationalize in terms of Wade's rules: Each has a total valence electron count of 4n + 12, where n is the number of vertices (60 and 52 for n = 12 and 10, respectively). Of these electrons, 10 can be assigned to the metal nd shell and the remaining 4n + 2, the characteristic electron count for a closo structure, to the cage [14]; the clusters are therefore straightforwardly formulated as a metal atom encapsulated in a Pb10/122– cage The partitioning of electrons into metal- and cage-based subsets becomes less clear-cut as we move to the left in the d block, where the metal atom typically has fewer nd electrons, but those that it does have are at higher energy. In some cases, transfer of charge density from the metal into the vacant orbitals of the cage can cause subtle distortions to the deltahedral structure, as for example in D2h-symmetric [MnPb12]3– [15] and C2v-symmetric [FeSn10]3– (Fig. 1, central column) [16], while in more extreme cases it can favor the adoption of entirely different, 3-connected architectures such as pentagonal prismatic [MGe10]3– (M = Fe, Co) [17,18], and bicapped pentagonal pyramids, [RuGe12]3– [19] and [TaGe4As8]3– (Fig. 1, left column) [20]. These 3-connected 'fullerene-like' architectures are typically associated with electron-precise counts (electron-precise here meaning a total valence electron count at the cage of 5n [21], which can only be reached if we assume that some or all of the metal d electrons also contribute to the count at the cluster. For [TaGe8As4]3–, for example, the count for the [Ge8As4]3– unit is 8 × 4 + 4 × 5 + 3 = 55, with the 5 valence electrons of Ta raising the total count to 5n = 60. This represents a marked departure from deltahedral cases such as [NiPb12]2– where the characteristic 4n + 2 electron count is reached without any contribution from the d electrons on the metal. The varying structural chemistry captured in Fig. 1 therefore signals a varying role for the metal d electrons, from core-like and inert in the deltahedra to clearly valence in the fullerene-like analogues.
The rich diversity of the ME10 and ME12 families stands in stark contrast to the relatively small number of examples in the literature of clusters containing two transition metals, M2Ex, or even larger metal fragments, M3Ex or M4Ex [22–26]. The potential for metal-metal bonding in these clusters introduces a further dimension to an already complex electronic landscape, in so much as covalent bonds between the metal centers may promote the release of electron density from M-M antibonding states onto the cage and vice versa. We and others have explored the electronic structure of silicon- and germanium-based clusters such as M2Si12 [27–30], but experimental characterization in these typically neutral or cationic cases is restricted to various flavors of gas-phase spectroscopy [31–35]. Attempts to establish whether the structural trends noted above for ME12 and ME10 are part of a more general pattern, and the extent to which metal-metal bonding plays a role, have been frustrated by the absence of a more extended family of endohedral M2 clusters, equivalent to ME12 and ME10 in single-metal analogues. The triple-decker E12 architecture found in [Ni2Bi12]4+ (Fig. 1, bottom row) [36], and also in the mixed Sn/Pb/Sb/Bi clusters [Ni2Sn7Sb5]3–, [Ni2Sn7Bi5]3–, [Ni2Pb7Bi5]3– and [Co2Sn5Sb7]3– [37–39] has the potential to fill this void, because the prolate architecture is ideally suited to accommodate an M2 unit, just as the icosahedron is ideally suited for a single metal atom. All of the Ni and Co clusters noted above are, however, rather similar: they share a common valence electron count of 76, and all show approximate D4h point symmetry (notwithstanding the disordered arrangement of group 14 and 15 elements in some cases) and similar M-M distances of ~2.45 Å. We report here the synthesis and structural characterization of two new open-shell members of the M2E12 family, [Fe2Sn4Bi8]3– and [Cr2Sb12]3–, both of which have a valence electron count of 75, one fewer than the Ni/Co clusters described above. [Fe2Sn4Bi8]3– retains the D4h-symmetric geometry typical of the 76-electron clusters but [Cr2Sb12]3– has a very different C4v-symmetric geometry where some of the Sb-Sb bonds are substantially elongated. We use density functional theory to place the diverse structural chemistry of this family into context, and argue that [Fe2Sn4Bi8]3– and [Cr2Sb12]3– are the M2 analogues of slightly distorted [MnPb12]3– and fullerene-like [RuGe12]3–, respectively, just as the 76-electron Ni and Co clusters are analogues of [PtPb12]2–.
All manipulations were performed in a nitrogen-filled glove box with a moisture level below 1 ppm. 4,7,13,16,21,24-Hexa-oxa-1,10-diazabicyclo[8.8.8]hexacosane (2.2.2-crypt, Sigma-Aldrich 98%), 18-crown-6 (Sigma-Aldrich 99%) and 1,1-bis(diphenylphosphino)ferrocene (DPPF, Sigma-Aldrich 99%) were dried under a vacuum for 12 h prior to use. Ethylene-diamine (en) (Aldrich, 99%) and toluene (tol) (Aldrich, 99.8%) were freshly distilled over sodium before use. K3Sb7 and KSnBi were synthesized by heating a stoichiometric mixture of the elements in a sealed niobium tube at 800 ℃ and 975 ℃, respectively [40]. [K(2.2.2-crypt)]2[Sn2Bi2] was prepared from the reaction of KSnBi with 2.2.2-crypt [41] while CrCp2 was prepared from the reaction of sodium cyclopentadienide with anhydrous chromium chloride [42].
Synthesis of [K(2.2.2-crypt)]3[Fe2Sn4Bi8] (1): [K(2.2.2–crypt)]2[Sn2Bi2] (148.6 mg, 0.1 mmol) and DPPF (110.0 mg, 0.2 mmol) were dissolved in en (2.5 mL) in a test tube. The mixture was then stirred for 3 h at room temperature and the resulting deep brown solution was centrifuged and transferred to a test tube, then layered carefully with toluene to crystallize. Black block-like crystals of [K(2.2.2-crypt)]3[Fe2Sn4Bi8] were isolated after two weeks in approximately 20% yield (based on [K(2.2.2-crypt)]2[Sn2Bi2]) along with [K(2.2.2-crypt)]2[Sn7Bi2].
Synthesis of [K(18-crown–6)]4[Cr2Sb12]·Cp (2): K3Sb7 (97 mg, 0.1 mmol) and 18-crown-6 (80 mg, 0.3 mmol) were weighed into a 10 mL vial inside a glovebox and dissolved in en (3 mL). After stirring for 1 h, the resulting dark brown solution was filtered into another vial of CrCp2 (15 mg, 0.1 mmol) and allowed to stir for a further 20 min at ambient temperature. The resulting red-brown solution was filtered through glass wool and transferred to a test tube, then layered carefully with toluene (3.5 mL) to allow for crystallization. Black rhombic flake-like crystals of [K(18-crown-6)]4[Cr2Sb12]·Cp were isolated after two weeks in approximately 31% yield in total (based on precursor CrCp2 used).
Suitable single crystals were selected for X-ray diffraction analyses. Crystallographic data were collected on the Rigaku XtalAB Pro-MM007 DW diffractometer with graphite monochromated Cu Kα radiation (λ = 1.54184 Å). Structures were solved using direct methods and then refined using SHELXL-2014 [43] and Olex2 [44] to convergence, in which all the non-hydrogen atoms were refined anisotropically during the final cycles. All hydrogen atoms of the organic molecule were placed by geometrical considerations and were added to the structure factor calculation. The crystal of compound 2 exhibits inversion twinning, the twin law (−1 0 0 0 −1 0 0 0 −1) was used for refinement. Both compounds are air and moisture-sensitive in solution and in the solid state. CCDC numbers are 2169859 and 1972524 for 1 and 2, respectively.
Electrospray ionization mass spectrometry (ESI-MS) was performed in negative-ion mode on an LTQ linear ion trap spectrometer by Agilent Technologies ESI-TOF-MS (6230). The spray voltage was 5.48 kV and the capillary temperature was kept at 300 ℃. The capillary voltage was 30 V and the sheath gas was maintained at 50 ℃. The samples were made up inside a glovebox under an N2 atmosphere and rapidly transferred to the spectrometer in an airtight syringe by direct infusion with a Harvard syringe pump at 0.2 mL/min.
Energy dispersive X-ray (EDX) analyses were performed using a scanning electron microscope (FE-SEM, JEOL JSM-7800F, Japan). Data acquisition was performed with an acceleration voltage of 15 kV and an accumulation time of 60 s.
All calculations described in this paper were performed with the Amsterdam Density Functional (ADF) package of program, version 2021.104 [45]. The exchange-correlation functional proposed by Perdew, Burke and Ernzerhof (PBE) was used throughout [46], and scalar relativistic effects were introduced using the Zeroth Order Regular Approximation (ZORA) [47]. The sensitivity of the results to choice of functional was tested by also performing calculations using the M06-L [48] and hybrid PBE0 [49] functionals (see supporting information). For all geometry optimizations, a triple-zeta quality basis set of Slater-type orbitals was used [50], supplemented by two sets of polarization functions ('TZ2P'). All electrons were included in the basis set. A 'good' setting of the numerical grid [51] was used in all calculations. The Conductor like Screening Model (COSMO, ε = 78.39) [52] was applied to simulate the confined effects of the crystalline environment. All SCF procedures were conducted using default setting of ADF: at convergence, the commutator of the Fock matrix and the density matrix is less than 10−6. The bond energies were calculated using the Morokuma-Ziegler energy decomposition scheme [53].
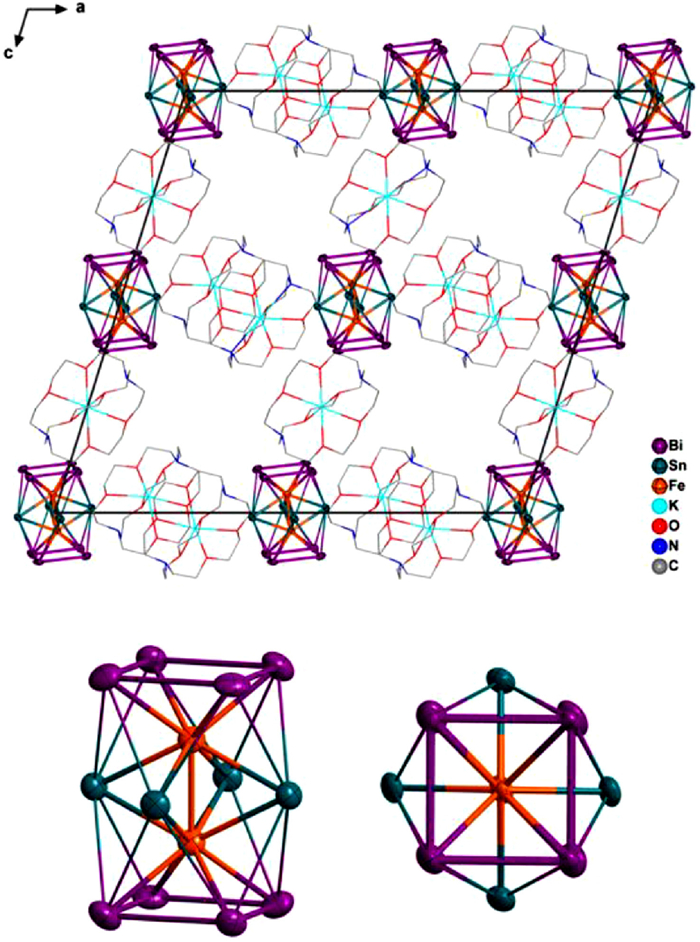
The asymmetric unit of [K(2.2.2-crypt)]3[Fe2Sn4Bi8] (1) and the structure of the [Fe2Sn4Bi8]3– anion are shown in Fig. 2, and key bond lengths are summarized in Table 1. The asymmetric unit contains a single [Fe2Sn4Bi8]3– anion along with three [K(2.2.2-crypt)]+ cations, with the shortest K+-Bi contacts in excess of 6.5 Å. The anion adopts a D4h-symmetric triple-decker architecture, very similar to those of the 76-electron Ni2 and Co2 clusters described in the introduction, structural data for which are also collected in Table 1. The Fe-Fe separation of 2.395(3) Å is shorter, albeit only marginally, than the Co-Co and Ni-Ni analogues [36–39], and the average Sn-Sn bond length in the equatorial plane is 3.373 Å, somewhat longer than the corresponding values of 3.264, 3.290 and 3.298 Å for [Ni2Sn7Sb5]3–, [Ni2Sn7Bi5]3– and [Co2Sn5Sb7]3–, respectively. The average Fe-Sn bond length of 2.669 Å is also ~0.03 Å longer than the corresponding values for the three 76-electron clusters. The Fe-Bi and Bi-Bi bond lengths (average 2.697 Å and 3.095 Å, respectively) are marginally longer than those in [Ni2Bi12]4+ (2.678 Å and 3.045 Å, respectively).
 DownLoad:
CSV
DownLoad:
CSV

|
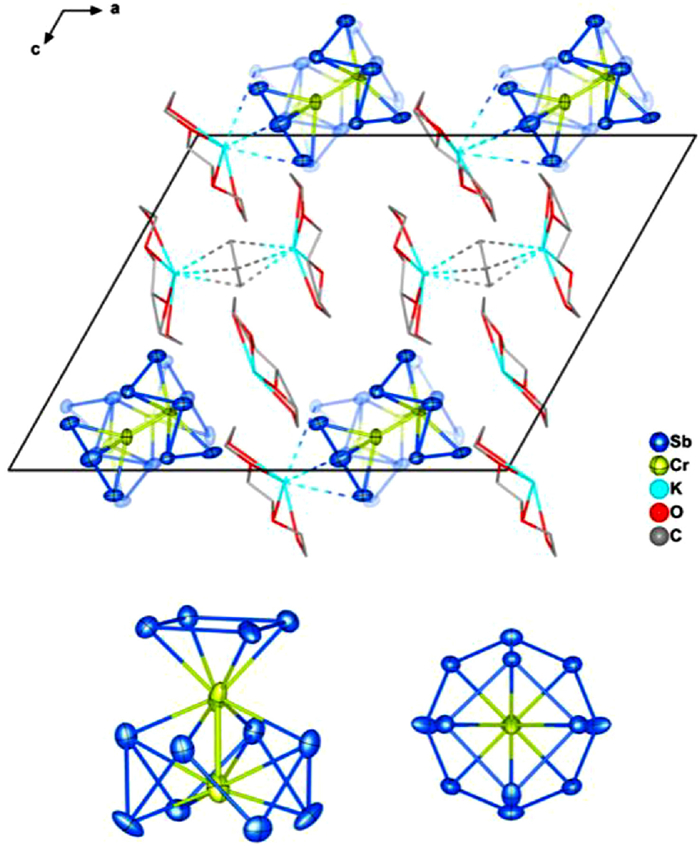
The asymmetric unit of [K(18-crown-6)]4[Cr2Sb12]·Cp (2) contains one [Cr2Sb12]3– anion along with a single Cp– unit and four [K(18-crown-6)]+ cations. The anions are disordered over two orientations (50% population of each) differing in the alignment of the polar axis, but with otherwise very similar bond lengths and angles - the two components on each lattice site are shown as full and shadowed purple/green spheres in Fig. 3. The bond lengths of one component are summarized in Table 1 while the second is summarized in Table S2 (Supporting information). The cluster anion in 2 is strikingly distorted from D4h to C4v symmetry and can be considered, to a first approximation, as separate CrSb8 and CrSb4 fragments, although the Sb-Sb distance of only 3.549 Å between the atoms of the Sb4 and Sb8 units suggests that this separation is not entirely clearcut. The CrSb8 unit is reminiscent of the [MAs8]z– and [MSb8]z– anions (M = Nb, Ta, Cr, Mo) which have the same crown-like E8 motif [54]. The Cr-Cr bond length is 2.319(17) Å, shorter again than the Fe-Fe bond in [Fe2Sn4Bi8]3–. The average Sb-Sb bond length within the Sb8 crown is 2.801 Å, typical of Sb-Sb single bonds, while the remaining Sb4 unit is a distorted square with bond lengths between 2.75 Å and 2.76 Å (average 2.755 Å); for comparison, the computed Sb-Sb distance for gas-phase cyclo-Sb42– is 2.82 Å [55]. The [Cr2Sb12]3– anion is sandwiched between two of the [K(18-crown-6)]+ cations which are aligned along the approximate 4-fold rotation axis, with K-Sb distances in the range of 3.23–3.72 Å (to CrSb4) and 4.26–4.66 Å (to the lower face of CrSb8). The alignment of cations along the principal axis is reminiscent of the packing in the one-dimensional 
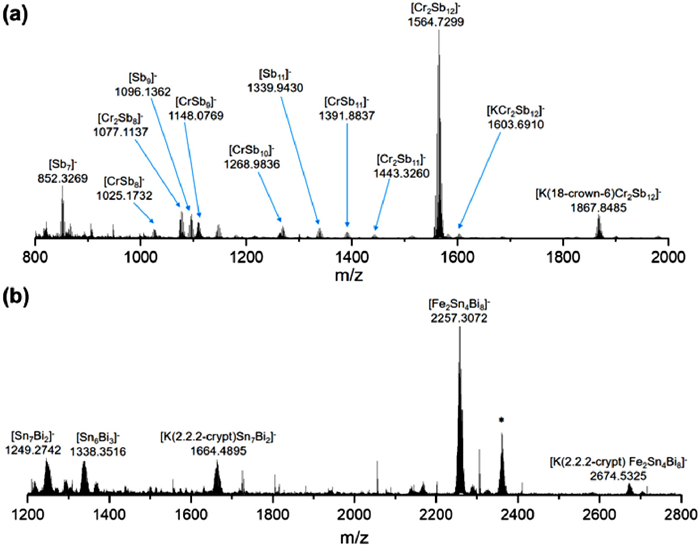
The ESI-MS of freshly prepared samples of 1 and 2, both taken in negative-ion mode, are compared in Fig. 4. In both cases the dominant peak is for the parent anion (carrying a 1– charge), with a further prominent peak at higher mass due to the cation-dianion pair (the cation being K(2.2.2-crypt)+ in the case of 1, K(18-crown-6)+ in the case of 2). The structural differences noted in the previous section are reflected in the fragmentation patterns of the two anions, with the [Cr2Sb12]3– cluster showing a marked tendency to cleave the Cr-Cr bond (leading to [CrSb8]–) and also the Cr-{Sb4} bond (leading to [Cr2Sb8]–). A number of peaks corresponding to Sb counts between 8 and 12 are evidence for substantial rearrangement following these initial fragmentations. In marked contrast, the mass spectrum of 1 is entirely devoid of fragments containing a single Fe atom, reflecting the more complete encapsulation of the Fe2 unit in a contiguous 12-vertex cage.
Despite numerous attempts on different carefully prepared samples of 1 and 2, we were unable to measure reproducible EPR spectra of either compound, and direct experimental proof of their paramagnetism therefore remains elusive. Paramagnetism is implied by the –3 charge of 1 and 2 because the total electron count must then be odd, and this, in turn, and this charge assignment rests on the presence of either three or four K+ cations per anion in the asymmetric units of 1 and 2, respectively. The assignment of charge in this way is, however, not without ambiguity - there is precedent, for example, for protonation of co-crystallized solvent molecules such as ethylenediamine, the solvent used in these experiments, in Zintl ion chemistry [57]. There are, however, no molecules of ethylenediamine incorporated into the unit cells of either 1 or 2. The Cp– unit in the unit cell of 2 could, in principle, be protonated but its planar structure and the tight binding of two K(18-crown-6)+ units appears to exclude such a possibility. An alternative explanation for the absence of a measurable EPR signal is disproportionation into distinct clusters with charges –2 and –4. Corbett and co-workers have argued for such a scenario in the compound [K(2.2.2-crypt)]6Ge92–Ge94–·2.5en [58], although a subsequent measurement of an EPR signal for the closely-related [K(2.2.2-crypt)]6Ge92–Ge94–·0.5en suggests that a formulation as Ge93– is more likely [59,60]. The challenge in distinguishing these possibilities using X-ray crystallography stems from the rather weak correlation between electron count and structure across the Ge9z– series (z = 2, 3, 4). As we show in the following section, the DFT-optimized geometry of [Fe2Sn4Bi8]z– is also rather insensitive to charge, and so we cannot rule out absolutely the possibility of a disproportionation into [Fe2Sn4Bi8]2– and [Fe2Sn4Bi8]4– based on structural data alone. However, we believe that the most likely explanation for our inability to measure reproducible EPR spectra for 1 and 2 is that the samples decomposed before the measurements could be made - this is a common problem in air-sensitive Zintl-ion chemistry [61], and is certainly consistent with the myriad of decomposition products observed in the ESI mass spectra (Fig. 4, below).
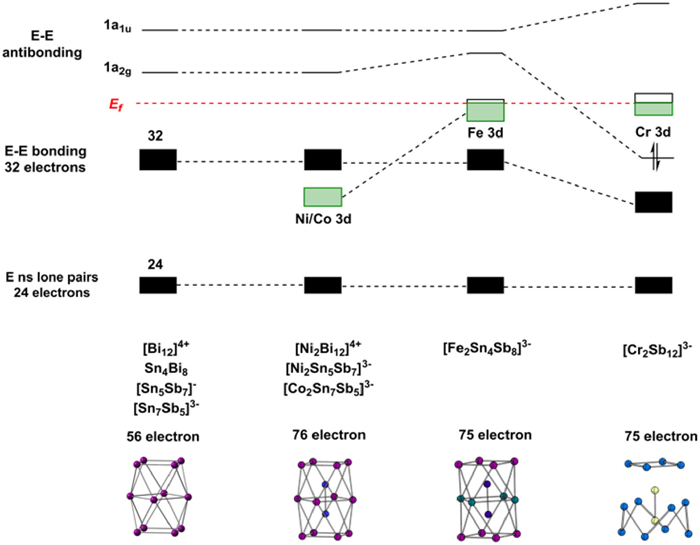
Our purpose in this section is to develop an electronic structure model that encompasses not just the two new clusters reported in this paper, but the whole family of closely-related examples set out in Table 1, where we have highlighted the total valence electron count, including the transition metal 3d electrons, as the central variable. By that measure, the two new 75-electron clusters reported here are clearly more electron-deficient, in an absolute sense, than their 76-electron analogues. That has to be set against the fact that the 3d orbitals will be less stable in the earlier transition metals, so what electron density there is on the M2 fragment will be more accessible to the cage for Fe and particularly for Cr, than for Ni. The effect of changing the transition metal from Fe to Cr is compounded by the change from a group 14 element (Sn) in the equatorial plane of 1 to a more electronegative group 15 element (Sb) in 2, which will also have the effect of drawing electron density away from the metal and towards the cage. A logical starting point for our analysis is therefore to consider the 76-electron Ni2 clusters, [Ni2Bi12]4+, [Ni2Sn7Bi5]3–, [Ni2Pb7Bi5]3– and [Ni2Sn7Sb5]3–, where the 3d electrons are the most core-like, in the same way that we have used [NiPb12]2– as a reference point in the icosahedral family [10,15,62]. We choose [Ni2Bi12]4+ to illustrate the key points, simply because it has the same point symmetry as [Fe2Sn4Bi8]3– (D4), and therefore affords the most transparent comparison.
The ground state of [Ni2Bi12]4+ is a spin-singlet with DFT-optimized structural parameters in close correspondence with the X-ray data and also the computational work reported in reference 36 (Table 1). In Fig. 5, we show three complementary perspectives on the electronic structure of this cluster: the Kohn-Sham molecular orbital array; the density of states (PDOS), projected onto Ni 3d, Bi 6s and Bi 6p; the overlap population based density of states (OPDOS), sometimes referred to as the crystal orbital overlap population (COOP) in a solid-state context. The OPDOS indicates whether states at a given energy have bonding (positive OPDOS) or anti-bonding (negative OPDOS) character with respect to specific pairs of atoms. The PDOS shows that almost all the Ni 3d character (green) accumulates below the Fermi level, and the Ni-Ni OPDOS shows that these levels range from π bonding (3eu with positive OPDOS) at the bottom to σ antibonding (4a2u) near the top, and the absence of significant Ni-Ni bonding is consistent with the conclusions of Ruck et al. in their original report of this cluster [36]. In short, all the above is consistent with a formulation as a Ni2° dimer encapsulated inside a Bi124+ cage, the direct analogue of [NiPb12]2–.
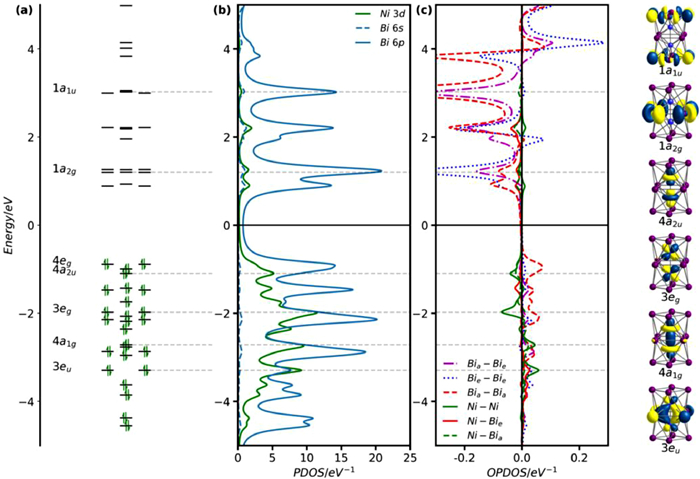
The HOMO-LUMO gap in [Ni2Bi12]4+ is large, and indeed it remains large in the isolated Bi124+ cation (Fig. S26 in Supporting information). This observation indicates that the triple-decker geometry is intrinsically suited to precisely 56 (i.e., 76 – 20) valence electrons. Possible reasons for the stability of this particular electron count were discussed by Lips and Dehnen in the context of the 76-electron anion [Ni2Sn7Bi5]3–, where they considered three distinct models of electronic structure, the Zintl-Klemm concept, a localized view of bonding and also the Wade-Mingos rules [37]. The Zintl-Klemm concept is not obviously applicable because the connectivity of the vertices is greater than three, while orbital localization generates 2-center-2-electron bonds around the upper and lower E4 units, but the remainder of the valence electrons remain heavily delocalized in multi-center orbitals with Sn, Bi and Ni character. A subsequent report on the lead analogue, [Ni2Pb7Bi5]3–, reached similar conclusions, and also confirmed the absence of any significant Ni-Ni bonding [38]. Finally, the authors noted that the Wade-Mingos rules predict a stable electron count of 4n + 6 = 54, two fewer than the actual count of 56, if the cluster is viewed as an arachno deltahedron (i.e., one derived from a 14-vertex structure with two atoms capping the terminal E4 squares). The authors concluded, therefore, that none of these three models can provide a simple explanation for the stability of the 56-electron count for the cage (in this case [Sn7Bi5]3–). Jemmis' 'mno' rules [63] offer an alternative perspective that is consistent with the observed electron count: if the cluster is viewed as two square antiprisms fused via a shared square face (rather than as a single 12-vertex polyhedron), the predicted count is 4n + 2m + 2o = 48 + 4 + 4 = 56, where m, n and o are the number of fused polyhedra (2), the number of vertices (12) and the number of open faces (2), respectively. The difference of 2 between the predictions of the Wade-Mingos and Jemmis electron-counting schemes arises from the assumed presence of two strongly bonding inwardly directed hybrids (one per polyhedron) in the Jemmis scheme but only one in the single polyhedron according to the Wade-Mingos scheme (where m, n and o are 1, 12 and 2, respectively). Seen in this light, the apparent break down of the Wade-Mingos rules is therefore a consequence of the prolate rather than spherical structure of the cluster. Ultimately, no matter which conceptual model we choose to rationalize the 56-electron count, the important point is that it is the 'natural' one for triple-decker E12 clusters because it generates a large HOMO-LUMO gap, just as 4n + 2 = 50 is the 'natural' count for closo clusters such as the icosahedron and 5n = 60 is the 'natural' count for the fullerene-like geometry in [TaGe4As8]3– [19,20].
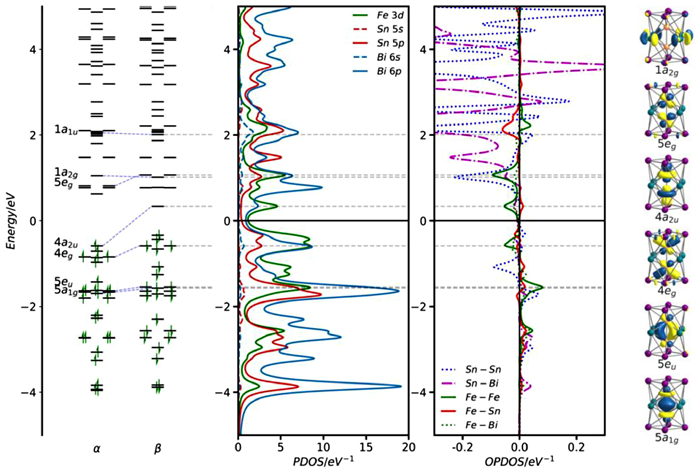
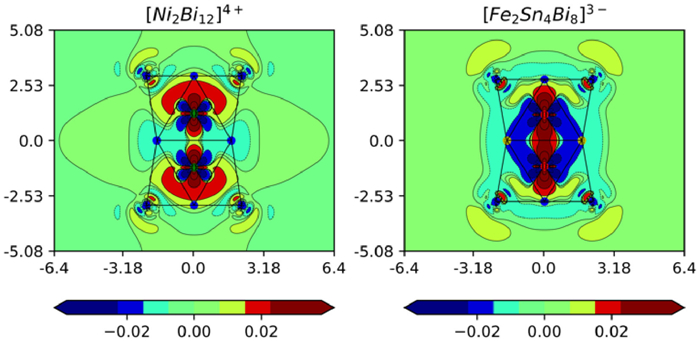
The ground state of the 75-electron [Fe2Sn4Bi8]3– cluster is a doublet (2A2u) with optimized Fe-Fe, Sn-Sn, Sn-Bi and Bi-Bi bond lengths that are within 0.03 Å of the crystallographically-determined values (Table 1). The spin-α and spin-β Kohn-Sham eigenvalues, PDOS and OPDOS plots are shown in Fig. 6. Compared to the [Ni2Bi12]4+ reference, one electron has been removed from the spin-β component of the Fe-Fe σ* orbital, 4a2u, which now lies above the Fermi level. This results in a formal Fe- Fe bond order of 0.5, consistent with the contraction of the M-M bond from 2.429 Å in [Ni2Bi12]4+ to 2.395 Å in [Fe2Sn4Bi8]3–. The PDOS and OPDOS plots in Fig. 7 indicate that a substantial amount of Fe π* character (green) is now found above the Fermi level, mixed with antibonding states on the cage in 5eg (note the negative peak in the Fe-Fe OPDOS at ~+1.0 eV). The Fe-Fe π character, in contrast, remains largely below the Fermi level (in 5eu at –1.8 eV), conferring a degree of Fe-Fe π character on the bond and indeed the calculated Fe-Fe Mayer bond order of 0.92 is substantially higher than the value of 0.30 computed for [Ni2Bi12]4+. The emergence of a π component to the Fe-Fe bond reflects the greater tendency to engage in back-bonding with the vacant levels of the E12 cage compared to the Ni2 analogues. This change is captured in the difference density plots shown in Fig. 7 (the difference between the self-consistent density and the sum of the pre-formed fragments). In both cases, a 56-electron configuration is imposed on the E12 cluster (Bi124+ and Sn4Bi8°, respectively), with 20 and 19-electron configurations on Ni2° and Fe23–, respectively. The plot on the left shows that redistribution of electron density upon formation of [Ni2Bi12]4+ from Ni2 and Bi124+ is limited to a redistribution of electron density between the 3dz2 and 4s orbitals on the Ni2 core. In contrast, charge transfer from the filled orbitals of the Fe23– fragment to the vacant levels of Sn4Bi8 is substantial, with charge accumulating at the poles of the cluster in [Fe2Sn4Bi8]3– (red regions just inside the top and bottom Bi4 rings) and a blue region of depletion around the Fe atoms, both of which carry the fingerprint of the Fe-Fe π orbital 5eg.
We have acknowledged above that we do not have definitive experimental proof of the paramagnetism of 1, and that it is therefore conceivable that the crystal instead contains a mixture of [Fe2Sn4Bi8]2– and [Fe2Sn4Bi8]4– rather than [Fe2Sn4Bi8]3–. The optimized structures of the 2– and 4– species are reported in Supporting Information, Table S6, where the most striking feature is the insensitivity to charge. The 4– anion is diamagnetic, with the vacancy in the Fe-Fe σ* orbital in Fig. 6 filled, while the most stable state of the 2– analogue is a triplet with a second electron removed from the Fe-Fe δ* orbital of b1u symmetry. Despite the variations in formal Fe-Fe bond order from 1.0 (2–) to 0.0 (4–), the bond lengths vary by less than 0.08 Å, reflecting the rigidity of the triple-decker E12 cage. We reiterate our belief that the most likely explanation for the failure to measure an EPR spectrum for 1 is decomposition of the paramagnetic 3– ion during sample preparation, but even if the 2–/4– scenario prevails, the key conclusion that Fe-Fe bonding is present remains secure, albeit only in the 2– component.
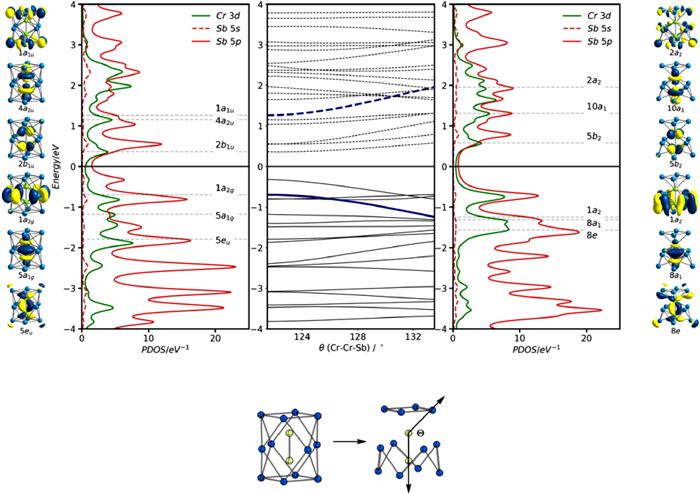
The ground state of [Cr2Sb12]3– is also a spin doublet, with C4v symmetry and an optimized Cr-Cr distance of 2.29 Å, somewhat shorter than the crystallographically determined value (2.319 Å). The symmetry of the ground state, 2B2, is also different from the isoelectronic [Fe2Sn4Bi8]3– cluster, with the unpaired electron now residing in the 5b2 orbital (Fig. 8a), a linear combination of Cr dx2−y2 orbitals with local Cr-Cr δ* symmetry. The Cr-Cr σ* orbital, 10a1, is vacant in both spin-α and spin-β manifolds (note that only the latter is shown in Fig. 8 and Fig. S28 in Supporting information), implying a formal σ bond order of 1.0 compared to the 0.5 for [Fe2Sn4Bi8]3–. The larger Mayer bond order of 1.382 is a direct measure of this increased Cr-Cr bonding.
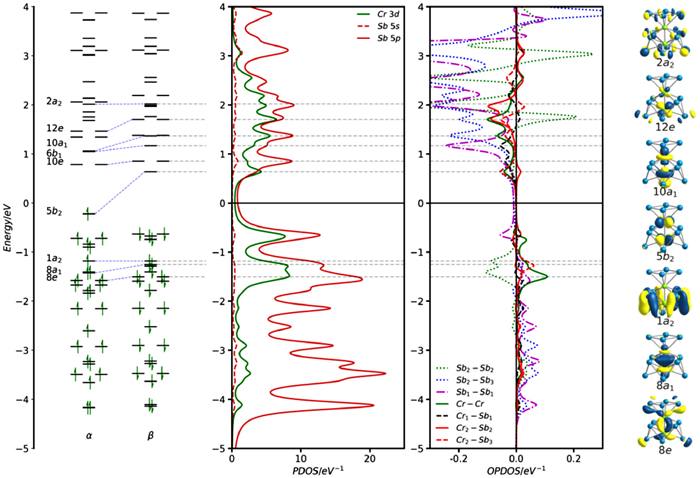
To fully understand the relationship between [Cr2Sb12]3– and [Fe2Sn4Bi8]3–, and the origin of the different structures and electronic configurations, we need to understand the driving force for the distortion from D4h to C4v symmetry in the former. In order to do so, we first constrain [Cr2Sb12]3– to the D4h-symmetric potential energy surface, where we again find a doublet ground state, and the unpaired electron again resides in an orbital with Cr-Cr δ* character (2b1u) (see left hand side of the Walsh diagram shown in Fig. 9). The Cr-Cr separation is even shorter, at 2.161 Å, and the equatorial Sb4 unit is very substantially expanded (Sb-Sb = 3.636 Å compared to Sn-Sn = 3.373 Å in [Fe2Sn4Bi8]3–). These structural differences can be traced to a change in electronic configuration: the 1a2g orbital that was vacant in both [Ni2Bi12]4+ and [Fe2Sn4Bi8]3– is occupied in [Cr2Sb12]3–, and its pronounced Sb-Sb antibonding character drives the expansion of the equatorial plane. At the same time two metal-based spin orbitals, 4a2uα and 2b1uβ, are depopulated and the M-M σ antibonding nature of the former, in particular, leads to the contraction of the Cr-Cr bond. In short, the Cr2 unit transfers two electrons from its uppermost, antibonding, levels into an Sb-Sb antibonding level localized on the equatorial ring, simultaneously strengthening the Cr-Cr bond while weakening the Sb-Sb bonds. The D4h-symmetric structure is, however, 0.86 eV less stable than the C4v global minimum, and the driving force for the distortion comes from the energetic proximity of the now-occupied 1a2g orbital and the vacant 1a1u orbital, which is a similar linear combination of Sb 5p orbitals, but localized on the terminal, rather than equatorial, Sb4 units. A distortion to C4v point symmetry allows mixing between 1a2g and 1a1u, both of which transform as a2 in the lower symmetry - a second-order Jahn-Teller distortion. The evolution of the valence orbitals along a distortion coordinate linking the D4h and C4v structures is shown in the Walsh diagram in Fig. 9 where the stabilization of 1a2 and concomitant destabilization of 2a2 is picked out in blue. The 1a2 orbital becomes bonding between the lower and middle Sb4 planes (Sb2 and Sb3 in Fig. 8) while 2a2 is Sb-Sb antibonding. In effect, the distortion allows the additional electron density that has accumulated on the equatorial Sb4 unit to be delocalized onto the two Sb4 caps. Note that when both 1a2g and 1a1u are vacant, as they are in [Ni2Bi12]4+ and [Fe2Sn4Bi8]3–, there is no equivalent driving force and therefore no distortion. The differences between [Fe2Sn4Bi8]3– and [Cr2Sb12]3– stem from a combination of two factors: (1) The higher energy of the 3d orbitals in Cr vs. Fe; (2) The lower energy of the 5p orbitals on Sb vs. Sn, both of which favor the occupation of the 1a2g orbital in the latter but not the former.
In conclusion, we have structurally characterized two new members of the M2E12 family, [Fe2Sn4Bi8]3– and [Cr2Sb12]3–, both of which have 75 valence electrons. Despite their common electron count, the clusters have quite different structures: [Fe2Sn4Bi8]3– is approximately D4h-symmetric, very similar to the 76-electron analogues [Ni2Bi12]4+, but [Cr2Sb12]3– has C4v symmetry, with a CrSb8 crown capped by a CrSb4 fragment. The structural landscape can be understood in terms of a progressive upward shift in the energies of the metal d orbitals relative to those on the cage as we move from Ni to Fe and then to Cr (Fig. 10). This leads to increased back-bonding in [Fe2Sn4Bi8]3–, and, in [Cr2Sb12]3–, to an orbital crossing that shifts two electrons from Cr-Cr anti-bonding states into the Sb-Sb antibonding 1a2g orbital. This then triggers a second-order Jahn-Teller distortion, leading to the observed C4v-symmetric structure.
The trends in geometry and electronic structure identified for the M2E12 family find exact analogues in the chemistry of their ME12 and ME10 counterparts: [Ni2Bi12]4+ is the direct analogue of [NiPb12]2– and [PtPb10]2–, [Fe2Sn4Bi8]3– is the analogue of [MnPb12]3– and [FeSn10]3– while [Cr2Sb12]3– is the analogue of [RuGe12]3– and [FeGe10]3–, in the sense that the nd electrons shift from being structurally inert in the first group to being a minor perturbation to the core deltahedral structure in the second and then to driving a switch to a lower connectivity architecture in the third.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 92161102 and 21971118) and the Natural Science Foundation of Tianjin City (Nos. 21JCZXJC00140 and 20JCYBJC01560) as well as the 111 Project (No. B18030) from Ministry of Education China. S. Mondal acknowledges the Indian government for a scholarship.
Supplementary material associated with this article can be found, in the online version, at doi:
B. Weinert, S. Dehnen, Binary and ternary intermetalloid Clusters, in: S. Dehnen (Ed.), Clusters – Contemporary Insight in Structure and Bonding, Springer International Publishing, Cham, 2017, pp. 99–134.
J. Zhao, Q. Du, S. Zhou, V. Kumar, Chem. Rev. 120 (2020) 9021–9163. doi: 10.1021/acs.chemrev.9b00651
B. Oelkers, M.V. Butovskii, R. Kempe, Chem. Eur. J. 18 (2012) 13566–13579. doi: 10.1002/chem.201200783
N. Lichtenberger, R.J. Wilson, A.R. Eulenstein, et al., J. Am. Chem. Soc. 138 (2016) 9033–9036. doi: 10.1021/jacs.6b04363
X. Min, I.A. Popov, F.X. Pan, et al., Angew. Chem. Int. Ed. 55 (2016) 5531–5535. doi: 10.1002/anie.201600706
A.R. Eulenstein, Y.J. Franzke, N. Lichtenberger, et al., Nat. Chem. 13 (2021) 149–155. doi: 10.1038/s41557-020-00592-z
C. Dong, Y. Li, D. Cheng, et al., ACS Catal. 10 (2020) 11011–11045. doi: 10.1021/acscatal.0c02818
K. Mayer, J. Weßing, T.F. Fässler, R.A. Fischer, Angew. Chem. Int. Ed. 57 (2018) 14372–14393. doi: 10.1002/anie.201805897
X. Jin, J.E. McGrady, Chapter eight - structure and bonding in endohedral transition metal clusters, in: R. van Eldik, R. Puchta (Eds.), Advances in Inorganic Chemistry, Academic Press, 2019, pp. 265–304.
J.E. McGrady, F. Weigend, S. Dehnen, Chem. Soc. Rev. 51 (2022) 628–649. doi: 10.1039/d1cs00775k
Y. Wang, J.E. McGrady, Z.M. Sun, Acc. Chem. Res. 54 (2021) 1506–1516. doi: 10.1021/acs.accounts.0c00876
R.J. Wilson, N. Lichtenberger, B. Weinert, S. Dehnen, Chem. Rev. 119 (2019) 8506–8554. doi: 10.1021/acs.chemrev.8b00658
E.N. Esenturk, J. Fettinger, B. Eichhorn, J. Am. Chem. Soc. 128 (2006) 9178–9186. doi: 10.1021/ja061842m
K. Wade, J. Chem. Soc. D: Chem. Commun. (1971) 792–793.
B. Zhou, T. Krämer, A.L. Thompson, J.E. McGrady, J.M. Goicoechea, Inorg. Chem. 50 (2011) 8028–8037. doi: 10.1021/ic200329m
T. Krämer, J.C.A. Duckworth, M.D. Ingram, et al., Dalton Trans. 42 (2013) 12120–12129. doi: 10.1039/c3dt50643f
J.Q. Wang, S. Stegmaier, T.F. Fässler, Angew. Chem. Int. Ed. 48 (2009) 1998–2002. doi: 10.1002/anie.200805511
B. Zhou, M.S. Denning, D.L. Kays, J.M. Goicoechea, J. Am. Chem. Soc. 131 (2009) 2802–2803. doi: 10.1021/ja900055j
G. Espinoza-Quintero, J.C.A. Duckworth, W.K. Myers, J.E. McGrady, J.M. Goicoechea, J. Am. Chem. Soc. 136 (2014) 1210–1213. doi: 10.1021/ja411280v
S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend, S. Dehnen, Nat. Commun. 7 (2016) 10480. doi: 10.1038/ncomms10480
D.M.P. Mingos, Pure Appl. Chem. 63 (1991) 807–812. doi: 10.1351/pac199163060807
F. Lips, R. Clérac, S. Dehnen, J. Am. Chem. Soc. 133 (2011) 14168–14171. doi: 10.1021/ja203302t
C. Liu, X. Jin, L.J. Li, et al., Chem. Sci. 10 (2019) 4394–4401. doi: 10.1039/c8sc03948h
H.W.T. Morgan, K.S. Csizi, Y.S. Huang, Z.M. Sun, J.E. McGrady, J. Phys. Chem. A 125 (2021) 4578–4588. doi: 10.1021/acs.jpca.1c02837
H.W.T. Morgan, C.C. Shu, Z.M. Sun, J.E. McGrady, J. Am. Chem. Soc. 144 (2022) 8007–8017. doi: 10.1021/jacs.1c10106
L. Qiao, C. Zhang, C.C. Shu, et al., J. Am. Chem. Soc. 142 (2020) 13288–13293. doi: 10.1021/jacs.0c04815
V. Khanna, R. Singh, P. Claes, et al., J. Phys. Chem. A 126 (2022) 1617–1626. doi: 10.1021/acs.jpca.1c10027
N.T. Mai, N.T. Tung, P.T. Thuy, N.T. Minh Hue, N.T. Cuong, Comput. Theoret. Chem. 1117 (2017) 124–129. doi: 10.1016/j.comptc.2017.08.012
H.T. Pham, T.T. Phan, N.M. Tam, et al., Phys. Chem. Chem. Phys. 17 (2015) 17566–17570. doi: 10.1039/C5CP02257F
V.T. Tran, Q.T. Tran, Int. J. Quant. Chem. 121 (2021) e26619. doi: 10.1002/qua.26619
X. Li, P. Claes, M. Haertelt, et al., Phys. Chem. Chem. Phys. 18 (2016) 6291–6300. doi: 10.1039/C5CP07298K
X.Q. Liang, X.J. Deng, S.J. Lu, et al., J. Phys. Chem. C 121 (2017) 7037–7046. doi: 10.1021/acs.jpcc.7b00943
S.J. Lu, L.R. Hu, X.L. Xu, et al., Phys. Chem. Chem. Phys. 18 (2016) 20321–20329. doi: 10.1039/C6CP00373G
M. Shibuta, T. Niikura, T. Kamoshida, H. Tsunoyama, A. Nakajima, Phys. Chem. Chem. Phys. 20 (2018) 26273–26279. doi: 10.1039/c8cp05580g
H. Tsunoyama, H. Akatsuka, M. Shibuta, et al., J. Phys. Chem. C 121 (2017) 20507–20516. doi: 10.1021/acs.jpcc.7b06449
M.F. Groh, U. Müller, A. Isaeva, M. Ruck, Zeitschrift für anorganische und allgemeine Chem. 645 (2019) 161–169. doi: 10.1002/zaac.201800441
F. Lips, S. Dehnen, Angew. Chem. Int. Ed. 50 (2011) 955–959. doi: 10.1002/anie.201005496
R. Ababei, J. Heine, M. Hołyńska, et al., Chem. Commun. 48 (2012) 11295–11297. doi: 10.1039/c2cc35318k
R.J. Wilson, F. Hastreiter, K. Reiter, et al., Angew. Chem. Int. Ed. 57 (2018) 15359–15363. doi: 10.1002/anie.201807180
S. Charles, B.W. Eichhorn, S.G. Bott, J. Am. Chem. Soc. 115 (1993) 5837–5838. doi: 10.1021/ja00066a067
S.C. Critchlow, J.D. Corbett, Inorg. Chem. 21 (1982) 3286–3290. doi: 10.1021/ic00139a008
L.O. Spreer, I. Shah, Inorg. Chem. 20 (1981) 4025–4027. doi: 10.1021/ic50225a093
G. Sheldrick, Acta Cryst. A 71 (2015) 3–8. doi: 10.1107/S2053273314026370
O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard, H. Puschmann, J. Appl. Crystal. 42 (2009) 339–341. doi: 10.1107/S0021889808042726
G. te Velde, F.M. Bickelhaupt, E.J. Baerends, et al., J. Comput. Chem. 22 (2001) 931–967. doi: 10.1002/jcc.1056
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865–3868. doi: 10.1103/PhysRevLett.77.3865
E. van Lenthe, A. Ehlers, E.J. Baerends, J. Chem. Phys. 110 (1999) 8943–8953. doi: 10.1063/1.478813
Y. Zhao, D.G. Truhlar, J. Chem. Phys. 125 (2006) 194101. doi: 10.1063/1.2370993
C. Adamo, V. Barone, J. Chem. Phys. 110 (1999) 6158–6170. doi: 10.1063/1.478522
E. Van Lenthe, E.J. Baerends, J. Comput. Chem. 24 (2003) 1142–1156. doi: 10.1002/jcc.10255
M. Franchini, P.H.T. Philipsen, L. Visscher, J. Comput. Chem. 34 (2013) 1819–1827. doi: 10.1002/jcc.23323
C.C. Pye, T. Ziegler, Theoret. Chem. Acc. 101 (1999) 396–408. doi: 10.1007/s002140050457
F.M. Bickelhaupt, E.J. Baerends, Rev. Comput. Chem. (2000) 1–86. doi: 10.1002/9780470125922.ch1
B. Kesanli, J. Fettinger, B. Eichhorn, J. Am. Chem. Soc. 125 (2003) 7367–7376. doi: 10.1021/ja034207e
Z. Li, W. Wu, S. Li, J. Mol. Struct. Theochem. 908 (2009) 73–78. doi: 10.1016/j.theochem.2009.05.006
H.G. von Schnering, J. Wolf, D. Weber, R. Ramirez, T. Meyer, Angew. Chem. Int. Ed. 25 (1986) 353–354. doi: 10.1002/anie.198603531
S. Dehnen, C. Zimmermann, Zeitschrift für anorganische und allgemeine Chem. 628 (2002) 2463–2469. doi: 10.1002/1521-3749(200211)628:11<2463::AID-ZAAC2463>3.0.CO;2-Y
C.H.E. Belin, J.D. Corbett, A. Cisar, J. Am. Chem. Soc. 99 (1977) 7163–7169. doi: 10.1021/ja00464a011
T.F. Fässler, Coord. Chem. Rev. 215 (2001) 347–377. doi: 10.1016/S0010-8545(01)00321-6
T.F. Fässler, U. Schütz, Inorg. Chem. 38 (1999) 1866–1870. doi: 10.1021/ic981157j
J. Rienmüller, A. Schmidt, N.J. Yutronkie, et al., Angew. Chem. Int. Ed. 61 (2022) e202210683. doi: 10.1002/anie.202210683
J.M. Goicoechea, J.E. McGrady, Dalton Trans. 44 (2015) 6755–6766. doi: 10.1039/C4DT03573A
E.D. Jemmis, M.M. Balakrishnarajan, P.D. Pancharatna, J. Am. Chem. Soc. 123 (2001) 4313–4323. doi: 10.1021/ja003233z

Figure 1 Structural trends within the ME10, ME12, M2E16 and M2E12 families. The two new clusters reported in this paper are highlighted in the box.

Figure 2 Unit cell of 1 and the structure of the 75-electron anion [Fe2Sn4Bi8]3– (side and top views, hydrogen atoms are removed for clarity).

Figure 3 Unit cell of 2 and the structure of the 75-electron anion [Cr2Sb12]3– (side and top views, hydrogen atoms are removed for clarity). The unit cell shows the two components on each lattice site (50:50 ratio), which differ in the orientation of the polar axis.

Figure 4 Negative ion ESI mass spectra of (a) [K(18-crown-6)]4[Cr2Sb12]·Cp and (b) [K(2.2.2-crypt)]3[Fe2Sn4Bi8]. Mass ranges are m/z 800–2000 and 1200–2800, respectively.

Figure 5 (a) Kohn-Sham orbitals, (b) PDOS and (c) OPDOS of [Ni2Bi12]4+. The Fermi level, Ef, is defined as the mid-point between the HOMO and the LUMO. Bia and Bie indicate axial and equatorial Bi atoms, respectively.

Figure 6 Kohn-Sham orbitals (α and β spins), PDOS and OPDOS for the spin-β-manifold of [Fe2Sn4Bi8]3–. The Fermi level, Ef, is defined as the mid-point between the HOMO and the LUMO of the spin-β set. Spin-α PDOS are shown for comparison in Fig. S27 (Supporting information).

Figure 7 Difference density plots for [Ni2Bi12]4+ and [Fe2Sn4Bi8]3–, showing the difference in electron density between the self-consistent solution and the sum of two separated fragments (Ni2 + Bi124+ or Fe23– + Sn4Bi8, respectively). Contours range from −3 × 10−3 e/au3 to 3 × 10−3 e/au3, and values on the x and y axes are in Å.

Figure 8 Kohn-Sham eigenvalue spectrum (α and β spins), PDOS and OPDOS for the spin-β manifold of [Cr2Sb12]3–. Spin-α PDOS are shown for comparison in Fig. S28 (Supporting information). The Fermi level, Ef, is defined as the mid-point between the HOMO and the LUMO of the spin-β set. The labels Cr1, Sb2, etc. are identified in the structural diagram.

Figure 9 Walsh diagram connecting the D4h- and C4v-symmetric limits in [Cr2Sb12]3–. 18 intermediate structures were generated by interpolating between the two optimized structures in Table 1. The orbitals and PDOS plots shown correspond to the spin-β set: Occupied levels are shown as full lines, virtual levels as dashed lines and the 1a2/2a2 pair is shown in blue.

Figure 10 Overview of the trends in electronic structure across the 75- and 76-electron M2E12 series.
Table 1. Total valence-electron count (VE) and crystallographic and DFT-optimized bond lengths for members of the M2E4E′8 family (all values in Å).
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: