
Scheme 1.
Study on ligand effects in Rh(Ⅲ)-catalyzed arene–alkene coupling reactions.
Elucidating ligand effects in rhodium(Ⅲ)-catalyzed arene–alkene coupling reactions
Kongchuan Wu , Dandan Lu , Jianbin Lin , Ting-Bin Wen , Wei Hao , Kai Tan , Hui-Jun Zhang

As powerful methods for C—C bond formation, C—H couplings of arenes with alkenes have been actively studied using a broad range of transition-metal catalysts (e.g., Pd, Rh, Ru and Ir) [1-5]. Nevertheless, many important mechanistic details with respect to ligand effects remain largely unknown [6-12]. Metal hydride species are often proposed as crucial intermediates relevant to product-determing steps in these transformations [13-22], yet in-depth investigations on their formation and reactivities are scarce [23,24]. In recent years, a variety of well-defined rhodium(Ⅲ) complexes with electronically and sterically tuned cyclopentadienyl (CpX) ligands have proven to be efficient catalysts for arene–alkene coupling reactions [25-33]. Subtle manipulation of the CpX ligands can result in significant changes in reactivity and selectivity. For instance, as depicted in Scheme 1, the reaction of arenes with vinyl esters under [Cp*RhⅢ] catalysis leads to the selective formation of vinyl arenes [29], while [CpRhⅢ] can catalyze dehydrogenative alkenylation of arenes [30]. The reason for such a dichotomy in product distribution remains unclear. We also question whether this kind of ligand-tuned selectivity is widespread in other arene–alkene coupling reactions.
Herein, we present a comprehensive mechanistic study of the C—H coupling reactions of benzamides with alkenes, such as vinyl acetate and methyl acrylate. These reactions were catalyzed by three distinct [CpXRhⅢ] catalysts (Scheme 1), where CpX represents Cp, CpCF3 and Cp* [34-36]. Our study demonstrated that the Cp ligand, which is more electron-deficient and less sterically hindered than Cp* ligand, can readily facilitate β-H elimination and the following reductive elimination of the corresponding rhodium hydride species. In contrast, rhodium hydride species containing the electron rich Cp* ligand tend to undergo reinsertion of the alkene, thereby enabling rhodium-walking. Based on these mechanistic findings, we have also developed several novel ligand-directed divergent arene–alkene coupling reactions.
We first synthesized and isolated three seven-membered rhodacycles 3-Cp*, 3-Cp and 3-Cp, as shown in Fig. 1. These complexes are believed to be crucial intermediates in the [CpXRhⅢ]-catalyzed C—H couplings of benzamides with vinyl acetate [5,27-33]. Stoichiometric reactions of [Cp*RhCl2]2, [CpCF3RhCl2]2 and [CpRhI2]n with benzamide 1a and vinyl acetate 2a were performed in acetone. We added 1.0 equiv. of NaOAc and 2.1 equiv. of AgSbF6 to the reaction systems to promote C—H rhodation [37,38] and enhance the reactivity of corresponding cyclometalated Rh intermediates [39,40]. After stirring the reaction systems at room temperature for 2–4 h, we were able to isolate 3-Cp*, 3-Cp and 3-Cp in high yields of 95%, 89% and 94%, respectively. Their 1H NMR spectra display ddd multiplets (δ 6.30 (3-Cp*), 6.75 (3-Cp) and 8.08 (3-Cp)) for methine groups that are connected to the rhodium centers. The corresponding 13C NMR signals appear at δ 105.5, 105.0 and 97.5 (doublet, 1Rh-C = 31.1, 28.1, 25.6 Hz). The 1H NMR signals of Cp* ligands in 3-Cp* and Cp ligand in 3-Cp appear at δ 1.29 (singlet) and 5.01 (doublet, 2JRh-H = 0.8 Hz), respectively. The CpCF3 ligand in 3-Cp displays an unusual set of four singlets, indicating four inequivalent methyl groups with different environments. The structures of 3-Cp*, 3-Cp and 3-Cp were all unambiguously confirmed by single-crystal X-ray diffraction analyses (Fig. 1b). The Rh-C and Rh-O distances decrease with an increase in the electron-deficiency of CpX ligands. These complexes are stable upon exposure to air for a few months perhaps due to extra coordination interactions between the rhodium centers and ester carbonyl groups. The molecular structures of 3-Cp*, 3-Cp and 3-Cp suggest a selective [1,2]-insertion of vinyl acetate into the Rh-C bonds of cyclometalated intermediates during the catalytic cycle.

The elimination reactions of the three alkene insertion intermediates 3-Cp were then investigated to determine the origin of selective vinylation or alkenylation (Fig. 2). Since the catalytic C—H couplings of arenes with alkenes are typically performed in the presence of Cu(OAc)2, acetate salts such as NBu4OAc and NaOAc were added (Fig. 2a, Figs. S8 and S13 in Supporting information) [5,27-33]. In the presence of highly soluble NBu4OAc, 3-Cp* quickly underwent elimination at room temperature to produce vinyl arene 4a and Cp*RhⅢ(OAc)2 (6-Cp*) in high yields (Fig. 2a). In contrast, treatment of 3-Cp with NBu4OAc mainly afforded alkenyl arene 5aa and a unique dimeric Rh(Ⅱ) complex 6′-Cp. Simultaneous formation of hydrogen gas was also observed by gas chromatography (Fig. S12 in Supporting information). The structure of 6′-Cp was confirmed by single-crystal X-ray diffraction analyses (Fig. 2a). We have also determined that 6′-Cp is a catalytically competent species in the arene–alkene couplings (Table S1 in Supporting information). These results indicate facile reductive elimination of corresponding [CpRh-H] intermediates in the catalytic cycle [41,42], although an acetate-promoted E2-type elimination cannot totally be excluded [43]. Similarly, addition of acetate salt also enabled the elimination of 3-Cp at room temperature, producing vinyl arene 4a and alkenyl arene 5aa in a ratio of 1:2. When the reaction was performed at 80 ℃, this ratio as well as the conversion of 3-Cp increased dramatically (Fig. S8 in Supporting information). We monitored these elimination processes by in situ 1H NMR spectroscopy. The kinetic profiles of the elimination reactions are displayed in Figs. 2b-d. The significant electronic effects of CpX ligands on product selectivities of the catalytic reactions were evident from the distinct initial rates of product formation (Fig. S9 in Supporting information).

Computational studies were also conducted on the elimination process using the Gaussian 16 program package [44]. Fig. 3 illustrates the energy profiles for 3-Cp, and additional energy values for the elimination processes of 3-Cp* and 3-Cp are also included. By exploring the potential energy surface, it is possible to locate stationary points and transition states that could give rise to rhodium-hydride intermediates IntB-Cp, which in turn can lead to the formation of both 5aa (path i) and 4a (path ii). The transition state TS1 for cis-β-hydride elimination of 3-Cp is markedly stabilized by the unsubstituted Cp ligand. In path i, the rhodium-hydride intermediate IntB-Cp can undergo reductive elimination via TS2 (with a free energy barrier of 4.72 kcal/mol) to generate 5aa and 6′-Cp. Our calculations indicate that the products lie 26.14 kcal/mol below the starting seven-membered rhodacycle 3-Cp. Alternatively, the rhodium-hydride intermediate can reinsert into the olefin to lead to the formation of a six-membered rhodacycle IntC—Cp as shown in path ii. The reaction free energy and energy barrier were calculated to be −18.88 and 7.16 kcal/mol (TS3), respectively. Subsequently, IntC—Cp undergoes β-acetate elimination, resulting in the formation of 4a and CpRhⅢ(OAc)2 (6-Cp). The free energy of the resulting products is predicted to be 4.92 kcal/mol higher than that of 5aa and 6′-Cp. Notably, the CpX ligand exerts a significant impact on the thermodynamic stability of the final products (Fig. 3a). In particular, replacing the Cp ligand with Cp* leads to a greater thermodynamic stability of the final products of path ii (−23.82 kcal/mol) than that of path i (−16.63 kcal/mol). This observation has been further confirmed by employing the CpCF3 ligand. On the other hand, the optimized structures of IntB-Cp complexes with Cp, CpCF3 and Cp* ligands are presented in Fig. S15 (Supporting information). The Rh-H bond lengths follow the order of Cp* (1.5544 Å) > CpCF3 (1.5421 Å) > Cp (1.5387 Å), suggesting that the Rh-H bond in IntB-Cp are stronger than that in IntB-Cp and IntB-Cp*. The HOMO and LUMO energy levels exhibit the trend of Cp═CpCF3 > Cp*, indicating that both Cp and CpCF3 ligands are more electron-deficient than Cp*, which further supports the relatively stronger Rh-H bonds in IntB-Cp and IntB-Cp. Overall, these findings could clearly explain the selective elimination of 3-Cp as illustrated in Fig. 2.

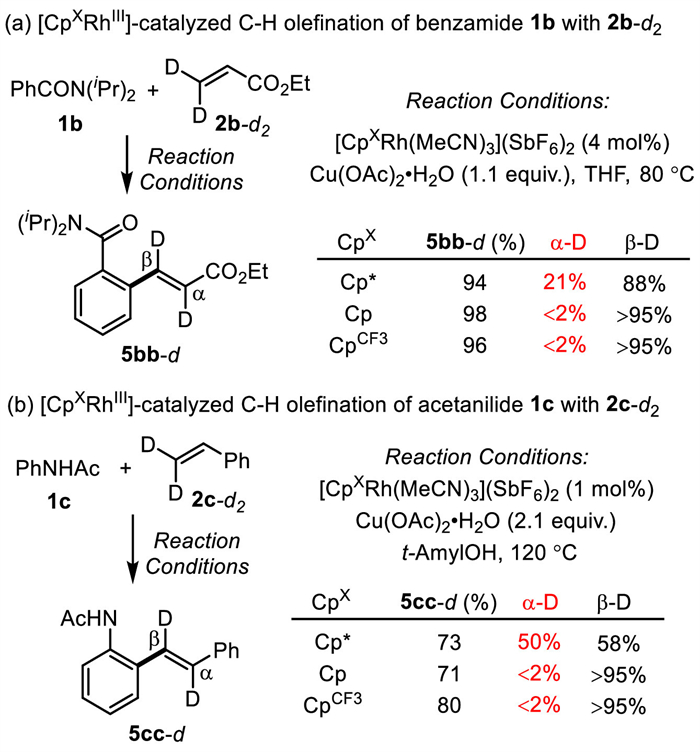
In order to investigate the effect of different CpX ligands on the C—H couplings of arenes with ethyl acrylate, catalytic reactions between benzamide 1b and β,β-dideuterated ethyl acrylate 2b-d were performed using three [CpXRh(MeCN)3](SbF6)2 precatalysts (CpX═Cp*, CpCF3 and Cp) (Fig. 4a). In the reaction catalyzed by electron-rich [Cp*RhⅢ], considerable migrations of β-deuterium onto α-position of the olefination products 5bb-d were observed, suggesting a β-H elimination/Rh-H olefin reinsertion sequence [45,46] in the catalytic cycle. On the contrary, no significant deuterium migrations were observed in reactions catalyzed by electron-deficient [CpCF3RhⅢ] and [CpRhⅢ]. Similar observations were made in [CpXRhⅢ]-catalyzed C—H olefination of electron-rich acetanilide 1c with β,β-dideuterated styrene 2c-d (Fig. 4b). Under catalytic reaction conditions, Rh-hydride olefin reinsertion was inhibited with electron-poor CpX ligands, possibly due to a facile β-H elimination/reductive elimination sequence.
To study the reaction mechanism, we thus conducted stoichiometric reactions of [Cp*RhCl2]2, [CpCF3RhCl2]2, and [CpRhI2]n with benzamide 1a and methyl acrylate 2d in the presence of AgSbF6 and NaOAc in acetone (Fig. 5a). Two six-membered rhodacycle complexes 7-Cp* and 7-Cp were isolated in good yields (73% and 70%). The 1H NMR spectra of 7-Cp* and 7-Cp showed ddd multiplets (δ 4.25 (7-Cp*) and 4.78 (7-Cp)) for methine groups connected to the rhodium centers. Along with these rhodacycles, the formation of the corresponding alkenylation product 5ad (26% for [Cp*Rh]; 40% for [CpCF3Rh]) were observed. The alkenylation reactions of 1a with 2d in the presence of 4 mol% of 7-Cp* and 7-Cp were then conducted, both of which resulted in the desired 5ad in good yields (Scheme S2 in Supporting information). However, the stoichiometric reaction of [CpRhI2]n with 1a and 2d directly afforded the alkenylation product 5ad in 80% yield (Fig. S6 in Supporting information). In this case, we did not observe any specific rhodium complexes related to the expected six-membered rhodacycle complex 7-Cp. The structures of 7-Cp* and 7-Cp were unambiguously confirmed by single-crystal X-ray diffraction analyses (Fig. 5b), which showed selective [1,1]-insertion of methyl acrylate 2d into the Rh-C bonds. Jones et al. previously established an analogous [1,1]-insertion process in reactions of cyclometalated rhodium complexes with ethylene and proposed a [1,2]-insertion/β-H elimination/Rh-H olefin reinsertion mechanism [45,46]. Here, the isolation of [1,1]-insertion products instead of the [1,2]-insertion counterparts could be attributed to the preference of six-membered-ring metallacycles over seven-membered-ring metallacycles. And the direct formation of 5ad instead of 7-Cp confirmed significant ligand effects on the reactivities of corresponding Rh-H intermediates.

The progress of the catalytic reactions of 1a and 2d with three [CpXRhⅢ] precatalysts (10 mol%) were further monitored utilizing 1H NMR spectroscopy (Fig. 5c and Table S4 in Supporting information). The results showed that the [CpRhⅢ]-catalyzed alkenylation is much faster than [Cp*RhⅢ]- or [CpCF3RhⅢ]-catalyzed reactions. And for the reaction catalyzed by [Cp*RhⅢ], 7-Cp* was the major rhodium species (7%) observed after 5 min, along with a minor amount of (μ2-H)Rh2 species. As shown in Fig. 5c, the (μ2-H)Rh2 species displays an evident triplet resonance (J = 26 Hz) in the hydride region (δ = −8.7), confirming its presence [47,48]. On the other hand, for the reaction catalyzed by [CpCF3RhⅢ], only 2% of 7-Cp was formed after 5 min, accompanied by several other unknown rhodium species (Supporting information). Because 7-Cp might be formed via the Rh-H olefin reinsertion step, they can be considered as alternate forms of corresponding rhodium hydride intermediates in the catalytic cycle. The concentrations of 7-Cp* and 7-Cp hardly varied with the reaction time. For the reaction catalyzed by [CpRhⅢ], we did not detect any signals belonging to 7-Cp or Rh-H species. These results confirmed more facile reductive elimination of the Rh-H intermediates with electron-deficient CpX ligands.
In Heck-type reactions, reinsertion of alkenes into metal hydride intermediates may result in undesired olefin isomerizations. However, in transformations like Sigman's asymmetric redox-relay oxidative Heck reactions [18,19], iterative β-hydride elimination/migratory insertion processes are promoted to achieve high selectivities. The knowledge of electronic ligand effects on β-H elimination/reductive elimination sequence can be utilized to customize catalytic systems for different needs. To this end, several arene–alkene coupling reactions were tested using different [CpXRh] catalysts (Scheme 2). Allylic surrogates, as unsaturated coupling partners, typically undergo direct C(sp2)-H allylation (via β-O or β-X elimination) instead of Heck-type olefination reaction (via β-H elimination) [49]. The catalytic reactions of benzamide 1a with allylic electrophile 2e- were conducted (Scheme 2a). [Cp*RhⅢ] led to 5ae' in 71% yield with excellent β-OCOPh elimination, while electron deficient [CpRhⅢ] selectively led to 5ae by β-H elimination. The deuterium-labelling results indicate that the reactions proceeded with complete γ-selectivity, ruling out the possibility of an oxidative addition of the allyl electrophile to RhⅢ centers. The [Cp*Rh]-catalyzed direct C(sp2)-H bond allylation with ally acetate have already been widely explored [50,51]. However, in the presence of [CpRhⅢ] catalyst, dehydrogenative C—H alkenylation reactions of benzamide 1a and acetanilide 1d with allyl acetate 2f proceeded smoothly (Scheme 2b), affording 5af and 5df in good yields (77% and 66%, respectively). [CpXRhⅢ]-catalyzed coupling reactions between benzamide 1b and methyl acrylate 2d were also tested using 3-methyl-1,4,2-dioxazol-5-one [17] as an oxidant (Scheme 2c). Under [Cp*RhⅢ] catalysis, the reaction afforded α-branched amine product 8 in 78% yield via an oxidative interception [52,53] of a [1,1]-insertion intermediate analogous to 7-Cp*. In contrast, under [CpCF3RhⅢ] or [CpRhⅢ] catalysis, alkenylation product 5ed was yielded selectively, indicating much more facile β-H elimination/reductive elimination of corresponding alkene insertion intermediates. The catalytic performance of [Cp*RhⅢ], [CpCF3RhⅢ] and [CpRhⅢ] in these reactions is consistent with the insights gained from the mechanistic studies.

In summary, we have investigated the impact of three representative CpX (Cp*, CpCF3 and Cp) ligands on rhodium(Ⅲ)-catalyzed arene–alkene coupling reactions. The successful isolation and characterization of seven-membered rhodacycles 3-Cp*, 3-Cp and 3-Cp, as well as six-membered rhodacycles 7-Cp* and 7-Cp, confirm the relationship between [1,2]-insertion and [1,1]-insertion processes. The experimental and theoretical studies on chemoselective eliminations of different 3-Cp, along with the isolation of corresponding Rh(Ⅲ) complex 6-Cp* and dimeric Rh(Ⅱ) complex 6′-Cp, demonstrates the significant impact of electronic ligand effects on product selectivity. These findings, together with the ligand-directed divergent coupling reactions of arenes with diverse alkenes, support the conclusions that (i) Rh(Ⅲ) centers with electron-deficient ligands can facilitate β-H elimination/reductive elimination sequences, (ii) Rh(Ⅲ) centers with electron-rich ligands can enable rhodium-migration. These mechanistic insights into ligand effects provide a roadmap for the rational design of increasingly efficient catalyst systems for C—C bond formation reactions.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the financial support from the National Natural Science Foundation of China (Nos. 21772162, 21772165, 22171237, 22071208) and Youth Innovation foundation of Xiamen (No. 3502Z20206058).
Supplementary material associated with this article can be found, in the online version, at doi:
J.L. Bras, J. Muzart, Chem. Rev. 111 (2011) 1170–1214. doi: 10.1021/cr100209d
C. Liu, J.W. Yuan, M. Gao, et al., Chem. Rev. 115 (2015) 12138–12204. doi: 10.1021/cr500431s
G.Y. Song, X.W. Li, Acc. Chem. Res. 48 (2015) 1007–1020. doi: 10.1021/acs.accounts.5b00077
C.S. Yeung, V.M. Dong, Chem. Rev. 111 (2011) 1215–1292. doi: 10.1021/cr100280d
R. Logeswaran, M. Jeganmohan, Adv. Synth. Catal. 364 (2022) 2113–2139. doi: 10.1002/adsc.202200193
R.D. Baxter, D. Sale, K.M. Engle, J.Q. Yu, D.G. Blackmond, J. Am. Chem. Soc. 134 (2012) 4600–4606. doi: 10.1021/ja207634t
M. Brasse, J. Campora, J.A. Ellman, R.G. Bergman, J. Am. Chem. Soc. 135 (2013) 6427–6430. doi: 10.1021/ja401561q
A. Deb, A. Hazra, Q. Peng, R.S. Paton, D. Maiti, J. Am. Chem. Soc. 139 (2017) 763–775. doi: 10.1021/jacs.6b10309
J. Kim, S.W. Park, M.H. Baik, S. Chang, J. Am. Chem. Soc. 137 (2015) 13448–13451. doi: 10.1021/jacs.5b09824
P. Wang, P.V. Erma, G.Q. Xia, et al., Nature 551 (2017) 489–493. doi: 10.1038/nature24632
S.L. Zhang, L. Shi, Y.Q. Ding, J. Am. Chem. Soc. 133 (2011) 20218–20229. doi: 10.1021/ja205294y
Y.T. Li, H. Shi, Chin. Chem. Lett. 35 (2024) 108650. doi: 10.1016/j.cclet.2023.108650
D. Fiorito, S. Scaringi, C. Mazet, Chem. Soc. Rev. 50 (2021) 1391–1406. doi: 10.1039/d0cs00449a
H.E. Bonfield, D. Valette, D.M. Lindsay, M. Reid, Chem. Eur. J. 27 (2021) 158–174. doi: 10.1002/chem.202002849
Y. Li, D. Wu, H.G. Cheng, G. Yin, Angew. Chem. Int. Ed. 59 (2020) 7990–8003. doi: 10.1002/anie.201913382
A. Vasseur, J. Bruffaerts, I. Marek, Nat. Chem. 11 (2016) 209–219. doi: 10.1038/nchem.2445
S. Maity, T.J. Potter, J.A. Ellman, Nat. Catal. 2 (2019) 756–762. doi: 10.1038/s41929-019-0330-7
W. Werner, T.S. Mei, A.J. Burckle, M.S. Sigman, Science 338 (2012) 1455–1458. doi: 10.1126/science.1229208
J.B. Liu, Q.J. Yuan, F.D. Toste, M.S. Sigman, Nat. Chem. 11 (2019) 710–715. doi: 10.1038/s41557-019-0289-7
C. Romano, D. Fiorito, C. Mazet, J. Am. Chem. Soc. 141 (2019) 16983–16990. doi: 10.1021/jacs.9b09373
N. Wagner-Carlberg, T. Rovis, J. Am. Chem. Soc. 144 (2022) 22426–22432. doi: 10.1021/jacs.2c10552
D. Wu, H.L. Pang, G.Y. Yin, Chin. Chem. Lett. 34 (2023) 108087. doi: 10.1016/j.cclet.2022.108087
S. Kim, F. Loose, M.J. Bezdek, X.P. Wang, P.J. Chirik, J. Am. Chem. Soc. 141 (2019) 17900–17908. doi: 10.1021/jacs.9b09540
Y.J. Wang, L. Zhu, Z.H. Shao, et al., J. Am. Chem. Soc. 141 (2019) 17337–17349. doi: 10.1021/jacs.9b09038
K. Tanaka, Rhodium Catalysis in Organic Synthesis: Methods and Reactions, Wiley-VCH, 2019.
T. Piou, T. Rovis, Acc. Chem. Res. 51 (2018) 170–180. doi: 10.1021/acs.accounts.7b00444
F.W. Patureau, T. Besset, F. Glorius, Angew. Chem. Int. Ed. 50 (2011) 1064–1067. doi: 10.1002/anie.201006222
Y. Takahama, Y. Shibata, K. Tanaka, Chem. Eur. J. 21 (2015) 9053–9056. doi: 10.1002/chem.201501232
K.D. Otley, J.A. Ellman, Org. Lett. 17 (2015) 1332–1335. doi: 10.1021/acs.orglett.5b00340
W. Lin, W. Li, D. Lu, et al., ACS Catal. 8 (2018) 8070–8076. doi: 10.1021/acscatal.8b01753
J. Tanaka, Y. Nagashima, K. Tanaka, Org. Lett. 22 (2020) 7181–7186. doi: 10.1021/acs.orglett.0c02499
Y. Nagashima, S. Ishigaki, J. Tanaka, K. Tanaka, ACS Catal. 11 (2021) 13591–13602. doi: 10.1021/acscatal.1c03454
Q. Zhang, H.Z. Yu, Y.T. Li, et al., Dalton Trans. 42 (2013) 4175–4184. doi: 10.1039/c3dt31898b
P.G. Gassman, J.W. Mickelson, J.R. Sowa Jr, J. Am. Chem. Soc. 114 (1992) 6942–6944. doi: 10.1021/ja00043a065
T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, et al., J. Am. Chem. Soc. 139 (2017) 1296–1310. doi: 10.1021/jacs.6b11670
N. Dunwoody, S.S. Sun, A.J. Lees, Inorg. Chem. 39 (2000) 4442–4451. doi: 10.1021/ic000395n
L. Li, W.W. Brennessel, W.D. Jones, J. Am. Chem. Soc. 130 (2008) 12414–12419. doi: 10.1021/ja802415h
L. Li, W.W. Brennessel, W.D. Jones, Organometallics 28 (2009) 3492–3500. doi: 10.1021/om9000742
Y. Li, X.S. Zhang, H. Li, et al., Chem. Sci. 3 (2012) 1634–1639. doi: 10.1039/c2sc01081j
M.E. Tauchert, C.D. Incarvito, A.L. Rheingold, R.G. Bergman, J.A. Ellman, J. Am. Chem. Soc. 134 (2012) 1482–1485. doi: 10.1021/ja211110h
Y. Hu, J.R. Norton, J. Am. Chem. Soc. 136 (2014) 5938–5948. doi: 10.1021/ja412309j
B.R. James, D. Mahajan, S.J. Rettig, G.M. Williams, Organometallics 2 (1983) 1452–1458. doi: 10.1021/om50004a036
C. Bruneau, P.H. Dixneuf, Ruthenium(Ⅱ)-Catalyzed Functionalisation of C–H Bonds With Alkenes: Alkenylation versus Alkylation, Topics in Organometallic Chemistry, Springer, 2015.
Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016.
L. Li, Y. Jiao, W.W. Brennessel, W.D. Jones, Organometallics 29 (2010) 4593–4605. doi: 10.1021/om100796q
Y.F. Han, G.X. Jin, Chem. Soc. Rev. 43 (2014) 2799–2823. doi: 10.1039/C3CS60343A
C. White, A.J. Oliver, P.M. Maitlis, Dalton Trans. (1973) 1901–1907.
F.L. Taw, H. Mellows, P.S. White, et al., J. Am. Chem. Soc. 124 (2002) 5100–5108. doi: 10.1021/ja0165990
S. Dutta, T. Bhattacharya, D.B. Werz, D. Maiti, Chem. 7 (2021) 555–605.
N.K. Mishra, S. Sharma, J. Park, S. Han, I.S. Kim, ACS Catal. 7 (2017) 2821–2847. doi: 10.1021/acscatal.7b00159
H. Wang, N. Schröder, F. Glorius, Angew. Chem. Int. Ed. 52 (2013) 5386–5389. doi: 10.1002/anie.201301165
A.D.S. Dipannita Kalyani, M.S. Sanford, J. Am. Chem. Soc. 132 (2010) 8419–8427. doi: 10.1021/ja101851v
T. Pinkert, T. Wegner, S. Mondal, F. Glorius, Angew. Chem. Int. Ed. 58 (2019) 15041–15045. doi: 10.1002/anie.201907269

Figure 1 (a) Syntheses of seven-membered rhodacycles 3-Cp*, 3-CpCF3 and 3-Cp. (b) X-ray crystal structures of 3-Cp*, 3-CpCF3 and 3-Cp. Thermal ellipsoids are drawn at 50% probability. H atoms and SbF6− anions have been omitted for clarity.

Figure 2 (a) Selective eliminations of rhodacycles 3-CpX. (b–d) Monitoring (1H NMR) of the elimination of 3-CpX.

Figure 4 Ligand-tuned product selectivity in Rh(Ⅲ)-catalyzed C—H olefinations with ethyl acrylate and styrene.

Figure 5 (a) Syntheses of six-membered rhodacycles 7-Cp* and 7-CpCF3. (b) X-ray crystal structures of 7-Cp* and 7-CpCF3. Thermal ellipsoids are drawn at 50% probability. H atoms and SbF6− anions have been omitted for clarity. (c) Detection of catalytic species relevant to [CpXRh-H] intermediates.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: