
Figure 1.
Synthetic host-guest pairs as novel non-covalent bioorthogonal tools to complement and further improve the bioorthogonal chemistry in pre-targeting.
Synthetic host-guest pairs as novel bioorthogonal tools for pre-targeting
Yan-Long Ma , Shi Yan , Xiong-Jie Xu , Hua Cao , Ruibing Wang

Two independent entities rapidly and selectively react with each other without reactivity towards endogenous species in living systems are collectively termed bioorthogonal chemistry [1]. At early 2000, Bertozzi and co-workers [2–5] firstly introduced the click reactions, such as staudinger ligation and strain-promoted [3 + 2] azide–alkyne cycloadditions (SPAAC), in a cellular environment for labeling the cell surface's glycoproteins, and proposed this concept [6,7]. Over the past two decades, bioorthogonal chemistry has become an important tool widely used in cell biology, chemical biology, molecular biology and biomedicine, and has already made a tremendous scientific impact on these disciplines [8]. Of note, Bertozzi was awarded the 2022 Nobel Prize in Chemistry due to her creative work in bioorthogonal chemistry [9].
To further clarify this concept, a bioorthogonal reaction should possess the following features: (1) quantitative yields in aqueous environments at physiological pH and temperature; (2) chemo-selectivity without reactivity towards endogenous functional species in living systems; (3) fast reaction rates even at low concentrations; (4) non-perturbing to biological systems and nontoxic under these conditions. To date, dozens of chemical reactions, such as staudinger ligation, Cu(Ⅰ)-catalyzed [3 + 2] azido–alkyne cycloadditions (CuAAC), SPAAC, photo-click 1,3-dipolar cycloadditions, strain-promoted alkyne-nitrone cycloadditions (SPANC), inverse electron demand Diels–Alder (IEDDA), hetero-Diels-Alder, are developed as bioorthogonal reactions [10–13]. Application of these bioorthogonal reactions in living systems typically involve two steps: First, a bioorthogonal group (e.g., azide or tetrazine) is incorporated into interested biomolecules (e.g., proteins, glycans, lipids and other biomolecules) via metabolic engineering or other methods. Second, an activated molecule which bears another complementary group (e.g., dibenzylcyclooctyne, trans-cyclooctene), is conjugated to the interested biomolecules via bioorthogonal reaction between the bioorthogonal group and its complementary group.
At the preliminary stage of its inception, many of these bioorthogonal reactions are used to image or label interested proteins and other biomolecules for profiling active enzymes [14,15], identifying drug targets [16,17], cell imaging [18–21], cell adhesion [22–27], understanding biomolecules and functions in complicated biological systems [8,28], etc. Subsequently, with the promotion of bioorthogonal chemistry, more and more chemical biologists began to seek its new pursuits in genetic code expansion, in vivo imaging, drug delivery, and disease therapy [8,12,28,29]. Until now, bioorthogonal chemistry has been applied in pharmaceutical chemistry, medicine, biology and their interdisciplinary areas. Its application in these disciplines is increasing year by year, showing an irreplaceable development trend [30]. The toolkit of bioorthogonal reactions has grown to meet the different requirements of application scenarios of the chemical biology community. Beyond all doubt, these covalent bioorthogonal reactions are very impressive and extremely useful, but these approaches are not without limitations. When conjugation needs to be done in pre-targeted delivery for imaging and therapy under dilute conditions in vivo, most of these covalent bioorthogonal reactions seem strikingly powerless due to their slow reaction rates [31,32].
A primary goal in present-day biomedicine is the pursuit of technologies to increase accumulations of active molecule (e.g., drug, probe) in targeted site and to improve its target-to-background ratios. The emergence of pre-targeted strategy can achieve this goal via two distinct steps: First, a vector (e.g., antibody, nanoparticles) bearing a party of the complementary pairs is injected and allowed to accumulate in the targeted sites over a sufficient period of time (hours or days). Second, a small active molecule bearing another complementary group is administered for highly-efficient conjugation to the vector within a short time (minutes or hours) at the targeted sites, and the free active molecule can be rapidly cleared from the body. Thus, molecular conjugation rate is particularly crucial in pre-targeting, especially in pre-targeted nuclear imaging and radiotherapy [33]. Although IEDDA with the fastest second order reaction rate constant (k2) among the presently available covalent bioorthogonal reactions [10,12] has been introduced in pre-targeting for imaging, therapy and drug delivery and release [31,34–39], it has fallen into a dilemma situation that the faster reaction rate, the more unstable is the trans-cyclooctene reaction partners [31,40,41]. This casus the low efficiency of IEDDA is in pre-targeting and fails to meet the expected need. Therefore, there is an urgent demand to develop the next-generation bioorthogonal approaches with extremely rapid rates and strong adaptability in pre-targeting.
Biological association pairs, such as (strept)avidin/biotin pairs, antibody/antigen pairs, oligonucleotide hybridization pairs, have a relatively fast binding rate, strong binding strength and high selectivity [32,33,42,43], and thus are used as a non-covalent approach for complementing the limitations of covalent bioorthogonal reactions in pre-targeted applications. For example, the most well known and widely used (strept)avidin/biotin pairs have a fast binding rate (kon ≈ 107 L mol-1 s-1) and high association constants (Ka = 1013–1015 L/mol) [44], and have been applied in clinical studies for pre-targeted radioimmunotherapy (BC8 antibody-streptavidin conjugate/111In-DOTA-biotin and 90Y-DOTA-biotin) [45]. Nevertheless, this natural binding pair, as well as other biological association pairs, also suffer from two drawbacks: large molecular weight and difficult modification; and biochemical fragility in vivo [32]. Synthetic host-guest pairs with small molecular sizes (~1 kDa) and strong chemical stability are not only without these problems of biological association pairs [32], but also have the features of fast conjugation rate, easy modification, chemo-selectivity and low-toxicity [46–50]. As a consequence, they can be used as a novel non-covalent bioorthogonal approach to complement and further improve the bioorthogonal chemistry in pre-targeting (Fig. 1).
The application of this novel bioorthogonal molecular recognition approach in pre-targeting is still on the preliminary stage of exploration, and there are not many research reports in this area. Nevertheless, we believe that the synthetic host-guest bioorthogonal recognition will become an important tool for bioorthogonal chemistry in the foreseeable future to compensate for the limitations of covalent bioorthogonal reactions and biological association pairs. By introducing and summarizing the progress achieved in pre-targeting based on host-guest bioorthogonal pairs during the past years, more and more researchers will see their value, and thus use them and seek their greatest value in pre-targeting applications.
Accordingly, this review will be divided into three parts: First, explaining the importance of molecular conjugation rate in pre-targeting via theoretical calculation of the conversion rates of covalent bioorthogonal reaction and host-guest bioorthogonal complex, respectively; Next, the recent research progress in the development of host-guest bioorthogonal pairs for pre-targeting, including cucurbit[7]uril (CB[7])/guest pairs, β-cyclodextrin (β-CD)/guest pairs and amide naphthotube (NT-anti)/guest pairs, will be systemically overviewed and summarized; Last, the future perspectives and limitations of host-guest bioorthogonal recognition will be discussed. The aim of this review is mainly focused on the applications of synthetic host-guest bioorthogonal pairs in pre-targeting, and the covalent bioorthogonal reactions [12,31,33–36,38,51] and non-covalent biological association pairs [33,44,52–55] applied in pre-targeting have been well discussed in other reviews, hence we will not discuss them in here. In addition, the applications of synthetic host-guest bioorthogonal pairs in vitro for labeling proteins [56–59], imaging proteins [60], purifying proteins [61], driving DNA strand displacement [62], regulating protein functions [63], imaging cells [64–66], imaging cellular processes [67,68], engineering cells [69,70], etc., are also not discussed in here, because molecular conjugation rates in these applications are not the main concern. This review will help researchers understand the pros and cons of synthetic host-guest bioorthogonal pairs in pre-targeted applications, and provide new insights for developing them to the more advanced level.
We mentioned above that the goal of pre-targeted strategy is as possible as to increase the accumulations of active molecule in targeted site and reduce its systemic exposure, and the molecular weight of the second administered active molecule should be as small as possible to facilitate fast diffusion to the targeted site and fast conjugation to the first injected vector (Fig. 2A). It is also reasonably clear that molecular conjugation rate is the decisive factor for whether expected results can be achieved in pre-targeting. So, in this part, we will theoretically calculate the conversion rates of covalent bioorthogonal reaction and host-guest bioorthogonal complex by means of analyzing their chemical kinetics and thermodynamics, respectively, to intuitively illustrate the importance of molecular conjugation rate in pre-targeting.

It is well kwon that the conversion rate of presently available covalent bioorthogonal reactions is governed by chemical kinetics, and these reactions are of second order (Fig. 2B). In the simplest case of a two-molecule reaction, the kinetic process of this reaction is shown as follows:
|
|
where [A] and [B] are the concentrations of the free substrates and [A-B] is the concentration of the reacted product. From the law of chemical kinetics, a second order reaction rate constant, k2, can be derived as follows:
|
|
(1) |
where A0 and B0 are the initial concentrations of the substrates. The units of k2 and t are L mol-1 s-1 and s, respectively. For calculating the conversion rate of [A-B], the above Eq. 1 is transformed as follows:
|
|
(2) |
here, we suppose that B0 is ≫ A0, so, B0 − [A-B] ≈ B0, B0 − A0 ≈ B0, and the above Eq. 2 is transformed as follows:
|
|
(3) |
|
|
(4) |
|
|
(5) |
then, the conversion rate of [A-B], y, can be derived as follows:
|
|
(6) |
According to the above Eq. 6, it is easy to conclude that if B0 is ≫ A0, improving k2 could decrease B0 at a certain time scale.
The propensity for a complex formed by host-guest pair is governed by thermodynamics (Fig. 2C). In the simplest case of a 1:1 monovalent recognition, the dynamic process of host-guest recognition is shown as follows:
|
|
where [H] and [G] are the concentrations of the free host and guest, respectively, and [G@H] is the concentration of the formed complex. From the law of thermodynamics, a binding constant, Ka, can be derived as follows:
|
|
(7) |
|
|
(8) |
where G0 and H0 are the initial concentrations of the host and guest, respectively. The unit of Ka is L/mol, and Ka is commonly used to evaluate the affinity of host-guest binding, a higher value of Ka signifies a more stable host-guest complex formed. To calculate the conversion rate of [G@H], we suppose that G0 is ≫ H0, so, G0 − [G@H] ≈ G0, and the above Eq. 8 is transformed as follows:
|
|
(9) |
|
|
(10) |
then, the conversion rate of [G@H], y, can be derived as follows:
|
|
(11) |
According to the above Eq. 11, we can conclude that if G0 is ≫ H0, y mainly depends on Ka. The higher Ka is, the higher y is. Moreover, Ka is also related to the rate constants of binding (kon) and dissociation (koff), as flows:
|
|
(12) |
For this 1:1 monovalent recognition, kon has the unit of L mol-1 s-1 and koff has the unit of s-1. The kon of synthetic host-guest pairs, such as CB[7]-guest pairs and β-CD-guest pairs, is controlled by diffusion rate (~108 L mol-1 s-1). Therefore, once these pairs are mixed, they can quickly form thermodynamic equilibrium complexes on a second time-scale [71,72]. This suggests a great advantage of synthetic host-guest pairs used in pre-targeting when compared to widely used covalent bioorthogonal reactions, such as SPAAC (~1–60 L mol-1 s-1) and IEDDA (~1–106 L mol-1 s-1) [28]. Apparently, a higher Ka of host-guest pair has a slower koff and has a more stable complex once formed. So, a higher Ka will result a higher y.
Although calculated the conversion rates of covalent bioorthogonal reaction and host-guest recognition in pre-targeting are very difficult and seem almost impossible, assumption was made that the human body is a simple homogeneous reaction reactor and we adopted the above theoretical methods to do this impossible work and give a certain reference value for applications of bioorthogonal chemistry in pre-targeting. Before using above Eqs. 6 and 11 doing this work, the following parameters are needed to be determined:
1) The effective concentration of antibody (Ab) bearing A or H localized to the tumor site (Ab-A0 or Ab-H0), which is equivalent to the above defined A0 or H0. According to the Eqs. 6 and 11, y is mainly decided by k2 or Ka, and seemly unrelate to A0 or H0. Nevertheless, this value of A0 or H0 predetermines the value range of B0 or G0, which is similar in pre-targeting for doing this work. Unfortunately, neither covalent bioorthogonal reaction or host-guest recognition has been clinically applied in pre-targeting, which causes it is difficult to define this effective concentration. But typically, antibody bearing a party of the bioorthogonal pairs is the first choice for the preclinical study in pre-targeted cancer. Research indicates that the amount of the administered dose of antibody finally localized to human tumor site is about 0.001%−0.01% [73,74]. Hence, the average of this ratio, 0.005%, is chose for subsequently computing the effective concentration (EC) of antibody localized to the tumor site. In addition, the amount of A or H borne on one antibody is roughly defined as 4 according to the average of drug–antibody ratio of FDA approved antibody–drug conjugates [75,76]. Following this, EC for an adult with 60 kg, 1.7 m2 body surface area and 5 L blood, can be obtained by administration dosage of antibody, as follows:
|
|
(13) |
For making this work more statistically significant, we analyze the adult dosage of FDA-approved 36 antibodies for cancer treatments, and then compute these antibodies’ EC using the above Eq. 13 (Table 1), and choose the average of these antibodies’ EC, approximately 0.17 nmol/L, as the value of Ab-A0/Ab-H0.
2) The concentrations of second administered active molecule bearing B or G (M-B0 or M-G0), which is equivalent to the above defined B0 or G0. According to previous suppositions (B0 ≫ A0 and G0 ≫ H0), M-B0 or M-G0 is assigned as different values from 1 nmol/L to 1 mmol/L in order to study the effect of molecular conjugation rate on conversion rate under these different values of M-B0 or M-G0.
3) The reaction time, t, of covalent bioorthogonal reaction. In pre-targeting, it would be best for bioorthogonal chemistry to be done within a short time (minutes or hours) at the targeted sites. Following this suggestion, t is roughly defined as 30 min.
Based on this simple computing model, the conversion rates of covalent bioorthogonal reaction and host-guest recognition in pre-targeting can be calculated by bringing these parameters into their corresponding Eqs. 6 and 11, and taking k2 and Ka as the independent variable. The results are presented in Figs. 2D and E. Fig. 2D plainly shows that faster k2 can result higher-efficient conjugation for covalent bioorthogonal reactions in pre-targeting, and the faster it is, the higher y is, and then the lower need of M-B0 is. In this computing model, if the expected need in pre-targeting wants to be well achieved (y = 99%, M-B0 = 1 nmol/L), k2 should be ≥ 2.6 × 106 L mol-1 s-1. Apparently, this is a difficult goal to achieve for presently available covalent bioorthogonal reactions, the faster k2 of IEDDA is although up to ~106 L mol-1 s-1, the trans-cyclooctene reaction partners suffer from instability and are easily converted into their deactivated syn-forms in living systems [31,40,41]. Besides, SPAAC is more unsuitable for achieving this expected need, due to its much lower k2.
However, it is very easy for synthetic host-guest recognition to achieve this expected need, because their kon is diffusion-controlled (~ 108 L mol-1 s-1), and is almost 20-flod higher than needed 2.6 × 106 L mol-1 s-1. But that does not mean that synthetic host-guest recognition has no limitation. It should be noted that kon is not the only decisive factor for synthetic host-guest pairs used in pre-targeting, it is also limited by koff. For achieving this expected need (y = 99%, M-G0 = 1 nmol/L), Ka also needs to be ≥ 1011 L/mol (koff ≤ 10–3 s-1), as shown in Fig. 2E. Although the lower Ka (108 L/mol ≤ Ka ≤ 1010 L/mol) can also faster form the complex, the complex once formed begins to dissociate into free host and guest due to its relatively quick koff (1 s-1 ≥ koff ≥ 10–2 s-1). This causes these lower-affinity host-guest pairs cannot form robust complex to eliminate the impact of koff and fail to meet the expected need. Nevertheless, these lower-affinity host-guest pairs may be with some advantages for pre-targeted drug release, because the dissociated lifetime of complex once formed maybe facilitate the accumulation and absorption of drugs at the target site. The higher-affinity host-guest pairs (Ka ≥ 1011 L/mol, koff ≤ 10–3 s-1) may be more suitable for pre-targeted nuclear imaging and radiotherapy due to their higher conversion rates, robust complex and lower need of the second administered active molecule meeting these application requirements. Anyway, the tunable Ka of synthetic host-guest pairs provides more choices for their pre-targeted applications.
In a word, this part not only intuitively illustrate the importance of molecular conjugation rate in pre-targeting, but also fully explain the advantages of synthetic host-guest pairs used in pre-targeting when compared to covalent bioorthogonal reactions. Additionally, the conversion rates of covalent bioorthogonal reaction and host-guest recognition will give a certain reference value for applications of these bioorthogonal approaches into pre-targeting. Of note, this theoretical calculation model does not consider the impact of endogenous species.
Host-guest recognition is featured as a host macrocycle encapsulating a small molecule guest in its cavity forming a non-covalent complex. The binding affinity (Ka) of host-guest recognition relies on the size matching degree between the host cavity and the guest and the non-covalent interactions between them. The systematic studies of synthetic calix[n]arenes [78], pillar[n]arenes [79], tetralactams [80,81], cyclodextrins [82], cucurbit[n]urils [83–86], amide naphthotubes [87–90] and many others [91,92], to their guests' Ka in water afford a sound fundamental for these host-guest pairs applied in pre-targeting. However, to the best of our knowledge, there are only three kinds of host-guest bioorthogonal pairs currently applied in pre-targeting, including CB[7]/guest pairs, β-CD/guest pairs and NT-anti/guest pairs. So, this part will systemically overviewed and summarized the recent research progress of these host-guest pairs for pre-targeted applications, in turn.
CB[7], one of the family of pumpkin-shaped cucurbit[n]uril (CB[n], n = 5–8, 10, 13–15) hosts, consists of 7 glycoluril units that are connected by 14 methylene groups (Fig. 3). The two hydrophilic carbonylated rims endow CB[7] with a certain solubility in water and electronegative property at the surface of the carbonyl-fringed portals, while a hydrophobic cavity provides CB[7] with encapsulating a small guest molecule. The driving force of CB[7] binding guests to form complexes comes from both the hydrophobic effects of hydrophobic cavity and non-covalent interactions (e.g., ion-dipole and hydrogen bonding interactions) of carbonyl-fringed portals [86]. Various cationic or neutral guest molecules with appropriate size can all be recognized by CB[7], and the Ka of most CB[7]-guest pairs is range from 103 L/mol to 1017 L/mol [83–86,93]. The nature and size of the guest plays a particularly important role in determining binding affinity and selectivity. In particular, ferrocenemethyl (Fc), adamantly (Ad), and diamantyl ammonium derivatives with the binding affinities to CB[7] are up to ~1012–1017 L/mol, which are comparable to or even higher than that of biological association pairs, such as (strept)avidin/biotin pairs (Ka = 1013–1015 L/mol) [44], antibody/antigen pairs (Ka = 107–1012 L/mol) [42], oligonucleotide hybridization pairs (Ka = 108–1010 L/mol) [32], and they can selectively recognize each other and form robust complexes in vitro [56–70]. Moreover, CB[7] has a good biocompatibility and no cytotoxicity at 1 mmol/L in vitro [94], and displays relatively low toxicities with the maximum tolerated doses of 5 g/kg (intragastrical administration), 150 mg/kg (intravenous administration) and 500 mg/kg (intraperitoneal administration) in vivo mice model [95]. Besides that, the derivatization methods of CB[7] have grown to maturity in recent years [57]. So, such these remarkable properties have paved the way for development of CB[7]/guest pairs as bioorthogonal tools for pre-targeting.

In 2018, Hooker and coworkers [96] investigated the biodistribution of the free 11C or 18F labeled adamantane derivatives, complexes of them with CB[7] and CB[7] with pre-administered them in vivo through positron emission tomography (PET) imaging. The Ka of these guests to CB[7] is in the range of 1010–15 L/mol [97]. The authors found that the biodistribution of the last two group were very similar, and were completely different from that of the free group, suggesting that the fast binding kinetic of CB[7] (kon = ~108 L mol-1 s-1) could ensure the effective and rapid formation of stable complex with adamantane derivative guests via monovalent molecular recognition in vivo. However, this work has not study the concern about how strong the binding affinity of CB[7]/guest pairs could be used as bioorthogonal tools for pre-targeted applications. It is widely known that endogenous amino acids, peptides, and proteins can also be recognized by CB[7], the binding affinity of some of enkephalin-type peptides to CB[7] is even up to 107 L/mol [84]. These competitors will inevitably affect the formation of CB[7]-guest complex in pre-targeting. So, the affinity in excess of these naturally present competitors is needed to make sure that CB[7] can selectively recognize its guests in pre-targeting.
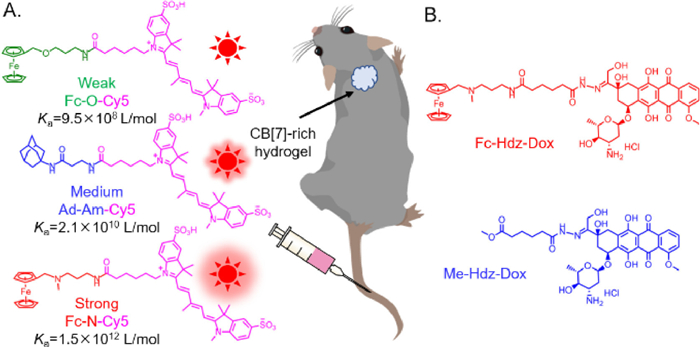
In 2019, Webber and coworkers [98] answered this concern. Three fluorescent probes, whose were synthesized by conjugating three guests of CB[7] with different binding affinity (~109 L/mol, ~1010 L/mol and ~1012 L/mol) to a near-infrared fluorescent dye (Cy5), were designed to evaluate the role of binding affinity for forming complex in pre-targeting via quantifying the fluorescence intensity of these probes homing to the mice dorsal subcutaneous site of the CB[7]-rich hydrogel (Fig. 4A). The probe with strong affinity guest (Fc-N: ~1012 L/mol) could be rapidly captured by CB[7] in situ within 30 min and accumulated at the CB[7]-rich hydrogel site, the formed complex could retain for a long time (~35 days). Whereas the probe with weak affinity guest (Fc-O: ~109 L/mol) showed almost no accumulation. The probe with medium affinity guest (Ad-Am: ~1010 L/mol) could also home to the CB[7]-rich hydrogel site, but the accumulation was 3-fold less than that of strong guest and the ratio of interest site-to-background was relatively low. These results clearly showed that the binding affinity of ~1012 L/mol could effectively make sure CB[7] and its guest selectively recognize each other and form robust complex in vivo, and eliminate the impact of natural competitors and dissociation rate (koff). Therefore, CB[7]/guest pairs as bioorthogonal tools using in pre-targeting should have a binding affinity of ~1012 L/mol. Subsequently, this CB[7]/guest pair with strong binding affinity was further applied in pre-targeting for treating cancer (Fig. 4B). A prodrug (Fc-Hdz-Dox) was created by conjugating the same strong affinity guest to the chemotherapeutic doxorubicin (Dox) to home to the CB[7]-rich hydrogel site near a tumor and form robust complex with CB[7], and then the complex slowly released the Dox to inhibit tumor growth by means of tumor acid pH environment rupturing the hydrazone linker. By comparing to another prodrug (Me-Hdz-Dox) without containing a guest, Fc-Hdz-Dox significantly reduced the tumor growth rate within 24 days following initial dosing. The results demonstrated that the strong binding affinity of ~1012 L/mol was the essential prerequisite and the basic guarantee for the complex of Fc-Hdz-Dox with CB[7] sustainedly releasing Dox to show a good therapeutic efficacy.
Taken together, these two studies fully prove that CB[7]/guest pairs with fast binding kinetic can be used as bioorthogonal tools for pre-targeted applications via monovalent molecular recognition, but the binding affinity of them should be up to or greater than ~1012 L/mol. These higher-affinity CB[7]/guest pairs (≥ 1012 L/mol, koff ≤ 10–4 s-1) can completely eliminate the impact of dissociation rate (koff) and efficiently form the robust complexes in vivo. By contrast, lower-affinity CB[7]/guest pairs (≤ 1010 L/mol, koff ≥ 10–2 s-1) cannot effectively or even at all recognize each other in vivo due to the impact of their quick koff and endogenous competitors, which causes these pairs are not used as bioorthogonal tools for pre-targeting in a monovalent recognition version. However, this issue can be tackled by using a multivalent recognition version [32]. Unfortunately, there is no systematic research report about this applied version, and are only two examples based on the application of this version. So, we will introduce this multivalent recognition version in these two examples, and not in here.
Antibody is commonly used as a vector for bearing a party of the bioorthogonal complementary pairs in pre-targeted tumor imaging and therapy. Although, there are four kinds of bioorthogonal approaches developed for pre-targeting, such as (strept)avidin/biotin, antibody/antigen, oligonucleotide hybridization and IEDDA approaches, these approaches have suffered from some limitations. CB[7]/guest pairs with strong binding affinity (≥ 1012 L/mol) as novel bioorthogonal tools doesn't have these limitations. So, in 2019, Kim and coworkers [99] firstly incorporated the CB[7]/Ad ammonium derivative pairs (Ka ~1014 L/mol) [85] into antibody system for pre-targeted tumor imaging. Conjugating CB[7] to cetuximab, which is an antibody for recognizing the epidermal growth factor receptor and treating colorectal, neck, and lung cancers, targeted to the tumor site allowed the post-injected guest vector (Ad ammonium guest conjugated with a Cy5) to home to the host vector (cetuximab-CB[7] conjugate) for tumor imaging. The selective imaging of a tumor site based on this method suggests that this novel bioorthogonal approach can act as an alternative for the developed four kinds of bioorthogonal approaches for pre-targeting.
Soon after that, Houghton and coworkers [100,101] explored the applications of this novel bioorthogonal approach in pre-targeted PET tumor imaging. Firstly, in 2021, the authors [100] investigated the feasibility of using carcinoembryonic antigen targeting antibody (M5A) based CB[7]-Fc bioorthogonal pair (Ka ~ 1012 L/mol) [85] for pre-targeted PET tumor imaging (Fig. 5A). M5A was conjugated with CB[7] by click chemistry to give a host vector (M5A-CB[7]) for targeting tumor, and Fc ammonium guest was labeled a radioactive isotope ligand (68Ga-NOTA) via PEG linker to give a Fc radioligand (68Ga-NOTA-PEG3-N-Fc, guest vector) for recognizing CB[7] and imaging tumor. The tumor mass of mice pre-treated with M5A-CB[7] of 72 h could be visualized by PET imaging at both 2 and 4 h timepoints after post-injected Fc radioligand. The maximum tumor uptake of Fc radioligand was at 2 h timepoints (3.3% ± 0.7% ID/g), which was significantly higher than that of free Fc radioligand (0.2% ± 0.1% ID/g). Next, in 2022, in order to improve the tumor uptake and clearance profile, the authors [101] optimized the pre-targeting parameters of this method, such as, radioligand's structure (Fig. 5B), interval time (lag time) and dosing of radioligand and M5A-CB[7]. Better PET tumor image quality could be obtained by adjusting these parameters. The lag time based on M5A-CB[7] and 64Cu-NOTA-PEG3-N-Fc radioligand for PET tumor image was even up to 9 days, meanwhile Fc radioligand was still able to retain tumor-specific uptake and good tumor delineation. So, this novel bioorthogonal approach for pre-targeted nuclear imaging and radiotherapy with long lag times will not require a clear agent which has been used successfully in clinical studies to remove the nontarget-bound antibody from the blood pool [102–105], and make the flexibility of radioligand in time of administration.

Inorganic nanoparticles (NPs), especially for Au NPs, with photothermal effect have been widely applied in biomedical science for tumor-specifc photothermal therapy (PTT) [106–108]. However, the rapid clearance of these small NPs often causes insufficient accumulation and short retention in the tumor site [109]. So, in 2022, Wang and coworkers [110] proposed a strategy to enhance the aggregation of Au NPs in the tumor tissue based on CB[7]-Fc multivalent recognition (Ka ~ 109 L/mol) inducing (Fig. 6) [85]. CB[7] was coated on the surface of Fe3O4 NPs to give a host vector (Fe3O4-CB[7] NPs) for magnetic localization at tumor site, and Au NPs were capped with Fc guest to give a guest vector (Au-Fc NPs) for recognizing CB[7] via multivalent interactions and aggregating in the tumor tissue for PTT. With the help of host-guest multivalent recognitions, Au-Fc NPs could highly accumulate in the tumor of mice pre-treated with Fe3O4-CB[7] NPs by CT imaging, and the tumor uptake of this nanoparticles were up to 10.06% ID/g, which was significantly higher than that of Au NPs group (1.87% ID/g) and mix group (Au NPs and Fe3O4 NPs, 2.06% ID/g). This highly accumulated Au-Fc NPs (aggregation group in the original article) could also significantly inhibit tumor growth under laser irradiation. These results suggest that CB[7]/guest pairs with low binding affinity (~109 L/mol) can be used as bioorthogonal tools in a multivalent recognition version for improving the accumulation and retention of nanomedicine for pre-targeted imaging and therapy.

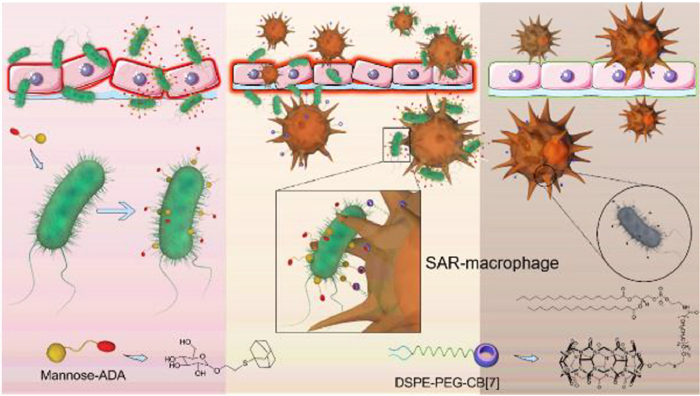
The insufficient capture of bacteria by immune cells, such as macrophage, often cause an inefficient clearance of pathogens and lead to a high infection rate. So, in 2022, Wang and coworkers [111] applied this multivalent host-guest recognition version to engineer macrophage for enhancing the recognition and capture of bacteria in living systems (Fig. 7). CB[7] was fabricated on the surface of macrophage by inserting DSPE-PEG-CB[7] into its membrane to give a SAR-macrophage for recognizing and capturing the E. coli, which was decorated with Ad guest by the strong and specific interactions between d-mannose-Ad (~1010 L/mol with CB[7]) [85] and FimH receptor [112]. SAR-macrophage could significantly recognize and catch this decorated E. coli both in vitro and in zebrafish via multivalent host-guest recognitions, and cleared it. Then, the authors extended this method to wound healing via local administration. SAR-macrophage could also clear the infected pathogens at the wound site of mice pre-treated with d-mannose-Ad for 1 h, and heal the wound. This work further confirms that CB[7]/guest pairs with low binding affinity (~1010 L/mol) can also be used as bioorthogonal tools in a multivalent recognition version to engineer immune cells for pre-targeted antibacterial applications.
β-CD, one of the family of cyclodextrins with truncated cone shape (α-, β-, γ-CDs), is cyclic oligosaccharides composed of 7 d-glucose units through α-(1, 4) glucosidic bonds linking in a head-to-tail manner (Fig. 8). The secondary 2- and 3‑hydroxyl groups are located on the wider side of the truncated cone, and the narrower side contains the primary 6‑hydroxyl groups. The hydrophilic portals provide a good water solubility for β-CD, and the hydrophobic interior cavity endows β-CD with an ability for binding small lipophilic molecules. Lipophilic guest species with suitable size can be all recognized and encapsulated by β-CD via hydrophobic effects, van der Waals interactions and hydrogen bonding interactions with its hydroxyl groups [113,114], and the Ka of most β-CD-guest pairs [42,82,115] is range from 10 L/mol to 105 L/mol. Moreover, β-CD has very good biocompatibility, and the oral and intravenous half lethal dose (LD50) of β-CD for rat are 19 g/kg and 1 g/kg, respectively [116]. β-CD has been approved by the U.S. Food and Drug Administration for use as food additives, as well as listed in the United States Pharmacopeia and National Formulary, European Pharmacopoeia and Japanese Pharmaceutical Codex as Inactive Pharmaceutical Ingredients [117,118]. So, β-CD is widely studied and applied in biomedicine.

Although Ad derivatives have the strongest binding affinity (Ka ~105 L/mol) among all the guests of β-CD [82], these pairs cannot be used as bioorthogonal tools for pre-targeted applications via monovalent molecular recognition. There are two reasons for this: firstly, their quick koff (103 s-1) will lead to a low conversion rate of complex formation at diluted concentrations according to the theoretical calculation model, it will be more than 50% only when the concentrations are larger 10 µmol/L, and these high concentrations obviously have no useful purpose in vivo; secondly, and worse than that, these pairs cannot even at all recognize each other under in vivo diluted concentrations due to the impact of endogenous competitors, including amino acids, monosaccharides, steroids [82,119,120], etc. In 2012, Weissleder and coworkers [121] proposed a multivalent recognition strategy to address this issue. Nanoparticles with surface modification of multiple Ad guests (Ad–MFNPs, ~63 Ads per MFNP) could selectively recognize the antibodies conjugated with multiple β-CD (β-CD–Abs, ~18–20 β-CDs per Ab) and form robust complexes between them via multivalent recognitions of β-CD and Ad in vitro. According to the analyses of surface plasmon resonance, the binding affinity and binding kinetics of β-CD and Ad guest in a multivalent recognition version are very different from their in a monovalent recognition version. In a multivalent recognition version, their kon drops to 8 × 106 L mol-1 s-1 which is lower than that of ~108 L mol-1 s-1 in a monovalent recognition version, but their Ka is up to 2 × 1010 L/mol, and their koff is down to 4 × 10–4 s-1. Moreover, the results of cell labeling using this multivalent recognition strategy based on β-CD/Ad guest pairs were superior to that of avidin/biotin pairs. So, β-CD/Ad derivative guest pairs can be used as bioorthogonal tools in a multivalent recognition version for pre-targeted applications in vivo.
The main drawback of current hepatic radioembolization therapies is the mismatch between the scout scan (diagnostic planning) and therapy delivery, which can result in adverse side-effects such as pulmonary shunting [122,123]. van Leeuwen and coworkers [124,125] proposed a nanotechnology-based pre-targeting strategy to overcome this problem via a multivalent β-CD/Ad guest recognition inducing the localization of therapeutic reagents in the liver pre-treated with diagnostic reagents. Firstly, in 2018, the authors [124] investigated the feasibility of this strategy by Single Positron Emission Computed Tomography (SPECT) imaging (Fig. 9A). Ad was coated on the surface of macro albumin aggregates (MAA) to give a guest vector (MAA-Ad) for localization at liver via local administration (spleen injection), and poly(isobutylene-alt-maleic-anhydride, PIBMA) polymers labeled with a radioactive isotope (99mTc) and Cy5 were conjugated with β-CD to give a host vector (99mTc-Cy50.5CD10PIBMA39) for recognizing Ad via multivalent interactions and accumulating in the liver. According to the results of SPECT imaging, MAA-Ad could significantly increase the biodistribution of the host vector in liver. For pre-locally treated with MAA-Ad, the accumulation of host vector in liver via systemic administration was 16.2% ± 0.7% ID/g, which was significantly higher than that of without pre-locally treated with MAA-Ad (1.0% ± 0.2% ID/g), systemic pre-treated with MAA (2.2% ± 0.9% ID/g), systemic pre-treated with MAA-Ad (5.7% ± 0.9% ID/g) and pre-locally treated with MAA (8.7% ± 1.0% ID/g). Furthermore, its accumulation in lung (2.6% ± 0.2% ID/g) was not much different from that in the without pre-locally with MAA-Ad (1.0% ± 0.2% ID/g), suggesting that this strategy could prevent the side effects due to pulmonary shunting.
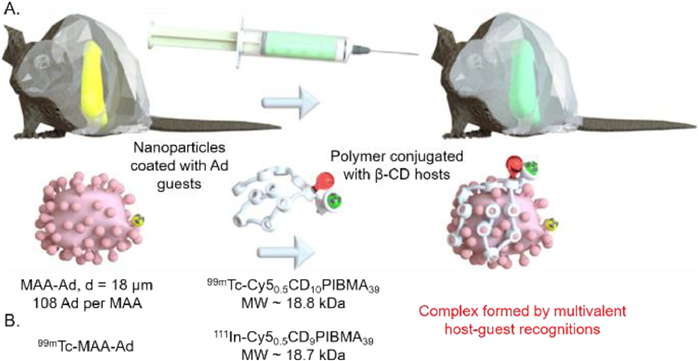
Next, in 2019, the authors [125] used the dual isotopic modified host-guest vectors (99mTc MAA-Ad and 111ln-Cy50.5CD9PIBMA39) to further validate that this strategy could match the timespan of the clinical radioembolization procedure (Fig. 9B). The accumulation of modified host vector in liver for pre-locally treated with 99mTc-MAA-Ad increased with the time from 2 h to 44 h, and reached the maximum at 20 h (27.0 ± 1.3 ID/g), suggesting that this strategy with shorter delay time (< 44 h) for liver radioembolization would be more patient-friendly compared to the current clinical liver radioembolization procedure performed several weeks apart. Thereby, β-CD/Ad derivative guest pairs as bioorthogonal tools for pre-targeted radioembolization can help address its unmet clinical needs.
What is more, both nanoparticles and polymers modified with β-CD or Ad can be acted as host vector or guest vector, whose can rapidly recognize each other and form stable complex via host-guest multivalent recognition in vivo, this β-CD/Ad derivative guests bioorthogonal tool can be utilized to achieve an in vivo pre-targeted delivery strategy of these cargoes by means of the pre-treated vector capturing the post-injected vector in situ and accumulating the latter at the site of former. In 2021, the authors [126] further demonstrated that the living cells (S. aureus bacteria) could also be used as a vector to realize this purpose effectively.
Although nanomedicine for treating tumor has achieved great success in clinical translation over the past 30 years, it also suffers from low accumulation in tumor site due to their various non-specific accumulation and relatively low targeting efficiency. In 2022, Zheng and coworkers [127] used a liposome (Lipo)-based pre-targeting delivery strategy to address this problem via a multivalent β-CD/Ad guest recognition improving the accumulation of therapeutic reagents (Dox@Ad-Lipo) in the tumor cell pre-locally treated with DSPE-PEG-β-CD (Fig. 10). Firstly, the authors used zebrafish model to demonstrate that a living cell, B16 tumor cell, anchored by DSPE-PEG-β-CD could also be acted as host vector to capture the post-injected guest vector (Dox@Ad-Lipo) high efficiently in vivo. Furthermore, this liposome could even bind to B16 tumor cells in bloodstream, and form robust complexes between them. The success achieved on host-guest interactions driven targeting process at the single-cell level encouraged these authors to next explore this strategy for tumor treating in mice model. Dox@Ad-Lipo could rapidly home to B16 tumor site pre-locally treated with DSPE-PEG-β-CD for 10 h, and their accumulations in tumor site were almost 10-flod more than that of without pre-locally treated with DSPE-PEG-β-CD. Hence, this highly accumulated Dox@Ad-Lipo could significantly inhibit tumor growth within 18 days compared with other groups. These results suggest that β-CD/Ad derivative guests in a multivalent recognition version utilized in drug delivery systems can achieve targeting delivery and targeting treatment.

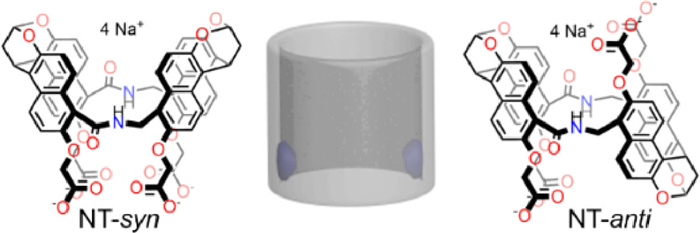
Amide naphthotubes (NT-syn and NT-anti) are a new category of endo-functionalized macrocycles, featuring a deep hydrophobic cavity with direct inward hydrogen bonding donors (N—H), as shown in Fig. 11. They were firstly reported in 2004 by Glass and coworkers [128], but their host-guest chemical properties have been systematically studied by Jiang in the years after 2016 [87–90,129]. Jiang and coworkers found that these naphthotubes could mimick the binding behaviors of bioreceptors to selectively recognize the highly hydrophilic molecules in water by combining the hydrophobic effect with hydrogen bonding shielded inside the hydrophobic cavity, such as solvent molecules [87,129], heterocycles [87], polyethylene glycols [130], epoxides [88,131], phosphate esters [132], urethane [133], benzene with different functional groups and drugs [90]. Ka of most guest to these naphthotubes is range from 102 L/mol to 106 L/mol, and Ka of NT-anti to guest is often larger than that of NT-syn. NT-anti is very soluble in water and its solubility is over 400 mg/mL (> 350 mmol/L), moreover, it also has a good biocompatibility and no effect on cell growth at 0.3 mmol/L in vitro [90]. Additionally, the mono-functionalized method of NT-anti has been developed by these authors [134], which means that NT-anti can be derived according to demand. So, these properties have laid a foundation for the applications of NT-anti in biomedicine.
As we mentioned in the theoretical calculation part, apart from the advantage of the faster kon, another advantage of synthetic host-guest pairs as bioorthogonal tools is the tunable Ka, which provides more choices for their applications in pre-targeting. The higher-affinity host-guest pairs can be used to meet the requirement of forming robust complexes and accumulating in the target site for diagnosis and treatment of diseases. Whereas, the lower-affinity host-guest pairs may be applied for drug release due to that the time span between their binding and dissociation maybe facilitate the accumulation and absorption of drugs at the target site. In achieving it, these lower-affinity host-guest pairs should be selectively recognized each other in vivo. In evidence, CB[7]/guest pairs and β-CD/guest pairs cannot do this due to their low selectivity. However, NT-anti can selectively recognize the hydrophilic guests, such as 2-phenyl pyrimidine derivatives (~106 L/mol). Thereby, in 2022, Jiang and coworkers [135] investigated the feasibility of applying such lower-affinity host-guest pairs as bioorthogonal tools for pre-targeting (Fig. 12). NT-anti were conjugated with tetraphenyl ethylene and its derivative by click chemistry to give artificial receptors (R1 and R2) for anchoring at the surface of tumor cell via intratumoral injection, and PEG labeled with a green fluorescent dye (4-nitro-2,1,3-benzoxadiazole, NBD) and liposome containing Cy5 were modified with 2-phenyl pyrimidine to give guest vectors (G2 and G5-modified liposome) for recognizing R2 and R1 via monovalent and multivalent interactions, respectively.

According to the results of fluorescent imaging, these guest vectors could be captured by artificial receptors in situ via monovalent or multivalent host-guest recognitions and accumulated at the tumor sites. For pre-locally treated with R2, the accumulation of G2 in tumor via systemic administration was almost 2-flod more than that of without pre-locally treated with R2. For pre-locally treated with R1, the accumulation of G5-modified liposome in tumor via systemic administration was almost 1.7-flod more than that of without pre-locally treated with R1 at any point within the whole 96 h. It has been proven that the binding affinity of host-guest pairs in multivalent recognition version is much stronger than that of in monovalent recognition version, and thus leading to a more accumulation. The low accumulation of G5-modified liposome in tumor was probably caused by the shunt to muscle tissue (left thigh), which was pre-locally treated with R1 to confirm the results from the tumor tissues. The average fluorescent intensity of the left thigh in the G5-modified liposome group was almost 2-fold of that of without pre-locally treated with R1. Surprisingly, the 2-phenyl pyrimidine unmodified G4 and PEG liposome could also be captured by these artificial receptors, but their accumulations in tumor were not significantly higher than that of without pre-locally treated with these artificial receptors. This might be due to the binding between the NT-anti and PEG2000 (ca. 1.6 × 104 L/mol) [130]. So, this work firstly demonstrates that the low-affinity host-guest pairs (~106 L/mol) with highly selectivity, such as NT-anti/2-phenyl pyrimidine derivative guest pairs, can also be used as bioorthogonal tools either in monovalent or multivalent recognition version, and these host-guest pairs may be applied in pre-targeting that do not need to form robust complex, such as drug release. It is a pity that the pre-targeted applications based on such host-guest pairs are not further reported.
For achieving the goal of increasing the accumulations of active molecule in targeted site and reducing its systemic exposure, the pre-targeted strategy can only seek the help of bioorthogonal tools. At present, there are only four kinds of bioorthogonal tools developed for pre-targeting, such as, (strept)avidin/biotin, antibody/antigen, oligonucleotide hybridization and IEDDA tools. Although these four bioorthogonal tools have made significant progress in the pre-targeted applications, especially for that (strept)avidin/biotin tools have been successfully applied in clinical studies for pre-targeted radioimmunotherapy, these biological association tools have to contend with the issue of the difficult modification, biochemical fragility in vivo and the pharmacokinetics caused by their larger molecular weight, and IEEDA also has to face the challenge of the chemical instability in vivo.
However, synthetic host-guest pairs with small sizes (~1 kDa) and strong chemical stability are not only without these drawbacks, but also have the features of fast conjugation rate (diffusion-controlled, ~108 L mol-1 s-1), tunable binding affinity, easy modification, chemo-selectivity and low-toxicity. Consequently, they can be used as a novel non-covalent bioorthogonal tool to complement the limitations of the presently available bioorthogonal tools for pre-targeted applications. But that does not mean this novel bioorthogonal tool is not without limitations in pre-targeted applications, they also suffer from the regulation of Ka and koff, and struggle with their endogenous competitors. For example, the presently developed three host-guest pairs, including CB[7]/guest pairs, β-CD/guest pairs and NT-anti/guest pairs, as bioorthogonal tools for pre-targeted applications must comply with the following using principles:
● CB[7]/guest pairs, for monovalent recognition version, their Ka should be ≥ 1012 L/mol; for multivalent recognition version, their Ka should be ≥ 109 L/mol according to the current research results.
● β-CD/guest pairs, they can be only used in a multivalent recognition version and their Ka should be ≥ 105 L/mol.
● NT-anti/guest pairs, they can be used both in a monovalent and multivalent recognition version, and their Ka should be ≥ 106 L/mol according to the current research results.
Based on using principles, these three host-guest pairs have successfully applied in pre-targeted tumor imaging, tumor treating, anti-bacteria and liver radioembolization, and with help of host-guest recognitions, the expected results of these applications could all be well achieved. But it should be also clearly aware that the applications of these novel bioorthogonal tools in pre-targeting are still on the preliminary stage of exploration. So, there are many challenges to be faced and many works to be done in the future for deeply understanding the nature of synthetic host-guest bioorthogonal tools in pre-targeted applications. For example, some works need to be done in future:
(ⅰ) Comparative studies of synthetic host-guest bioorthogonal tools and presently developed bioorthogonal tools in pre-targeting should be systematically done. Currently, the advantages of synthetic host-guest bioorthogonal tools compared with others all come from the theoretical deduction and analysis, and have no experimental data to support. Thence, the comparative studies between them in pre-targeting will help researchers fully appreciate the pros and cons of synthetic host-guest bioorthogonal tools and their adaptability.
(ⅱ) Enriching the synthetic host-guest bioorthogonal toolkits should be done. Up to now, there are only three kinds of host-guest pairs, including CB[7]/guest pairs, β-CD/guest pairs and NT-anti/guest pairs, developed as bioorthogonal tools for pre-targeted applications, and the binding affinity and selectivity of them cause difference to their adaptability in pre-targeted applications. The higher-affinity host-guest bioorthogonal pairs, such as CB[7]/guest and β-CD/guest bioorthogonal pairs, are more suitable for the requirement of forming robust complexes in pre-targeted applications, such as nuclear imaging, radiotherapy, etc. The lower-affinity host-guest pairs with highly selectivity, such as NT-anti/guest pairs, may be used as novel non-covalent "click-to-release" [30,34–36] bioorthogonal tools to facilitate the accumulation and absorption of drugs at the target site in pre-targeted drug release. Thereby, enriching the synthetic host-guest bioorthogonal toolkits will provide more choices for meeting the different requirements of application scenarios in pre-targeting.
(ⅲ) Broadening the applications of synthetic host-guest bioorthogonal tools in pre-targeting should be done. Since the first reported case of the application of synthetic host-guest bioorthogonal tools in pre-targeting appeared in 2018, their applications mainly focus on tumor imaging and treating, anti-bacteria and liver radioembolization in the presently several examples. Quite evidently, the synthetic host-guest bioorthogonal pairs based on their using principles are also available for other applications as similar as the developed bioorthogonal tools in pre-targeting, such as tumor's radiotherapy, radioimaging, chemotherapy, photodynamic therapy, and even extending other disease's diagnosis and therapy [28–39,43,44]. In particular, the short-lived radionuclides, such as 11C (20.4 min), 68Ga (67.8 min) and 18F (110 min), require the fast conjugation rate in the process of pre-targeting [33], synthetic host-guest pairs maybe well achieve this purpose due to their eximious predominance in binding rate (~108 L mol-1 s-1). For another, the lower-affinity host-guest pairs with highly selectivity as novel non-covalent "click-to-release" bioorthogonal tools in pre-targeted drug release also need to be validated. Therefore, broadening the applications of synthetic host-guest bioorthogonal tools in pre-targeting will help to cognize and discover their potential application values.
(ⅳ) Improving the amounts of a party of the bioorthogonal pairs bore by the first vector at the targeted site should be done. Antibody and nanoparticle are commonly used as the first injected vector in pre-targeting to bear a party of the bioorthogonal pairs, but they have the following limitations in improving the amounts of a party of the bioorthogonal pairs they bear at the targeted sites. First, their accumulations at the targeted site are particularly low. For example, only 0.7% of nanoparticles coated with cancer cell recognizing ligands (trastuzumab and folic acid) were delivery to the tumor tissue, and only 0.0014% injected dose were delivered to cancer cells [136,137], and only 0.001%−0.01% the administered dose of antibody finally reached human tumor site [73,74]. Second, the amounts of a party of the bioorthogonal pairs they bear are not only restricted by their own sizes, and also need to consider the balance with effectiveness and economy. Third, these vectors are difficult to penetrate to the interior of solid tumors due to the tumor microenvironment barriers of dense tissue stroma, high intratumoural pressure [138,139], etc. Fourth, the internalization of these vectors at the targeted site in the pre-targeted process will reduce the amounts of a party of the bioorthogonal pairs bore by these vectors. These limitations will be detrimental to improve the pre-targeted efficiency. Although, the methods of local delivery [140,141], magnetic targeting [142,143] and others can significantly improve the amounts of these vectors at the targeted sites, these methods are not with general applicability, especially for the early diagnosis of tumors and treatment of advanced metastatic tumors. So, there is an urgent need to develop new vectors to overcome these limitations for further improving the pre-targeted efficiency and achieving the precise diagnosis and therapy of tumor and even other disease. Living cells, such as mesenchymal stem cells, immune cells, bacterium and virus, are not only with abilities of homing into pathological sanctuary sites and distributing throughout the tissue [144–146], but also can bear more amounts of a party of the bioorthogonal pairs via methodology of cell surface engineering [147] based on their large sizes. As a result, these living cells may be used as new vectors to achieve it, but further exploration is also needed in future.
The above proposed research topics are key concerns of synthetic host-guest bioorthogonal tools for pre-targeted applications from the preliminary development stage into the rapid development stage. Addressing these concerns will make researchers see their value, and attract more and more researchers to use them and realize their final clinical applications.
Although synthetic host-guest pairs as novel bioorthogonal tools for pre-targeted applications have been developed for only 5 years and are currently still in an embryonic stage of exploration, the presently developed threes novel host-guest based bioorthogonal tools, such as CB[7]/guest pairs, β-CD/guest pairs and NT-anti/guest pairs, have begun to showcase in several applications of pre-targeting and shown promising results. Fundamentally, the comparison of the conversion rates of covalent bioorthogonal reaction with host-guest bioorthogonal complex via theoretical calculation shows clearly the unique advantages of host-guest pairs as novel bioorthogonal tools in pre-targeted applications in the binding rates (kon = ~108 L mol-1 s-1) and tunable binding affinity (Ka). We anticipate that this review provides an important overview with sufficient analysis of fundamental aspect and experimental cases of the development of synthetic host-guest pairs as novel bioorthogonal tools in pre-targeted applications, and ultimately attract the attention of chemists, medicinal chemists, pharmacologists, and preclinical researchers to expand their applications in biomedicine and realize its final practical applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Science and Technology Program of Guangzhou (No. 202103000089), the National Natural Science Foundation of China (Nos. 22271323 and 22071275), the Innovation Team Project of Universities in Guangdong Province (No. 2020KCXTD009), the Scientific and Technological Innovation Leading Talent Project of Zhongshan City (No. LJ2021009), the Key Projects of Social Welfare and Basic Research of Zhongshan City (No. 2021B2012).
J.A. Prescher, C.R. Bertozzi, Nat. Chem. Biol. 1 (2005) 13–21. doi: 10.1038/nchembio0605-13
E. Saxon, C.R. Bertozzi, Science 287 (2000) 2007–2010. doi: 10.1126/science.287.5460.2007
H.C. Hang, C. Yu, D.L. Kato, C.R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 14846–14851. doi: 10.1073/pnas.2335201100
J.A. Prescher, D.H. Dube, C.R. Bertozzi, Nature 430 (2004) 873–877. doi: 10.1038/nature02791
N.J. Agard, J.A. Prescher, C.R. Bertozzi, J. Am. Chem. Soc. 126 (2004) 15046–15047. doi: 10.1021/ja044996f
E.M. Sletten, C.R. Bertozzi, Angew. Chem. Int. Ed. 48 (2009) 6974–6998. doi: 10.1002/anie.200900942
E.M. Sletten, C.R. Bertozzi, Acc. Chem. Res. 44 (2011) 666–676. doi: 10.1021/ar200148z
S.L. Scinto, D.A. Bilodeau, R. Hincapie, et al., Nat. Rev. Methods Prim. 1 (2021) 30. doi: 10.1038/s43586-021-00028-z
C. Bertozzi, ACS Cent. Sci. 9 (2022) 558–559.
D.M. Patterson, L. Nazarova, J. Prescher, ACS Chem. Biol. 9 (2014) 592–605. doi: 10.1021/cb400828a
M.L. Smeenk, J. Agramunt, K.M. Bonger, Curr. Opin. Chem. Biol. 60 (2021) 79–88. doi: 10.1016/j.cbpa.2020.09.002
D. Wu, K. Yang, Z. Zhang, et al., Chem. Soc. Rev. 51 (2022) 1336–1376. doi: 10.1039/D1CS00451D
A. Battigelli, B. Almeida, A. Shukla, Bioconjugate Chem. 33 (2022) 263–271. doi: 10.1021/acs.bioconjchem.1c00564
L.I. Willems, W.A. van der Linden, N. Li, et al., Acc. Chem. Res. 44 (2011) 718–729. doi: 10.1021/ar200125k
D.K. Nomura, M.M. Dix, B.F. Cravatt, Nat. Rev. Cancer 10 (2010) 630–638. doi: 10.1038/nrc2901
K.S. Yang, G. Budin, C. Tassa, O. Kister, R. Weissleder, Angew. Chem. Int. Ed. 52 (2013) 10593–10597. doi: 10.1002/anie.201304096
A. Rutkowska, D.W. Thomson, J. Vappiani, et al., ACS Chem. Biol. 11 (2016) 2541–2550. doi: 10.1021/acschembio.6b00346
N.K. Devaraj, R. Weissleder, S.A. Hilderbrand, Bioconjug. Chem. 19 (2008) 2297–2299. doi: 10.1021/bc8004446
N.K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek, R. Weissleder, Angew. Chem. Int. Ed. 49 (2010) 2869–2872. doi: 10.1002/anie.200906120
H. Wu, J. Yang, J. Šečkutė, N.K. Devaraj, Angew. Chem. Int. Ed. 53 (2014) 5805–5809. doi: 10.1002/anie.201400135
J. Yang, J. Seckute, C.M. Cole, N.K. Devaraj, Angew. Chem. Int. Ed. 51 (2012) 7476–7479. doi: 10.1002/anie.201202122
Z.J. Gartner, C.R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 4606–4610. doi: 10.1073/pnas.0900717106
G.A. Hudalla, W.L. Murphy, Langmuir 25 (2009) 5737–5746. doi: 10.1021/la804077t
C.H. Kim, J. Axup, A. Dubrovska, et al., J. Am. Chem. Soc. 134 (2012) 9918–9921. doi: 10.1021/ja303904e
Z.Y. Guan, C.Y. Wu, J.T. Wu, et al., ACS Appl. Mater. Interfaces 8 (2016) 13812–13818. doi: 10.1021/acsami.6b03908
H. Koo, S.K. Hahn, S.H. Yun, Bioconjug. Chem. 27 (2016) 2601–2604. doi: 10.1021/acs.bioconjchem.6b00546
H. Koo, M. Choi, E. Kim, et al., Small 11 (2015) 6458–6466. doi: 10.1002/smll.201502972
E. Kim, H. Koo, Chem. Sci. 10 (2019) 7835–7851. doi: 10.1039/C9SC03368H
N.K. Devaraj, ACS Cent. Sci. 4 (2018) 952–959. doi: 10.1021/acscentsci.8b00251
R.E. Bird, S.A. Lemmel, X. Yu, Q.A. Zhou, Bioconjugate Chem. 32 (2021) 2457–2479. doi: 10.1021/acs.bioconjchem.1c00461
R. Rossin, M.S. Robillard, Curr. Opin. Chem. Biol. 21 (2014) 161–169. doi: 10.1016/j.cbpa.2014.07.023
C.L. Schreiber, B.D. Smith, Nat. Rev. Chem. 3 (2019) 393–400. doi: 10.1038/s41570-019-0095-1
E.J.L. Stéen, P.E. Edem, K. Nørregaard, et al., Biomaterials 179 (2018) 209–245. doi: 10.1016/j.biomaterials.2018.06.021
G. Liu, E.A. Wold, J. Zhou, Curr. Top. Med. Chem. 19 (2019) 892–897. doi: 10.2174/1568026619666190510091921
W. Yi, P. Xiao, X. Liu, et al., Sig. Transduct. Target. Ther. 7 (2022) 386. doi: 10.1038/s41392-022-01250-1
M. Handula, K.T. Chen, Y. Seimbille, Molecules 26 (2021) 4640. doi: 10.3390/molecules26154640
T. Liang, Z. Chen, H. Li, Z. Gu, Trends Chem. 4 (2022) 157–168. doi: 10.1016/j.trechm.2021.11.008
B.L. Oliveira, Z. Guo, G.J.L. Bernardes, Chem. Soc. Rev. 46 (2017) 4895–4950. doi: 10.1039/C7CS00184C
R. Zhang, J. Gao, G. Zhao, et al., Nanoscale 15 (2023) 461–469. doi: 10.1039/D2NR06056F
R. Rossin, S.M. van den Bosch, W. ten Hoeve, et al., Bioconjugate Chem. 24 (2013) 1210–1217. doi: 10.1021/bc400153y
R. Selvaraj, J.M. Fox, Curr. Opin. Chem. Biol. 17 (2013) 753–760. doi: 10.1016/j.cbpa.2013.07.031
K.N. Houk, A.G. Leach, S.P. Kim, X. Zhang, Angew. Chem. Int. Ed. 42 (2003) 4872–4897. doi: 10.1002/anie.200200565
A. Rondon, F. Degoul, Bioconjug. Chem. 31 (2020) 159–173. doi: 10.1021/acs.bioconjchem.9b00761
R.J. McMahon, Avidin-Biotin Interactions: Methods and Applications, Humana Press, Totowa, NJ, 2008.
G. Yu, K. Jie, F. Huang, Chem. Rev. 115 (2015) 7240–7303. doi: 10.1021/cr5005315
X. Ma, Y. Zhao, Chem. Rev. 115 (2015) 7794–7839. doi: 10.1021/cr500392w
M.A. Beatty, F. Hof, Chem. Soc. Rev. 50 (2021) 4812–4832. doi: 10.1039/D0CS00495B
J. Zhou, G. Yu, F. Huang, Chem. Soc. Rev. 46 (2017) 7021–7053. doi: 10.1039/C6CS00898D
W.C. Geng, J.L. Sessler, D.S. Guo, Chem. Soc. Rev. 49 (2020) 2303–2315. doi: 10.1039/C9CS00622B
L. Taiariol, C. Chaix, C. Farre, E. Moreau, Chem. Rev. 122 (2022) 340–384. doi: 10.1021/acs.chemrev.1c00484
M. Altai, R. Membreno, B. Cook, V. Tolmachev, B.M. Zeglis, J. Nucl. Med. 58 (2017) 1553–1559. doi: 10.2967/jnumed.117.189944
F.C.J. van de Watering, M. Rijpkema, M. Robillard, W.J.G. Oyen, O.C. Boerman, Front. Med. 1 (2014) 44.
C.J. Addonizio, B.D. Gates, M.J. Webber, Bioconjugate Chem. 32 (2021) 1935–1946. doi: 10.1021/acs.bioconjchem.1c00326
S.M. Cheal, S.K. Chung, B.A. Vaughn, N.K.V. Cheung, S.M. Larson, J. Nucl. Med. 63 (2022) 1302–1315. doi: 10.2967/jnumed.121.262186
J. Murray, J. Sim, K. Oh, et al., Angew. Chem. Int. Ed. 56 (2017) 2395–2398. doi: 10.1002/anie.201611894
S.K. Ghosh, A. Dhamija, Y.H. Ko, et al., J. Am. Chem. Soc. 141 (2019) 17503–17506. doi: 10.1021/jacs.9b09639
G. Sung, S.Y. Lee, M.G. Kang, et al., Chem. Commun. 56 (2020) 1549–1552. doi: 10.1039/C9CC09699J
W. Cao, X. Qin, T. Liu, ChemBioChem 22 (2021) 2914–2917. doi: 10.1002/cbic.202100357
K.L. Kim, G. Sung, J. Sim, et al., Nat. Commun. 9 (2018) 1712. doi: 10.1038/s41467-018-04161-4
J. An, S. Kim, A. Shrinidhi, et al., Nat. Biomed. Eng. 4 (2020) 1044–1052. doi: 10.1038/s41551-020-0589-7
D.V.D.W. Kankanamalage, J.H.T. Tran, N. Beltrami, et al., J. Am. Chem. Soc. 144 (2022) 16502–16511. doi: 10.1021/jacs.2c05726
W. Cao, X. Qin, Y. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 11196–11200. doi: 10.1002/anie.202100916
A. Kataki-Anastasakou, S. Hernandez, E.M. Sletten, ACS Chem. Biol. 16 (2021) 2124–2129. doi: 10.1021/acschembio.1c00494
R. Sasmal, N.D. Saha, M. Pahwa, et al., Anal. Chem. 90 (2018) 11305–11314. doi: 10.1021/acs.analchem.8b01851
A. Som, M. Pahwa, S. Bawari, et al., Chem. Sci. 12 (2021) 5484–5494. doi: 10.1039/D0SC06860H
M. Li, A. Lee, K.L. Kim, et al., Angew. Chem. Int. Ed. 57 (2018) 2120–2125. doi: 10.1002/anie.201711629
A. Lee, M. Li, Y.H. Ko, et al., Chem. Commun. 57 (2021) 12179–12182. doi: 10.1039/D1CC04779E
B.D. Gates, J.B. Vyletel, L. Zou, M.J. Webber, Bioconjug. Chem. 33 (2022) 2262–2268. doi: 10.1021/acs.bioconjchem.2c00242
F. Huang, J. Liu, Y. Liu, Chem. Sci. 13 (2022) 8885–8894. doi: 10.1039/D2SC02797F
H. Tang, D. Fuentealba, Y.H. Ko, et al., J. Am. Chem. Soc. 133 (2011) 20623–20633. doi: 10.1021/ja209266x
A. Douhal, Chem. Rev. 104 (2004) 1955–1976. doi: 10.1021/cr020669j
A.A. Epenetos, D. Snook, H. Durbin, P.M. Johnson, J. Taylor-Papadimitriou, Cancer Res. 46 (1986) 3183–3191.
G.G. Bornstein, AAPS J. 17 (2015) 525–534. doi: 10.1208/s12248-015-9738-4
I. Mahmood, Antibodies 10 (2021) 20. doi: 10.3390/antib10020020
Y. Matsuda, B.A. Mendelsohn, Chem. Pharm. Bull. 69 (2021) 976–983. doi: 10.1248/cpb.c21-00258
S.Y. Wu, F.G. Wu, X. Chen, Adv. Mater. 34 (2022) 2109210. doi: 10.1002/adma.202109210
X. Shu, K. Xu, D. Hou, C. Li, Isr. J. Chem. 58 (2018) 1230–1240. doi: 10.1002/ijch.201800115
T. Ogoshi, T.-a. Yamagishi, Y. Nakamoto, Chem. Rev. 116 (2016) 7937–8002. doi: 10.1021/acs.chemrev.5b00765
W. Liu, E.M. Peck, K.D. Hendzel, B.D. Smith, Org. Lett. 17 (2015) 5268–5271. doi: 10.1021/acs.orglett.5b02633
C.F.A. Gómez-Durán, W. Liu, M.L. Betancourt-Mendiola, B.D. Smith, J. Org. Chem. 82 (2017) 8334–8341. doi: 10.1021/acs.joc.7b01486
M.V. Rekharsky, Y. Inoue, Chem. Rev. 98 (1998) 1875–1918. doi: 10.1021/cr970015o
J. Lagona, P. Mukhopadhyay, S. Chakrabarti, L. Isaacs, Angew. Chem. Int. Ed. 44 (2005) 4844–4870. doi: 10.1002/anie.200460675
A. Urbach, V. Ramalingam, Isr. J. Chem. 51 (2011) 664–678. doi: 10.1002/ijch.201100035
K.I. Assaf, W.M. Nau, Chem. Soc. Rev. 44 (2015) 394–418. doi: 10.1039/C4CS00273C
S.J. Barrow, S. Kasera, M.J. Rowland, J. del Barrio, O.A. Scherman, Chem. Rev. 115 (2015) 12320–12406. doi: 10.1021/acs.chemrev.5b00341
H. Yao, H. Ke, X. Zhang, et al., J. Am. Chem. Soc. 140 (2018) 13466–13477. doi: 10.1021/jacs.8b09157
L.L. Wang, Z. Chen, W.E. Liu, et al., J. Am. Chem. Soc. 139 (2017) 8436–8439. doi: 10.1021/jacs.7b05021
L.P. Yang, X. Wang, H. Yao, W. Jiang, Acc. Chem. Res. 53 (2020) 198–208. doi: 10.1021/acs.accounts.9b00415
Y.L. Ma, M. Quan, X.L. Lin, et al., CCS Chem. 3 (2021) 1078–1092. doi: 10.31635/ccschem.020.202000288
Z. Liu, S.K.M. Nalluri, J.F. Stoddart, Chem. Soc. Rev. 46 (2017) 2459–2478. doi: 10.1039/C7CS00185A
H. Yao, S.Y. Li, H. Zhang, et al., Chem. Commun. 59 (2023) 5411–5414. doi: 10.1039/D2CC06622J
L. Cao, M. Šekutor, P.Y. Zavalij, et al., Angew. Chem. Int. Ed. 53 (2014) 988–993. doi: 10.1002/anie.201309635
G. Hettiarachchi, D. Nguyen, J. Wu, et al., PLoS One 5 (2010) e10514. doi: 10.1371/journal.pone.0010514
X. Zhang, X. Xu, S. Li, et al., Sci. Rep. 8 (2018) 8819. doi: 10.1038/s41598-018-27206-6
M.G. Strebl, J. Yang, L. Isaacs, J.M. Hooker, Mol. Imaging 17 (2018), doi: 10.1177/1536012118799838.
S. Moghaddam, C. Yang, M. Rekharsky, et al., J. Am. Chem. Soc. 133 (2011) 3570–3581. doi: 10.1021/ja109904u
L. Zou, A.S. Braegelman, M.J. Webber, ACS Cent. Sci. 5 (2019) 1035–1043. doi: 10.1021/acscentsci.9b00195
M. Li, S. Kim, A. Lee, et al., ACS Appl. Mater. Interfaces 11 (2019) 43920–43927. doi: 10.1021/acsami.9b16283
V.I.J. Jallinoja, B.D. Carney, M. Zhu, et al., Bioconjugate Chem. 32 (2021) 1554–1558. doi: 10.1021/acs.bioconjchem.1c00280
V.I.J. Jallinoja, B.D. Carney, K. Bhatt, et al., Mol. Pharmaceut. 19 (2022) 2268–2278. doi: 10.1021/acs.molpharmaceut.2c00102
A. Forero, P.L. Weiden, J.M. Vose, et al., Blood 104 (2004) 227–236.
S. Shen, A. Forero, A.F. LoBuglio, et al., J. Nucl. Med. 46 (2005) 642–651.
W. Wei, Z.T. Rosenkrans, J. Liu, et al., Chem. Rev. 120 (2020) 3787–3851. doi: 10.1021/acs.chemrev.9b00738
V.I.J. Jallinoja, J.L. Houghton, J. Nucl. Med. 62 (2021) 1200–1206. doi: 10.2967/jnumed.120.260687
H.C. Huang, S. Barua, G. Sharma, S.K. Dey, K. Rege, J. Control. Release 155 (2011) 344–357. doi: 10.1016/j.jconrel.2011.06.004
N. Fernandes, C.F. Rodrigues, A.F. Moreira, I.J. Correia, Biomater. Sci. 8 (2020) 2990–3020. doi: 10.1039/D0BM00222D
H. Zhu, B. Li, C.Y. Chan, et al., Adv. Drug Deliver. Rev. 192 (2023) 114644. doi: 10.1016/j.addr.2022.114644
G. Yang, S.Z.F. Phua, A.K. Bindra, Y. Zhao, Adv. Mater. 31 (2019) 1805730. doi: 10.1002/adma.201805730
Q. Cheng, L. Yue, J. Li, et al., Small 17 (2021) 2101332. doi: 10.1002/smll.202101332
Q. Cheng, M. Xu, C. Sun, et al., Mater. Horiz. 9 (2022) 934–941. doi: 10.1039/D1MH01813B
W. Li, K. Dong, H. Wang, et al., Biomaterials 217 (2019) 119310. doi: 10.1016/j.biomaterials.2019.119310
G. Crini, Chem. Rev. 114 (2014) 10940–10975. doi: 10.1021/cr500081p
Y. Chen, Y. Liu, Chem. Soc. Rev. 39 (2010) 495–505. doi: 10.1039/B816354P
K.A. Connors, Chem. Rev. 97 (1997) 1325–1357. doi: 10.1021/cr960371r
M.E. Brewster, T. Loftsson, Adv. Drug Deliver. Rev. 59 (2007) 645–666. doi: 10.1016/j.addr.2007.05.012
S.V. Kurkov, T. Loftsson, Int. J. Pharmaceut. 453 (2013) 167–180. doi: 10.1016/j.ijpharm.2012.06.055
T. Loftsson, M.E. Brewster, J. Pharm. Pharmacol. 62 (2010) 1607–1621. doi: 10.1111/j.2042-7158.2010.01030.x
C. Kahle, U. Holzgrabe, Chirality 16 (2004) 509–515. doi: 10.1002/chir.20068
Z. Yang, R. Breslow, Tetrahedron Lett. 38 (1997) 6171–6172. doi: 10.1016/S0040-4039(97)01427-5
S.S. Agasti, M. Liong, C. Tassa, et al., Angew. Chem. Int. Ed. 51 (2012) 450–454. doi: 10.1002/anie.201105670
E. Garin, Y. Rolland, S. Laffont, J. Edeline, Eur. J. Nucl. Med. Mol. I. 43 (2016) 559–575. doi: 10.1007/s00259-015-3157-8
J.F. Prince, R. van Diepen, R. van Rooij, M.G. Lam, Nucl. Med. Commun. 37 (2016) 218–219. doi: 10.1097/MNM.0000000000000424
S.J. Spa, M.M. Welling, M.N. van Oosterom, et al., Theranostics 8 (2018) 2377–2386. doi: 10.7150/thno.23567
M.M. Welling, S.J. Spa, D.M. van Willigen, et al., J. Control. Release 293 (2019) 126–134. doi: 10.1016/j.jconrel.2018.11.020
M.M. Welling, N. Duszenko, D.M. van Willigen, et al., Bioconjug. Chem. 32 (2021) 607–614. doi: 10.1021/acs.bioconjchem.1c00061
M. Xu, J. Tao, Z. Wei, et al., Nano Today 43 (2022) 101450. doi: 10.1016/j.nantod.2022.101450
B.J. Shorthill, C.T. Avetta, T.E. Glass, J. Am. Chem. Soc. 126 (2004) 12732–12733. doi: 10.1021/ja047639d
G.B. Huang, S.H. Wang, H. Ke, L.P. Yang, W. Jiang, J. Am. Chem. Soc. 138 (2016) 14550–14553. doi: 10.1021/jacs.6b09472
H. Ke, L.P. Yang, M. Xie, et al., Nat. Chem. 11 (2019) 470–477. doi: 10.1038/s41557-019-0235-8
H. Chai, Z. Chen, S.H. Wang, et al., CCS Chem. 2 (2020) 440–452. doi: 10.31635/ccschem.020.202000160
W.E. Liu, Z. Chen, L.P. Yang, H.Y. Au-Yeung, W. Jiang, Chem. Commun. 55 (2019) 9797–9800. doi: 10.1039/C9CC04603H
L.M. Bai, H. Yao, L.P. Yang, W. Zhang, W. Jiang, Chin. Chem. Lett. 30 (2019) 881–884. doi: 10.1016/j.cclet.2018.11.033
H. Yao, X. Wang, M. Xie, et al., Org. Biomol. Chem. 18 (2020) 1900–1909. doi: 10.1039/D0OB00290A
Y.L. Ma, C. Sun, Z. Li, et al., CCS Chem. 4 (2022) 1977–1989. doi: 10.31635/ccschem.021.202101178
S. Wilhelm, A.J. Tavares, Q. Dai, et al., Nat. Rev. Mater. 1 (2016) 16014. doi: 10.1038/natrevmats.2016.14
Q. Dai, S. Wilhelm, D. Ding, et al., ACS Nano 12 (2018) 8423–8435. doi: 10.1021/acsnano.8b03900
J. Liu, M. Li, Z. Luo, et al., Nano Today 15 (2017) 56–90. doi: 10.1016/j.nantod.2017.06.010
K.T. Xenaki, S. Oliveira, P.M.P. van Bergen en Henegouwen, Front. Immunol. 8 (2017) 1287. doi: 10.3389/fimmu.2017.01287
J.M. Mejia Oneto, I. Khan, L. Seebald, M. Royzen, ACS Cent. Sci. 2 (2016) 476–482. doi: 10.1021/acscentsci.6b00150
Q. Jin, Z. Liu, Q. Chen, J. Control. Release 329 (2021) 882–893. doi: 10.1016/j.jconrel.2020.10.019
Y.L. Liu, D. Chen, P. Shang, D.C. Yin, J. Control. Release 302 (2019) 90–104. doi: 10.1016/j.jconrel.2019.03.031
M. Saadat, M.K.D. Manshadi, M. Mohammadi, et al., J. Control. Release 328 (2020) 776–791. doi: 10.1016/j.jconrel.2020.09.017
L.A.L. Fliervoet, E. Mastrobattista, Adv. Drug Deliv. Rev. 106 (2016) 63–72. doi: 10.1016/j.addr.2016.04.021
A.S. Timin, M.M. Litvak, D.A. Gorin, et al., Adv. Healthc. Mater. 7 (2018) 1700818. doi: 10.1002/adhm.201700818
T. Zhang, R. Lin, H. Wu, X. Jiang, J. Gao, Adv. Drug Deliv. Rev. 185 (2022) 114300. doi: 10.1016/j.addr.2022.114300
S. Abbina, E.M.J. Siren, H. Moon, J.N. Kizhakkedathu, ACS Biomater. Sci. Eng. 4 (2018) 3658–3677. doi: 10.1021/acsbiomaterials.7b00514

Figure 1 Synthetic host-guest pairs as novel non-covalent bioorthogonal tools to complement and further improve the bioorthogonal chemistry in pre-targeting.

Figure 2 Bioorthogonal chemistry used in pre-targeting. (A) The process of pre-targeting via covalent bioorthogonal reactions and host-guest bioorthogonal recognition. (B) Chemical kinetics of covalent bioorthogonal reaction. (C) Thermodynamics of host-guest bioorthogonal recognition. (D, E) Theoretical calculation of percent conversion of covalent bioorthogonal reactions and host-guest bioorthogonal complexes in pre-targeting, respectively.

Figure 4 (A) Methodology to evaluate the binding affinity needed for forming complex in pre-targeting. (B) Methodology to treat tumor for integrating with CB[7]/guest pair. This figure inspired by [98].

Figure 5 (A) Methodology of antibody as vector for host−guest pre-targeted tumor nuclear imaging. Reproduced with permission [100]. Copyright 2021, American Chemical Society. (B) Optimization of tumor nuclear imaging based CB[7]/guest pair.

Figure 6 Methodology of nanoparticle as vector for host−guest pre-targeted tumor treating via nanoparticles conjugation. Reproduced with permission [110]. Copyright 2021, Wiley Publishing Group.

Figure 7 Methodology of engineering macrophage for anti-bacteria via multivalent host-guest recognition. Copied with permission [111]. Copyright 2022, Royal Society of Chemistry.

Figure 9 (A) Methodology of β-CD/Ad derivative guest pairs as bioorthogonal tools in a multivalent recognition version for pre-targeted liver radioembolization. Reproduced with permission [125]. Copyright 2019, Elsevier. (B) Validation of this method matching the timespan of the clinical radioembolization procedure via dual-isotope SPECT imaging.

Figure 10 Methodology of β-CD/Ad derivative guest pairs as bioorthogonal tools in a multivalent recognition version for pre-targeted tumor treating. Reproduced with permission [127]. Copyright 2022, Elsevier.

Figure 12
Methodology to evaluate the NT-anti/2-phenyl pyrimidine derivative guest pairs as bioorthogonal tools in a monovalent or multivalent recognition version for pre-targeting [135]. This figure has been published in CCS Chemistry [2022], [Biomimetic recognition-based bioorthogonal host–guest pairs for cell targeting and tissue imaging in living animals] is available online at [DOI:

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



 下载:
下载: