Citation:
Sen Qiao, Kunquan Chen, Qiang Liu, Zhixiang Wang, Xiangyu Chen. Charge transfer complex enabled photoreduction of ether phosphonium salts for the selective oxyalkylation of enamides[J]. Chinese Chemical Letters,
2024, 35(2): 108979.
doi:
10.1016/j.cclet.2023.108979
Charge transfer complex enabled photoreduction of ether phosphonium salts for the selective oxyalkylation of enamides
English
Charge transfer complex enabled photoreduction of ether phosphonium salts for the selective oxyalkylation of enamides
Received Date:
31 May 2023 Accepted Date:
24 August 2023 Revised Date:
09 August 2023 Available Online:
15 February 2024
Abstract:
A charge transfer complex (CTC)-enabled photoreduction of ether phosphonium salts for the generation of oxyalkyl radicals was described. The photoreduction provides a convenient method to achieve selective oxyalkylation of enamides with broad substrate scope. The method features operational simplicity, mild and inherent green conditions.
Enamides are essential building blocks for the synthesis of pharmaceuticals and natural products [1-4]. Thus, the direct functionalization of enamides to obtain high value-added complex compounds is of great importance and has remained a long-standing interest in organic synthesis [5-23]. The development of synthetic strategies for the β-C(sp2)–H alkylation of enamides has gained considerable efforts [24-26]. Transition-metal-catalyzed cross coupling reactions have been proven to be effective for the regio- and stereoselective synthesis of β-alkylated enamides [27-32]. Meanwhile, visible-light-catalysis provides a powerful and sustainable alternative for radical alkylation reactions [33-40]. In this context, Yu and co-workers disclosed a photoredox-catalyzed alkylation reaction of enamides with alkyl bromides as radical precursors [41]. The group of Zhao, Loh [42,43] and Shang, Fu [44] developed photocatalytic decarboxylative coupling reactions of enamides with N-hydroxyphthalimide (NHPI) esters to prepare alkylated enamides, respectively. Our group demonstrated that a charge transfer complex (CTC) between NHPI ester and enamide could lead to the alkylation of enamides [45]. Lu and Xiao and co-workers described an elegant deaminative alkylation of enamides with Katritzky salts by using an Ir-based photocatalyst (Fig. 1A) [46]. Despite these advances, these elegant methods suffered from the use of expensive photocatalysts or stoichiometric amounts of additive and the limited radical precursors. The development of new alkylating agents to achieve the selective functionalization of enamides under mild condition is still highly desirable.
Figure 1
Figure 1.
(A) photocatalysis for the alkylation of enamides; (B) photoredox-catalyzed CDC reactions for the oxyalkylation of enamides; (C) phosphonium salts as precursors generating organic radical species; (D) CTC-enabled photoreduction of ether phosphonium salts for the oxyalkylation of enamides.
Ethers are important scaffolds appearing in natural products and drug molecules [47-49]. Specifically, tetrahydrofuran (THF), one of the common solvents, has been widely used as a basic chemical feedstock in synthetic chemistry and industrial processes [50]. Great efforts have been devoted to the development of novel methods for their transformations [51,52]. Surprisingly, the photoinduced oxyalkylation of enamides from ether compounds was rarely explored. Very recently, Zhao and co-workers described an Ir/Eosin Y photocatalyst-catalyzed cross-dehydrogenative coupling (CDC) reactions for the ether functionalization of enamides (Fig. 1B) [53]. However, the photocatalyst, excess oxidant, and stoichiometric additive were all indispensable. In the past decade, CTC-enabled photoreaction has emerged as an effective alternative for radical generation under transition metal-, photocatalyst-, and oxidant-free conditions [54-56]. Our group demonstrated that the phosphonium salts could form charge transfer complexes (CTCs) with suitable electron donors to generate organic radical species under blue light irradiation without using photocatalysts and transition metals (Fig. 1C) [57-63]. We wondered whether we could introduce this strategy to realize the selective oxyalkylation of enamides. If so, a new and efficient method to generate oxyalkyl radicals would be established. Herein, we disclosed a blue light induced CTC-enabled photoreduction of ether phosphonium salts for the selective oxyalkylation of enamides without using photocatalyst, transition metal, and stoichiometric amounts of additive (Fig. 1D).
To verify the feasibility of our method, we first examined whether the CTC strategy could promote the oxyalkylation of enamides with THF phosphonium salt 1, which was prepared from the reaction of perfluorobutyl iodide, PPh3, and THF under reflux. To our delight, treatment of 1, enamide 2 with TMEDA as the additive and DMA as the solvent under irradiation of blue light gave the desired product 3 in 84% yield with E/Z = 6.4:1 (entry 1, Table 1). Previous study found that the deprotonation of the iminium ion intermediate significantly affect the E/Z ratios of the products [53]. Likewise, we evaluated various bases including inorganic bases and organic bases, such as DIPEA, DBU, DABCO, DMAP, K2CO3, Cs2CO3, and NaOEt (Table S1 in Supporting information for more results), but these bases had no obvious effects on the E/Z ratios (entries 2-8). Solvent screening revealed that DMA was the best choice (entry 9). Notably, the reaction worked as well in the absence of TMEDA, affording the desired product in 55% yield (entry 10). Changing the equivalent of TMEDA was carried out but no better result was delivered (entry 11). The control experiment confirmed the necessity of visible light (entry 12).
With the optimized conditions in hand, the reaction scope was investigated. As presented in Scheme 1, the results revealed that the current strategy turned out to be a general and efficient method for the synthesis of oxyalkylated enamides. The gram-scale reaction between 1 and 2 gave rise to 3 in 71% yield with 5.9:1 E/Z ratio (Supporting information for details). Both electron-donating (MeO) and electron-withdrawing (F, Br, CF3 and CO2Et) groups on the phenyl ring at the para-, ortho-, or meta-position of enamides were tolerable and furnished the corresponding products 4–10 in moderate to good yields with good E/Z selectivity. Multi-substituted enamides were also used and provided the target products 11 and 12 with good results. Naphthyl-substituted substrate was also used and delivered the desired enamide 13 in 70% yield with 3.3:1 E/Z ratio. In addition, the R1 and R2 substituents were also examined. The desired oxyalkylated enamides 14 and 15 were obtained in 75% and 72% yields, respectively. The N-alkyl enamides without a Bn group were also tolerable and gave good yields of the products 16 and 17, with good E/Z ratios. To further examine the generality of the strategy, other reaction partners, such as vinylnaphthalene, N-arylacrylamide, and N-methacryloyl-N-methylbenzamide, were introduced to the reaction system and the desired oxyalkylated alkene, oxindole, and isoquinolinedione were obtained with good reaction efficiency (18–20). Next, we studied the versatility of the reaction with various ether phosphonium salts, which could be easily prepared from the corresponding commercially available halides. Tetrahydropyran derived phosphonium salt worked well to afford the desired product in 78% yield with 4.6:1 E/Z ratio (21). Acyclic ether, including dimethyl ether, ethyl methyl ether, benzyl methyl ether, and dimethoxyethane, derived phosphonium salts could be readily employed to forge a variety of enamides with satisfactory results (22–25). In addition, TMS-substituted ether phosphonium salt also reacted smoothly to give the desired product (26). It was noteworthy that phosphonium salt prepared from ester was also effective to furnish the oxyalkylated enamides (27).
Scheme 1
Scheme 1.
Substrate scope. Yields of isolated products are given. The E/Z ratios were determined by 1H NMR spectroscopy of the reaction mixtures. a4 mmol scale reaction.
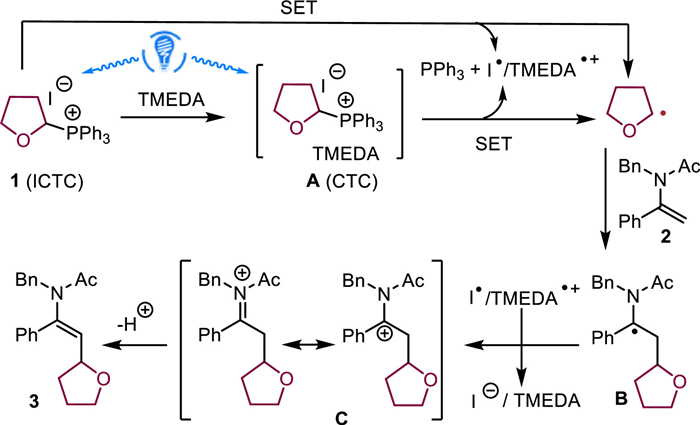
To better understand the reaction, a series of mechanistic experiments were carried out (Scheme 2). The UV-vis experiments were conducted to identify the possible pathways for the photoactivation of phosphonium salts (Scheme 2A). The UV-vis spectroscopy studies showed that phosphonium iodide salt 1 exhibited good photoactivity. Its peak tail deeply extended to the visible light region and reached a wavelength of over 600 nm, indicating that the salt could be activated by blue light irradiation. The absorption of the mixture of 1 and TMEDA shifted bathochromically compared to 1 and TMEDA, while the mixture of 1 with 2 exhibited no redshifted absorption. These results suggested that a more photoactive charge transfer complex could be formed between 1 and TMEDA. Nevertheless, the photolysis experiments of the salt also demonstrated that the phosphonium iodide salt alone was sufficiently photoactive as an intramolecular charge transfer complex (ICTC) to generate THF radical, as well as the byproduct PPh3 (Scheme 2B). In addition, the radical inhibition study also suggested the presence of a free THF radical in this transformation (Scheme 2C). In addition, the chloride salt 28 was employed as the phosphonium salt for the reaction. In the absence of TMEDA, only trace amount of product 22 could be obtained, but the addition of TMEDA obviously increased the yield of the product (Scheme 2D). These results imply an important role of I− counter anion, but TMEDA could also serve as an electron donor to enable single electron transfer process for the generation of the oxyalkyl radical.
According to the above results and our previous reports [59-62], we proposed a possible mechanism in Fig. 2. First, the photoactive complexes, including the intramolecular ICTC (1) and intermolecular CTC A, could be excited by blue light to undergo a SET process, where I− and TMEDA serve as the electron donor, respectively. The SET processes generate the oxyalkyl radical/I• and the oxyalkyl radical/TMEDA•+, respectively. Then the addition of the THF radical to the C=C double bond of enamide 2 afforded radical intermediate B, which could be sequentially oxidized by I•/TMEDA•+ to provide a cationic intermediate C featuring two key resonance structures. Finally, the deprotonation of C furnished the product 3.
According to the mechanism, we propose that the reaction could be benefited from the addition of TMEDA due to the following reasons: (ⅰ) The reaction involves deprotonation, which could be facilitated by stronger base TMEDA, (ⅱ) the reaction produces HI as the byproduct which could be stabilized by TMEDA, and (ⅲ) TMEDA serves as an electron donor to form photoactive CTC, thus enhancing the photoactivity of the system. Nevertheless, these roles of TMEDA are not essential, because (ⅰ) and (ⅱ) could be fulfilled by DMA solvent and the salt itself is photoactive. As such, the reaction could give 55% yield in the absence of TMEDA (entry 10 in Table 1).
In conclusion, we have developed a simple and efficient strategy for the direct oxyalkylation of enamides with ether phosphonium salts as the oxyalkylating reagents. The ether phosphonium salts themselves or their combinations with TMEDA could serve as photoactive ICTCs or CTCs to generate oxyalkyl radicals under visible light irradiation. With this strategy, enamides, vinylnaphthalene, oxindole, and isoquinolinedione, bearing ether groups can be easily prepared without using transition metal, photocatalyst, and stoichiometric additive. It is expected that the synthetic method could have potential applications in the synthesis of diverse enamides.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (No. 22001248) and the Fundamental Research Funds for the Central Universities and University of Chinese Academy of Sciences.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108979.
[1]
T. Kuranaga, Y. Sesoko, M. Inoue, Nat. Prod. Rep. 31 (2014) 514–532. doi: 10.1039/c3np70103d
[2]
T. Courant, G. Dagousset, G. Masson, Synthesis 47 (2015) 1799–1856. doi: 10.1055/s-0034-1378706
Q. Liu, B.B. Zhang, H. Sheng, et al., Angew. Chem. Int. Ed. (2023) e202305088.
[62]
F. Tan, P. Zheng, Q. Liu, X.Y. Chen, Org. Chem. Front. 9 (2022) 5469–5472. doi: 10.1039/d2qo01079h
[63]
Q. Liu, Y. Lu, H. Sheng, et al., Angew. Chem. Int. Ed. 60 (2021) 25477–25484. doi: 10.1002/anie.202111006
Figure 1
(A) photocatalysis for the alkylation of enamides; (B) photoredox-catalyzed CDC reactions for the oxyalkylation of enamides; (C) phosphonium salts as precursors generating organic radical species; (D) CTC-enabled photoreduction of ether phosphonium salts for the oxyalkylation of enamides.
Scheme 1
Substrate scope. Yields of isolated products are given. The E/Z ratios were determined by 1H NMR spectroscopy of the reaction mixtures. a4 mmol scale reaction.


 DownLoad:
DownLoad:




 下载:
下载:





 下载:
下载:
