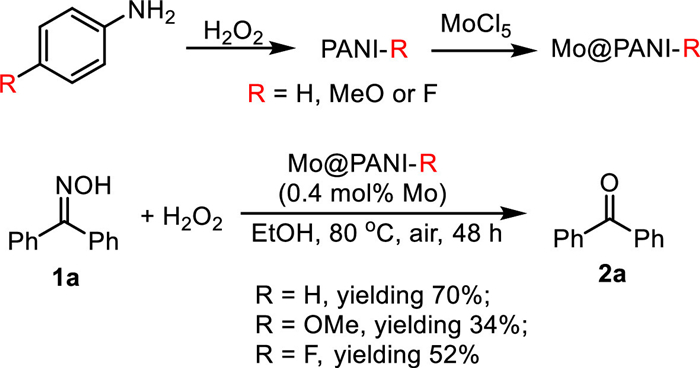
Scheme 1.
Catalyst preparation & screenings.
Mo@PANI-catalyzed oxidative deoximation reaction
Yiyang Zhang , Wen Li , Zuofeng Hu , Xiaobi Jing , Lei Yu

Deoximation is an important transformation in synthetic industry [1,2]. Since oximes are usually crystals with stable melting points, the oximation-deoximation strategies can be employed to protect, characterize or purify carbonyls in the total synthesis of medicines or natural products. Early in 1979, Cory et al. have employed this protocol in the total synthesis of erthronolide A, a macro-cyclic antibiotic molecule with endo cyclic carbonyl [3]. In industrial production of watermelon ketone, oximation-deoximation processes are employed for product purification [4]. In addition, deoximation reaction can be employed in the production of ketones from non-carbonyl molecules. For example, the high-value-added spice carvone can be prepared from accessible limonene, and the processes include the addition of the endo cyclic C═C of limonene with NOCl, the elimination of HCl, and the last step deoximation reaction [5–7]. Catalytic oxidative deoximation reaction is now the major deoximation method at present. In comparison with the traditional acidic deoximation method, it can utilize the driving force of the unit reaction of oxidation so that the transformation can occur under relatively mild conditions free of hazardous additives or halogen-containing solvents [8–11], and is in accordance with the developing trend of industrial synthesis [12–14]. Currently, a series of elements such as Se, Te, Fe, have been employed to develop the catalysts for oxidative oximation reactions [15–19]. The reaction can also be catalyzed by organic molecules, but suffer from the narrow substrate scope and high catalyst loadings [20,21].
On the other hand, polyanilines (PANIs) are recently found to be nice catalyst supports [22]. Although anilines are toxic, their polymers are nontoxic and safe to the environments [23]. The nitrogen groups in PANIs can well coordinate with catalytic metals, i.e., PANIs are not only catalyst supports, but also ligands that may adjust the catalytic activities of the materials by using a varieties of easily available substituted anilines as their monomers [24–26]. During the last decade, a variety of PANI-supported metal catalysts using Pd [27], Au [28], Pt [29], Ni [30], Cu [31], etc., as the catalytic metals have been developed. These materials are phosphorus-free catalysts friendly to natural water sources, and the reactions can be performed under mild conditions with high catalyst turnover numbers (TONs). In our cases, we found that polyaniline-supported tungsten (W@PANI) could catalyze the oxidative deoximation [32]. Owing to the strong coordination of nitrogen with tungsten, metal residues in the produced ketones were very low, and this feature might in accordance with the requirements of pharmaceutical industry. Recently, we designed and prepared the polyaniline-supported molybdenum (Mo@PANI) catalyst and successfully applied it in oxidative deoximation reactions. Since Mo is a necessary trace element for both animals and plants, this method is environment-friendly and is suitable for large-scale preparation.
PANIs were prepared via the oxidative polymerization of the aniline monomers (Scheme 1). Three typical monomers such as aniline (R = H), p-anisidine (R = OMe) and p-fluoroaniline (R = F) were employed to produce the related PANIs marked as PANI-H, PANI-OMe and PANI-F respectively. In this work, we employed H2O2 as the clean oxidant so that the generation of solid wastes could be avoided [33]. Mo@PANIs were prepared just by immersing the related PANIs into the 60 vol% aqueous ethanol solution of MoCl5.
The prepared Mo@PANIs were then employed in the oxidative deoximation reaction of diphenylmethanone oxime (1a) to evaluate their catalytic activities (Scheme 1). The deoximation reactions also employed H2O2 as the clean oxidant and were performed in green solvent ethanol. Mo@PANIs containing 0.4 mol% of Mo vs. 1a were employed. The reaction catalyzed by Mo@PANI-H afforded the desired ketone 2a in 70% yield, while for the reactions being catalyzed by Mo@PANI-OMe and Mo@PANI-F, 2a was produced in 34% and 52% yields respectively. Clearly, Mo@PANI-H was screened out to be the preferable catalyst. Introducing electron-donating or -withdrawing groups onto the aromatic ring of PANI could not improve the catalyst activity.
A series of parallel experiments were then performed to optimize the reaction conditions. Without catalyst, the blank reaction of 1a with H2O2 in ethanol at 80 ℃ could afford 2a in 31% yield (Fig. 1a). The yield gradually increased along with the enhanced catalyst amount, and it could reach the peak at 77% when Mo@PANI-H containing 0.3 mol% of Mo vs. 1a was employed (Fig. 1a). Further increasing the catalyst amount led to decreased 1a yield contrarily, possibly due to the fact that Mo might led to the decomposition of the oxidant H2O2 (Fig. 1a). Without H2O2, 2a was produced in only 5% yield (Fig. 1b). Elevated H2O2 dosage could enhance the yield of 2a, and using 100 mol% of H2O2 vs. 1a was screened out to be the most favorable condition (Fig. 1b). Using excess H2O2 resulted in the generation of over oxidation by-products and led to the decrease of the 2a yield (Fig. 1b).

Catalyzed by Mo@PANI-H containing 0.3 mol% of Mo, and using 100 mol% of H2O2 as oxidant, parallel reactions of 1a at different reaction temperatures were performed and the results were given in Table 1. It was found that 60 ℃ was the preferable reaction temperature (Table 1, entry 2 vs. entries 1, 3 and 4). Low reaction temperature resulted in the incomplete reaction, while high reaction temperature might lead to the decomposition of H2O2, and all of them resulted in decreased product yields (Table 1, entry 2 vs. entries 1, 3 and 4). High polar solvents, such DMF and CH3CN, were not fit for the reaction (Table 1, entries 5 and 6). Reactions in 1,4-dioxane, DMC, EtOAc and toluene led to 2a in 56%–70% yields, less than the yield of the reaction in EtOH (Table 1, entries 2 vs. 7–10). Thus, it could be concluded that performing the reaction in ethanol at 60 ℃ should be the favorable reaction condition.
Under the optimized conditions described in Table 1, entry 2, a series of ketoximes 1 were employed as substrate to examine the scope of the reaction (Table 2). Besides 1a, diaryl ketoximes with electron-withdrawing or -donating substituents were also suitable substrates for the reaction (Table 2, entries 1–8). Notably, in regardless of the oxidative reaction conditions, substrates with reductive functional groups, such as phenol (1i) and aniline (1j), were fit for the reaction, leading to the desired ketones 2i and 2j in moderate yields (Table 2, entries 9 and 10). Besides diary ketoximes, the reaction could employ ketoximes bearing aliphatic groups as substrates, affording the related ketones in moderate to excellent yields (Table 2, entries 11–21). Moreover, the reaction showed certain degree tolerance to heterocycles in substrates, such as pyridine, thiophene and furan, indicating that the method might be applicable for medicine intermediate synthesis (Table 2, entries 22–24). Reaction of (E)−1-(naphthalen-2-yl)ethan-1-one oxime (1y) could produce 2y in 95% yield, in regardless of the steric hindrance of the substrate bearing naphthyl (Table 2, entry 25). Finally, cyclohexanone oxime (1z) was employed as the example of aliphatic substrate and its reaction led to 2z in 40% GC yield (Table 2, entry 26).
Materials characterizations as well as control experiments were performed to get sufficient information for reaction mechanism study. X-ray photoelectron spectroscopy (XPS) analysis showed that Mo existed as Mo6+ species in fresh Mo@PANI-H (Fig. 2). The result indicated that the nitrogen-coordinated Mo was easily oxidized by air under ambient conditions. Moreover, in the XPS spectrum of the used catalyst, both Mo5+ and Mo6+ species could be observed, attesting that the high valent Mo species could be reduced by the organic substrates during the deoximation reaction process, i.e., the valent of Mo shifted between +5 and +6 during the catalysis process.

MoCl5 could also catalyze the reaction. However, in comparison with Mo@PANI-H, the catalytic activity of MoCl5 was poor (Table 3, entry 2 vs. 1). Thus, it is supposed that PANI is a support that can enhance the activity of the metal owing to the coordination effect of its nitrogen group, and this has been discussed by our previous works [24–26]. Performing the reaction in N2 resulted in decreased product yield (Table 3, entry 3 vs. 1). Without H2O2 oxidant, the reaction in air could also produce 2a in 9% yield (Table 3, entry 4), and the product yield could be enhanced to 31% when O2 was employed as the oxidant (Table 3, entry 5). In contrast, no reaction occurred when heating 1a and Mo@PANI-H in N2 without H2O2 (Table 3, entry 6). The experimental results in Table 3, entries 3–6 could support the hypothesis that the Mo-catalyzed oxidative deoximation reaction could employ H2O2/air as the hybrid oxidant, so that no excess H2O2 was required. Addition of free radical scavenger TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) into the system could restrain the reaction, showing that it proceeded via a free radical reaction mechanism (Table 3, entry 7) [34]. Furthermore, the salicylic acid trapping experiment verified the existence of hydroxyl radical during the reaction progress (Fig. S1 in Supporting information). Cr@PANI can also catalyze the reaction, but the product yield is low (Table 3, entry 8).
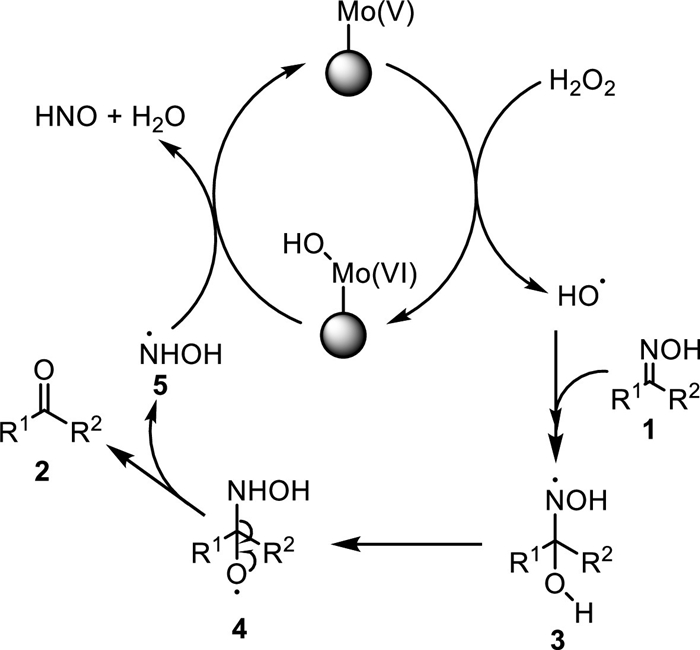
On the basis of the experimental results as well as the literature reports, a plausible mechanism of the reaction was supposed in Scheme 2. Since the reaction was performed in organic solvents such as ethanol, the Mo(Ⅵ) species in catalyst could be reduced to Mo(Ⅴ). Single electron transfer reaction of Mo(Ⅴ) with H2O2 could afford the hydroxyl radical, while Mo was oxidized into the Mo(Ⅵ) species bearing an addition hydroxyl group [35]. The free radical addition of hydroxyl radical with substrate 1 led to the intermediate 3 [36], which rearranged to give the radical intermediate 4 [36]. Decomposition of 4 produced the ketone 2, i.e. the deoximation product, and led to the radical 5 [37]. Shifting of the hydroxyl in Mo(Ⅵ) species to 5 led to HNO and H2O [38], and regenerated the Mo(Ⅴ) species that could restart the catalysis circle.
In conclusion, we designed and prepared Mo@PANI catalyst for the oxidative deoximation reaction. The material was easily prepared, while the deoximation process was clean. Since Mo is a necessary element for animals and plants, using this metal as catalyst is more environment friendly. Thus, this work could afford a practical method for deoximation reaction, which is an important transformation in synthetic industry. Continuous investigations are ongoing in our laboratory to expand the application scope of Mo-based catalysts in more useful reactions.
We thank Priority Academic Program Development of Jiangsu Higher Education Institutions for support. This project was also financially supported by the fund of the joint-laboratory of Shanghai Dingya Pharmaceutical Chemical Technology Co., Ltd. with Yangzhou University (No. 2022-2027).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Zhang, X. Wen, Y. Wang, W. Mo, C. Ding, Prog. Chem. 24 (2012) 361–369.
Y. Zheng, A. Wu, Y. Ke, H. Cao, L. Yu, Chin. Chem. Lett. 30 (2019) 937–941. doi: 10.1016/j.cclet.2019.01.012
E.J. Corey, P.B. Hopkins, S. Yoo, et al., J. Am. Chem. Soc. 101 (1979) 71311–77134.
Q. Tong, Y. Liu, X. Gao, et al., Adv. Synth. Catal. 361 (2019) 3137–3145. doi: 10.1002/adsc.201900202
E.E. Royals, S.E. Horne Jr, J. Am. Chem. Soc. 73 (1951) 5856–5857. doi: 10.1021/ja01156a119
A. Grirrane, A. Corma, H. Garcia, J. Catal. 268 (2009) 350–355. doi: 10.1016/j.jcat.2009.10.005
C. Isart, D. Bastida, J. Burés, J. Vilarrasa, Angew. Chem. Int. Ed. 50 (2011) 3275–3279. doi: 10.1002/anie.201007269
P.K. Pradhan, S. Dey, P. Jaisankar, V.S. Giri, Synth. Commun. 35 (2005) 913–922. doi: 10.1081/SCC-200051681
M.H. Lin, H.J. Liu, C.Y. Chang, W.C. Lin, T.H. Chuang, Molecules 17 (2012) 2464–2473. doi: 10.3390/molecules17032464
L. Du, J. Gao, S. Yang, et al., Russ. J. Gen. Chem. 84 (2014) 2200–2204. doi: 10.1134/S1070363214110267
Y. Liu, N. Yang, C. Chu, R. Liu, Chin. J. Chem. 33 (2015) 1011–1014. doi: 10.1002/cjoc.201500325
C. Chen, Y. Yao, X. Wu, et al., Chin. Chem. Lett. 31 (2020) 1078–1082. doi: 10.1016/j.cclet.2019.12.019
X. Xiao, Z. Shao, L. Yu, Chin. Chem. Lett. 32 (2021) 2933–2938. doi: 10.1016/j.cclet.2021.03.047
J. Liu, Y. Cai, H. Pang, et al., Chin. Chem. Lett. 33 (2022) 4061–4063. doi: 10.1016/j.cclet.2021.12.068
X. Jing, D. Yuan, L. Yu, Adv. Synth. Catal. 359 (2017) 1194–1201. doi: 10.1002/adsc.201601353
C. Chen, X. Zhang, H. Cao, et al., Adv. Synth. Catal. 361 (2019) 603–610. doi: 10.1002/adsc.201801163
X. Deng, H. Cao, C. Chen, H. Zhou, L. Yu, Sci. Bull. 64 (2019) 1280–1284. doi: 10.1016/j.scib.2019.07.007
X. Deng, R. Qian, H. Zhou, L. Yu, Chin. Chem. Lett. 32 (2021) 1029–1032. doi: 10.1016/j.cclet.2020.09.012
F. Wang, C. Yang, Y. Shi, L. Yu, Mol. Catal. 514 (2021) 111849. doi: 10.1016/j.mcat.2021.111849
H. Li, X. Jing, Y. Shi, et al., React. Chem. Eng. 6 (2021) 119–124. doi: 10.1039/d0re00333f
F. Wang, T. Chen, Y. Shi, L. Yu, Asian J. Org. Chem. 10 (2021) 614–618. doi: 10.1002/ajoc.202000675
Z. Zeng, Y. Chen, X. Zhu, et al., Chin. Chem. Lett. 34 (2023) 107728. doi: 10.1016/j.cclet.2022.08.008
H. Zhao, B. Zhu, J. Sekine, S.C. Luo, H.H. Yu, ACS Appl. Mater. Interfaces 4 (2012) 680–686. doi: 10.1021/am2012905
Y. Liu, D. Tang, K. Cao, et al., J. Catal. 360 (2018) 250–260. doi: 10.1016/j.jcat.2018.01.026
X. Meng, Y. Zhang, H. Hong, et al., ACS Sustain. Chem. Eng. 10 (2022) 7658–7663. doi: 10.1021/acssuschemeng.2c01540
Y. Chen, L. Yu, H. Zhou, J. Phys. Chem. C 126 (2022) 17084–17092. doi: 10.1021/acs.jpcc.2c05290
L. Yu, Y. Huang, Z. Wei, et al., J. Org. Chem. 80 (2015) 8677–8683. doi: 10.1021/acs.joc.5b01358
A. Vijayakumar, Y. Zhao, J. Zou, et al., ChemSusChem 13 (2020) 5023–5030. doi: 10.1002/cssc.202001248
M.J. García-Fernández, M.M. Pastor-Blas, F. Epron, Appl. Catal. B: Env. 225 (2018) 162–171. doi: 10.1016/j.apcatb.2017.11.064
M. Sun, Z. Gong, J. Yi, et al., Chem. Commun. 56 (2020) 8798–8801. doi: 10.1039/d0cc03410j
Y. Chen, X. Jing, L. Yu, Chin. J. Org. Chem. 40 (2020) 2570–2574. doi: 10.6023/cjoc202003044
W. Li, F. Wang, Y. Shi, L. Yu, Chin. Chem. Lett. 34 (2023) 107505. doi: 10.1016/j.cclet.2022.05.019
G. Gao, J. Han, L. Yu, Q. Xu, Synlett 30 (2019) 1703–1707. doi: 10.1055/s-0037-1612088
Y. Chen, C. Chen, Y. Liu, L. Yu, Chin. Chem. Lett. 34 (2023) 108489. doi: 10.1016/j.cclet.2023.108489
Y. Yuan, Z. Wang, Y. Su, J. Chem. Eng. Chin. Univ. 20 (2006) 548–553.
K.R. Huber, R. Sridhar, E.H. Griffith, et al., Biochim. Biophys. Acta 915 (1987) 267–276. doi: 10.1016/0167-4838(87)90309-8
G. Wood, C.J. Easton, A. Rauk, et al., J. Phys. Chem. A 110 (2006) 10316–10323. doi: 10.1021/jp062916j
H. Mikami, S. Saito, S. Yamamoto, J. Chem. Phys. 94 (1991) 3415–3422. doi: 10.1063/1.459764

Figure 1 Screenings of the catalyst (a) and H2O2 (b) dosage for the reaction of 1a in ethanol at 80 ℃ for 48 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:




 下载:
下载:


 下载:
下载: