
Citation:
Yanan Shang, Yujiao Kan, Xing Xu. Stability and regeneration of metal catalytic sites with different sizes in Fenton-like system[J]. Chinese Chemical Letters,
2023, 34(8): 108278.
doi:
10.1016/j.cclet.2023.108278

Stability and regeneration of metal catalytic sites with different sizes in Fenton-like system
English
Stability and regeneration of metal catalytic sites with different sizes in Fenton-like system
Abstract:
Metal-based catalysts with different site sizes (e.g., metal nanoparticles (NPs) and single atom catalysts (SACs)) demonstrated outstanding catalytic activities in versatile Fenton-like reactions. However, the surface/structural instability is a critical issue, which will result in rapid passivation in Fenton-like reaction and fail in long-term operation. The catalytic stability of the catalysts with different metal sizes considering versatile peroxides (H2O2, peroxymonosulfate (PMS), and peroxodisulfate (PDS)) should be analyzed. In addition, strategies for catalyst regeneration and recyclability improvement are also important to realize the metal-based catalysts for practical applications. In this review, catalytic stability of catalysts with different metal sizes in the backgrounds of versatile peroxides and water matrixes in Fenton-like reactions were first evaluated. Regeneration of metal catalytic sites with different methods were also reviewed. Finally, major challenges and development of methods concerning the stability and regeneration of metal catalytic sites with different sizes were discussed to understand the future researches of metal catalytic sites in Fenton-like reactions.
-
Key words:
- Size effect
- / Metal nanoparticles catalysts
- / Single atom catalysts
- / Fenton-like
-
1. Introduction
Recently, the design of cost-effective and stable Fenton-like based catalysts with highly active metal sites for the environmental remediations is critical. The activations of various Fenton-like systems using different peroxides (e.g., H2O2, peroxymonosulfate (PMS), and peroxodisulfate (PDS)) via the heterogeneous metal-based catalysts have received more and more attentions due to their high-efficiency in oxidizing versatile refractory organics [1-4]. Substantial effort has been devoted recently to exploiting the highly active metal-based catalysts. Since the metal centers are the dominant active sites for Fenton-like reactions, a series of metal-based nanoparticles (NPs) have been developed for the activations of various peroxides. Various metal NPs catalysts (e.g., Co3O4, FeMnOn, FeCoOn, Fe0) as well as their carbon hybrids have showed extraordinary excellent catalytic performances towards various peroxides [5-7]. However, most metal NPs catalysts exhibited a wide range of particle size distributions and surface features; this posed the serious challenges to the rational designs of active metal sites for different Fenton-like reactions. In addition, since most of the metal atoms in the metal NPs catalysts are embedded inside the catalysts, the atomic utilization of metal NPs catalysts was mainly controlled on the surface of catalysts [5,6]. Therefore, many recent studies have focused on the downside of particle size and engineering surface topography to obtain more surface metal catalytic sites.
Single-atom catalysts (SACs) with fully exposure of metal sites have showed superior selectivity and efficiency in Fenton-like reactions as compared with those of metal NPs catalysts [8-10]. The superior catalytic activities of SACs were mainly ascribed to the atomically-dispersion of metal atoms. In addition, the geometric, electronic properties and metal-support interactions of SACs were closely related to the single-atom metal sites and supporting matrixes, and ultimately affect their activation pathways towards different peroxides and catalytic performances [11-14].
Although metal NPs and SACs demonstrate outstanding catalytic activities in versatile Fenton-like reactions, the surface/structural instability of both metal NPs and SACs is a critical issue, which will result in rapid passivation in catalysis and fail in long-term operation. Previous reviews always focused on the catalytic activities and mechanisms of different metal catalytic sites in various Fenton-like systems. The catalytic stability of the catalysts with different metal sizes considering the versatile peroxides (H2O2, PMS, and PDS) were not considered previously, which can be well analyzed to understand the potential relationships. In fact, the water matrixes (e.g., coexisting anions) were also the important factors that affected the catalytic stability of different metal catalytic sites. In addition, strategies for catalyst regeneration and to improve the recyclability are also important to realize the metal-based catalysts for practical applications, but they were also less reviewed.
2. Stability of different metal catalytic sites for versatile peroxides
Multiple peroxides (H2O2, PMS and PDS) should be considered when analyzing the effect of metal size on catalyst stability [1-4]. The difference in the catalytic stabilities between the SACs and metal NPs was relatively lower for H2O2 activation as compared with those for PMS/PDS activation [2-4]. The H2O2 activated by the metal-based catalysts (metal NPs and SACs) was always based on the HO•-based pathways, which mean the high mineralization degree of organic pollutants with less intermediate products. Therefore, the coverage of catalytic sites by reaction intermediates was retarded in the metal-based catalysts/H2O2 systems leading to the less inactivity of catalysts. In contrast, both radical or nonradical pathways always occurred in the PMS/PDS systems with lower mineralization of organic pollutants for both metal NPs and SACs. For example, low-valent Co species (e.g., Co0 and CoO) and high-valent species (Co3O4) showed significant different contributions to the HO•/SO4•− generation via PMS activation (Fig. 1a) [5]. Catalytic mechanism of MnOx with PMS activation for phenol removal showed three kinds of complicated mechanisms (Fig. 1b), which resulted in different catalytic capacities and stabilities [6]. PMS/PDS can be activated into nonradical/radical reactive species by the metal NPs, which were regulated by their physicochemical features, such as crystalline structure and defective sites (oxygen and iodine vacancies). In fact, PMS/PDS activated by SACs always showed more complicated mechanisms since these SACs can be anchored onto the various substrate matrixes, which exhibited both metal and substrate properties in PMS/PDS catalytic systems [1,9,11-13]. In addition, the intermediate products would be generated and adhered onto the catalytic sites of both metal NPs and SACs, which resulted in the catalyst poisoning [11,13]. Recent studies also reported that some catalytic sites would form the complexes with PMS/PDS, which would further hinder the recovery of catalytic sites [1,11,13].
Figure 1

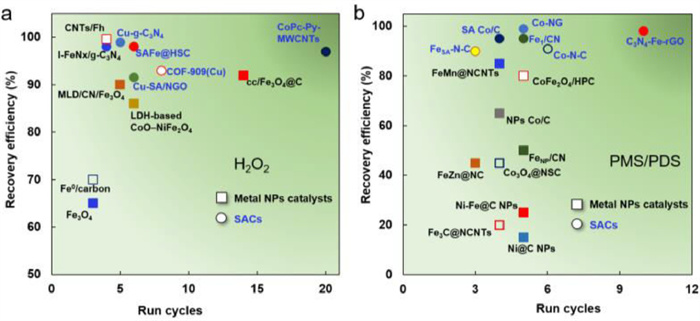
Considering that both metal NPs and SACs can be used in versatile peroxide systems, their superiorities in stability can be summarized. A comparison of the catalytic stability of metal NPs catalysts and SACs in H2O2 activation and PMS/PDS activation system was summarized in Fig. 2. It was obvious that the recovery efficiency of most SACs in PMS/PDS activation system can be stable over 90% (Fig. 2), while most metal NPs counterparts only achieved 50%−80% of recyclability in PMS/PDS system [15]. Qi et al. fabricated both Co-SACs and Co NPs catalyst by using lignin as carbon precursor, and compared their catalytic stabilities in PMS activation system [1]. The Co-SACs showed excellent recyclability after four cycles with only 3% loss of catalytic activity. This is in stark contrast to the Co NPs catalyst that the efficiency of Co NPs catalyst decreased by over 30% after running four times (Fig. 3a). As a result, the SACs exhibit their superiorities in the high stability, which can be attributed to the high dispersion of single metal sites in the supporting matrixes as well as the strong metal-support interactions, which endowed the metal sites with high catalytic activity and protected the active sites from metal leaching (Fig. 3b). Although the catalytic stabilities of SACs as well as their structural stabilities (e.g., metal leaching, substrates decomposition) seemed to be inevitable in most reported work and almost 2%−10% of single-atom metal leaching can be observed in some cases [16-20]. This can be ascribed to several well-known factors, summarized in Fig. 4 [21]. As for the metal NPs, although their recyclability was lower than those of SACs, the relatively mature catalytic membrane immobilized with metal NPs can effectively improve the stability of the catalytic system, while single-atom catalytic membranes are still in their infancy [9].
Figure 2
 Figure 2. Comparison of the catalytic stability of metal NPs catalysts and SACs by (a) H2O2 activation and (b) PMS/PDS activation.
Figure 2. Comparison of the catalytic stability of metal NPs catalysts and SACs by (a) H2O2 activation and (b) PMS/PDS activation.Figure 3
 Figure 3. (a) Stability of SA Co-N/C and NPs Co/C. (b) N 1s of original and used SA Co-N/C. Reprinted with permission [1], Copyright 2020, Elsevier.
Figure 3. (a) Stability of SA Co-N/C and NPs Co/C. (b) N 1s of original and used SA Co-N/C. Reprinted with permission [1], Copyright 2020, Elsevier.Figure 4
 Figure 4. Factors affecting the catalytic stabilities of SACs.
Figure 4. Factors affecting the catalytic stabilities of SACs.Based on the above analysis, increasing the density of metal active sites in the SACs or stabilizing the atomically dispersed metal sites could effectively improve the stability of SACs in versatile peroxide systems [19]. Li et al. reported that the SACs with 8.0 wt% of single-atom Co loading could achieve almost 100% of recovery after 5 cycles [22]. To obtained the stabilized catalytic sites, Zuo et al. showcased a strategy to stabilize the atomically dispersed Fe catalysts by anchoring the Fe atoms in a C3N4-rGO double-layered support (C3N4-Fe-rGO) [23]. The double-layered support could significantly prohibit metal leaching and agglomeration of Fe atoms while maintaining activity of the reaction sites. The layered C3N4-Fe-rGO exhibited stable catalytic efficiency (> 95%) for several reaction cycles, while the efficiency of single-layered C3N4-Fe decreased by over 50% only after five cycles. They also found that the populations of the Fe sites as well as their reductive and oxidative potentials were essentially unaltered, indicating the stable bonding environment for Fe atoms in the ternary C3N4-Fe-rGO. As for the metal NPs, the promotion of catalytic stability of metal NPs catalysts can be achieved by stabilizing the catalytic sites with the supports after specific modification [24]. For example, Fe3O4 magnetic nanoparticles stabilized on the carbon substrates after poly(catechol) modification exhibited exceptional high stability and catalytic activity towards refractory organics [24]. The poly(catechol) could not only stabilize the Fe3O4 to avoid the loss of metal sites, but also promoted the regeneration of Fe(Ⅱ) via the strong metal-support interaction to promote the activation of peroxides. As a result, negligible recovery loss were observed after 8 cycles with little iron leaching during all running cycles [24]. This also provides a strategy for improving the stabilities of both metal NPs and SACs.
3. Stability of different metal catalytic sites for water matrixes
Inorganic anions (e.g., H2PO4−, CO32−, HCO3−, NO3−, Cl−) and natural organic matters (NOMs) that exist in water bodies have a great influence on the activation of peroxides [25-27]. These substances could react with HO• and SO4•– to form weaker radicals or compete with pollutant molecules with the active sites, resulting in the inferior degradation stability [27,28]. As a result, effect of water matrixes on the catalytic stability of metal-based catalysts (metal NPs and SACs) can be analyzed. A comparison of the catalytic performance of SACs/PMS and metal NPs/PMS systems with different types of coexisting ions and NOMs indicated that SACs show stronger anti-interference performance to coexisting substances than that of metal NPs [29,30]. Luo and co-workers reported that the single iron atom anchored in C3N4 (FeSA/CN) was more stable than the FeNP/CN during the activation of PMS with coexisting inorganic ions, and NOM [18]. Because the FeSA/CN only produced 1O2 via activating PMS, which exhibited strong resistance to environmental factors with excellent cycle stability [18]. In contrast, radicals-dominated pathways induced by FeNP/CN can be easily quenched by the anions or NOMs in the PMS systems [1,31]. Similarly, Qi et al. reported that degradation of NPX by the Co-SAC/PMS system with the background of H2PO4− and NO3− can be restrained due to the quenching of active radicals to form the secondary radicals with lower activity [1]. The inhibition caused by humic acid was most likely attributed to the attachment with catalytic sites on surface of Co-SAC catalyst as well as the competition with NPX for catalytic sites, thus hindering the ROS formation in Co-SAC/PMS system [27,32].
Generally, the coexisting CO32− and HCO3− could transform the high-oxidative radicals (HO• and SO4•–) into the lower oxidative carbonate radicals (e.g., HCO3•, and CO3•–), which reduced the catalytic activity in the radical-dominated systems [33,34]. However, the HCO3•, and CO3•– were more selective than HO• and SO4•– for targeted pollutants with electron-rich moieties [35-37], and recent studies reported that the nonradical-dominated reaction systems induced by both metal NPs and SACs were insensitive to the coexisting CO32− and HCO3− [35-38]. It was also reported the existence of dual-effects of Cl− for PMS activation via metal NPs and SACs [1,38]. Low-concentrations (2–5 mmol/L) of Cl− had a slight inhibitory effect on the degradation of organics, which can be attributed to the reaction between Cl− and free radicals (HO• and SO4•–), producing the Cl• and Cl2•− with lower redox potentials [2,39]. In contrast, catalytic process can be greatly facilitated with high concentrations (500 mmol/L) of Cl−, which was due to the generation of high HOCl as well as the nonradical pathways insensitive to the Cl− [1,38]. Such dual-effects were mainly affected by the concentrations of Cl− existed in the water matrixes rather than the sizes of metal sites. As a result, the dual-effects of Cl− on the PMS activation were universal and have been reported in both metal NPs/PMS and SACs/PMS systems [2,38]. Peng et al. reported the ultrafast paracetamol (PCM) degradation (0.534 min−1) in the seawater (salinity of 3.6%) by Co3O4-based catalyst as compared with that (0.162 min−1) in tap water [2]. Oxidization of NPX by Co-SACs/PMS system with coexisting 50 mmol/L of Cl− was 2.5 times higher than that of lower Cl− background (5 mmol/L) [1]. Such results unveiled the potential applications of oxidization of refractory organic pollutants in saline waters for both metal NPs and SACs.
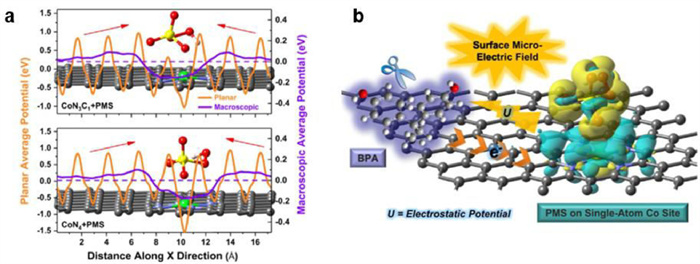
Although the stability of metal catalytic sites can be affected by coexisting anions no matter their sizes, many studies have further evaluated the decomposition of organics by the SACs and metal NPs catalysts in different in actual water bodies [30,40]. Peng and co-workers reported the strong anti-interference Fenton-like reaction due to the construction of surface micro-electric fields on hollow Co-SAC (Fig. 5) [40]. Zou and co-workers reported the negligible impact of organics degradation operated in Poyang lake water and Yangtze river water for PMS activation by FeSA/CN [18], indicating its potential application for practical waters. There are also observations that the Fenton-like activities of SACs decreased in the actual water bodies, mainly due to the coverage and blockage of the catalytic sites by the degradation intermediates and impurities. In particular, most reported metal NPs catalysts always showed a higher loss of degradation activity in the actual water environments [41,42], which further indicated the superior catalytic stability of SACs over metal NPs catalysts.
Figure 5
 Figure 5. (a) Macroscopic average electrostatic potentials of different SAC/PMS complexes. (b) Mechanism of surface micro-electric fields on BPA oxidation by PMS activation. Reprinted with permission [40], Copyright 2020, Elsevier.
Figure 5. (a) Macroscopic average electrostatic potentials of different SAC/PMS complexes. (b) Mechanism of surface micro-electric fields on BPA oxidation by PMS activation. Reprinted with permission [40], Copyright 2020, Elsevier.4. Regeneration of metal catalytic sites
Regeneration is another major concern for the metal-based catalysts (metal NPs catalysts and SACs) to be available in practical applications. The reduction of recyclability of catalysts was mainly due to the deactivation of the catalytic sites resulted from the coverage of product intermediates and unreacted peroxides, and the loss of catalytic sites. Therefore, the regeneration of deactivated catalysts can be achieved by a variety of methods (e.g., calcination, solvent washing, metal impregnation, and UV irradiation), which were universal to both metal NPs catalysts and SACs [1,30,43-48].
Generally, calcination of the deactivated catalysts at a medium temperature (350–500 ℃) is the most widely used method for catalyst regeneration and shows a very promising recovery capacity [11,13,38]. This is partially due to the thermal decomposition of the product intermediates attached on the surface of catalysts, which could re-expose the catalytic sites for use. Shang et al. reported the severe deactivation (75% of catalytic performance loss) of Fe3C@NCNTs catalysts after four running cycles. The exhausted catalysts were calcinated at 350 ℃ and the catalytic activity could be recovered from 25% to more than 98%. Ma and co-workers also achieved 100% of recovery of the biochar loaded with CoFe2O4 nanoparticles (80% of performance left after 5 cycles) via calcining at 400 ℃ [44]. Calcination of the exhausted ACs also showed almost complete recovery of the catalytic activity [1]. Deactivated single-atom cobalt sites anchored on the lignin-based carbon frameworks can be effectively regenerated by heating treatment at 400 ℃ with the same kinetic rate as the original sample [1,30,43]. In addition, the morphologies of these recovered catalysts remain intact after calcination, indicating that the heat-treatment was a promising method for exhausted catalysts with both high recovery capacity and high stability.
The facile solvent washing can achieve the recovery of some exhausted metal-based catalysts [46,48,49]. Pan and co-workers reported that the Fe2O3-based catalyst was slightly deactivated after five consecutive MB degradation due to the cumulative degradation products occupying the active sites [49]. The catalytic activity could be fully recovered after ethanol washing. The same group further examined the reusability of the single-atom Fe anchored carbon nanotubes for MB degradation [46]. Methanol washing was also proved to be efficient to recover the metal NPs and SACs. In addition to these organic solvents, Oladipo et al. reported the desorption of the attached dye intermediates from the exhausted CoO–NiFe2O4 by using 0.01 mol/L of NaOH [48]. The Fenton-like performance of the NaOH-eluted hybrid catalyst after six cycles can be recovered ~90%. However, the solvent method has a narrow application, since it can only be used in catalytic systems in which the degradation products can be dissolved by these solvents.
Since catalyst deactivation can also be ascribed to the loss of metal catalytic sites, some attempts have been made to re-impregnate the metal sites onto the used catalysts [47,50,51]. Successful attempt to regenerate an exhausted iron-coated Fenton catalyst was described by Akinremi et al. [47]. The novel iron-based catalyst was deactivated due to the iron loss caused by acid hydrolysis in a Fenton-like system. The deactivated catalyst was modified by a hydroxylamine reaction and then the iron(Ⅲ) salt was impregnated onto the hydroxylaminated iron-based catalyst. Li et al. reported the recovery of the V and W active sites in the V2O5-WO3/TiO2 catalyst via the impregnation of V and W elements from the salts of ammonium metavanadate (NH4VO3) and ammonium paratungstate hydrate (H40N10O41W12·xH2O) [51]. The catalytic performance of regenerated V2O5-WO3/TiO2 catalyst can be almost completely recovered as compared with the fresh catalyst. Although the preliminary results suggest that this method can be used for catalyst recovery, the regeneration process is relatively complicated and can only be used in cases of severe metal sites loss. Other regeneration methods, e.g., ultrasonic regeneration, UV irradiation, are sporadically reported in some cases [45,52], which can be used in both metal NPs and SACs.
5. Conclusions and perspectives
Metal-based catalysts with both metal NPs and SACs can be used in versatile peroxide systems, while the difference in the catalytic stabilities between the SACs and metal NPs was relatively lower for H2O2 activation as compared with those for PMS/PDS activation. The H2O2 activated by the metal-based catalysts was always based on the HO•-based pathways, which mean the high mineralization degree of organic pollutants with less intermediate products. In contrast, both radical or nonradical pathways always occurred in the PMS/PDS systems with lower mineralization of organic pollutants. Recovery efficiency of most SACs in PMS/PDS activation system can be higher than those of most metal NPs counterparts. Basically, the high stability of the SACs can be attributed to the high dispersion of single metal sites in the supporting matrixes as well as the strong metal-support interactions, which endowed the metal sites with high catalytic activity and protected the active sites from metal leaching. In addition, the stability of metal catalytic sites can be affected by coexisting anions no matter their sizes, while most reported metal NPs catalysts always showed a higher loss of degradation activity in the actual water environments, which further indicated the superior catalytic stability of SACs over metal NPs catalysts.
The regeneration of deactivated catalysts can be achieved by a variety of methods (e.g., calcination, solvent washing, metal impregnation, and UV irradiation), which were universal to both metal NPs catalysts and SACs. In future, more efforts can be donated to understand the deactivation pathways of catalysts in different reaction environments. With increased understanding of deactivation mechanisms, effective strategies to prolong the lifetimes of highly active and selective catalysts can be expected. Developing catalysts with self-healing abilities is another intriguing approach to addressing these stability issues.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The work was supported by National Natural Science Foundation of China (No. 52170086). The authors also want to thank Conghua Qi from Shiyanjia Lab (
http://www.shiyanjia.com ).
-
-
[1]
Y. Qi, J. Li, Y. Zhang, et al., Appl. Catal. B Environ. 286 (2021) 119910. doi: 10.1016/j.apcatb.2021.119910
-
[2]
L. Peng, Y. Shang, B. Gao, X. Xu, Appl. Catal. B Environ. 282 (2021) 119484. doi: 10.1016/j.apcatb.2020.119484
-
[3]
Q. Zhao, C.C. Wang, P. Wang, Chin. Chem. Lett. 33 (2022) 4828–4833. doi: 10.1016/j.cclet.2022.01.033
-
[4]
N. Li, Y. Wang, C. Wu, et al., Appl. Surf. Sci. 434 (2018) 1112–1121. doi: 10.1016/j.apsusc.2017.11.048
-
[5]
J. Liang, L. Fu, K. Gao, X. Duan, Appl. Catal. B Environ. 315 (2022) 121542. doi: 10.1016/j.apcatb.2022.121542
-
[6]
Y. Yang, P. Zhang, K. Hu, et al., Appl. Catal. B Environ. 315 (2022) 121593. doi: 10.1016/j.apcatb.2022.121593
-
[7]
D. Guo, L.X. Zhao, H. Zhang, Chin. Chem. Lett. 33 (2022) 1263–1266. doi: 10.1016/j.cclet.2021.07.051
-
[8]
Z. Shen, Y. Zhou, Y. Guo, et al., Chin. Chem. Lett. 32 (2021) 2524–2528. doi: 10.1016/j.cclet.2021.01.044
-
[9]
Y. Shang, X. Duan, S. Wang, et al., Chin. Chem. Lett. 33 (2022) 663–673. doi: 10.1016/j.cclet.2021.07.050
-
[10]
Y. Su, M. Lu, R. Su, et al., Chin. Chem. Lett. 33 (2022) 2573–2578. doi: 10.1016/j.cclet.2021.08.078
-
[11]
L. Peng, X. Duan, Y. Shang, B. Gao, X. Xu, Appl. Catal. B Environ. 287 (2021) 119963. doi: 10.1016/j.apcatb.2021.119963
-
[12]
H. Zheng, Y. Hou, S. Li, et al., Chin. Chem. Lett. 33 (2022) 5013–5022. doi: 10.1016/j.cclet.2022.01.048
-
[13]
Y. Shang, X. Xu, B. Gao, S. Wang, X. Duan, Chem. Soc. Rev. 50 (2021) 5281–5322. doi: 10.1039/d0cs01032d
-
[14]
M. Yang, Z. Hou, X. Zhang, et al., Environ. Sci. Technol. 56 (2022) 11635–11645. doi: 10.1021/acs.est.2c01261
-
[15]
Y. Li, T. Yang, S. Qiu, et al., Chem. Eng. J. 389 (2020) 124382. doi: 10.1016/j.cej.2020.124382
-
[16]
Q.Y. Wu, J. Wang, Z.W. Wang, et al., Mater. Chem. A 8 (2020) 13685–13693. doi: 10.1039/d0ta04943c
-
[17]
C. Chu, J. Yang, X. Zhou, et al., Environ. Sci. Technol. 55 (2021) 1242–1250. doi: 10.1021/acs.est.0c06086
-
[18]
L.S. Zhang, X.H. Jiang, et al., Angew. Chem. Int. Ed. 60 (2021) 21751–21755. doi: 10.1002/anie.202109488
-
[19]
X. Li, X. Huang, S. Xi, et al., J. Am. Chem. Soc. 140 (2018) 12469–12475. doi: 10.1021/jacs.8b05992
-
[20]
K. Qian, H. Chen, W. Li, et al., Environ. Sci. Technol. 55 (2021) 7034–7043. doi: 10.1021/acs.est.0c08805
-
[21]
Y. Gao, X.G. Duan, B. Li, et al., J. Mater. Chem. A 9 (2021) 14793–14805. doi: 10.1039/d1ta02446a
-
[22]
H. Li, J. Qian, B. Pan, Chem. Eng. J. 403 (2021) 126395. doi: 10.1016/j.cej.2020.126395
-
[23]
S. Zuo, X. Jin, X. Wang, et al., Appl. Catal. B Environ. 282 (2021) 119551. doi: 10.1016/j.apcatb.2020.119551
-
[24]
Y. Hua, C. Wang, S. Wang, J. Xiao, Environ. Sci. Pollut. Res. 28 (2021) 62690–62702. doi: 10.1007/s11356-021-15088-7
-
[25]
G. Wang, X. Nie, X. Ji, et al., Environ. Sci. Nano 6 (2019) 399–410. doi: 10.1039/c8en01231h
-
[26]
R. Bai, W. Yan, Y. Xiao, et al., Chem. Eng. J. 397 (2020) 125501. doi: 10.1016/j.cej.2020.125501
-
[27]
W. Du, Q. Zhang, Y. Shang, et al., Appl. Catal. B Environ. 262 (2020) 118302. doi: 10.1016/j.apcatb.2019.118302
-
[28]
S. Yanan, X. Xing, Q. Yue, B. Gao, Y. Li, Environ. Sci. Nano 7 (2020) 1444–1453. doi: 10.1039/d0en00050g
-
[29]
Y. Yin, R. Jia, W. Zhang, et al., J. Clean. Prod. 319 (2021) 128680. doi: 10.1016/j.jclepro.2021.128680
-
[30]
X. Peng, J. Wu, Z. Zhao, et al., Chem. Eng. J. 429 (2022) 132245. doi: 10.1016/j.cej.2021.132245
-
[31]
J.C.E. Yang, Y. Lin, H.H. Peng, et al., Appl. Catal. B Environ. 268 (2020) 118549. doi: 10.1016/j.apcatb.2019.118549
-
[32]
C. Chen, T. Ma, Y. Shang, et al., Appl. Catal. B Environ. 250 (2019) 382–395. doi: 10.1016/j.apcatb.2019.03.048
-
[33]
H. Zheng, J. Bao, Y. Huang, et al., Appl. Catal. B Environ. 259 (2019) 118056. doi: 10.1016/j.apcatb.2019.118056
-
[34]
Y. Liu, H. Guo, Y. Zhang, et al., Environ. Pollut. 252 (2019) 1042–1050. doi: 10.1016/j.envpol.2019.05.157
-
[35]
Y. Fan, Y. Ji, D. Kong, J. Lu, Q. Zhou, J. Hazard. Mater. 300 (2015) 39–47. doi: 10.1016/j.jhazmat.2015.06.058
-
[36]
J. Luo, T. Liu, D. Zhang, et al., Water Res. 159 (2019) 102–110. doi: 10.1016/j.watres.2019.05.019
-
[37]
Y. Wang, P. Yan, X. Dou, et al., Appl. Catal. B Environ. 290 (2021) 120048. doi: 10.1016/j.apcatb.2021.120048
-
[38]
F. Chen, L. Liu, J. Chen, et al., Water Res. 191 (2021) 116799. doi: 10.1016/j.watres.2020.116799
-
[39]
Z. Zhang, Y. Zheng, W. Hang, X. Yan, Y. Zhao, Talanta 85 (2011) 779–786. doi: 10.1016/j.talanta.2011.04.078
-
[40]
C. Zhu, Y. Nie, S. Zhao, Z. Fan, F. Liu, A. Li, Appl. Catal. B Environ. 305 (2022) 121057. doi: 10.1016/j.apcatb.2021.121057
-
[41]
X. Peng, J. Wu, Z. Zhao, X. Wang, H. Dai, Y. Li, Y. Wei, G. Xu, F. Hu, et al., Environ. Res. 205 (2022) 112538. doi: 10.1016/j.envres.2021.112538
-
[42]
Z. Wu, Y. Wang, Z. Xiong, et al., Appl. Catal. B Environ. 277 (2020) 119136. doi: 10.1016/j.apcatb.2020.119136
-
[43]
X. Peng, J. Wu, Z. Zhao, et al., Chem. Eng. J. 427 (2022) 130803. doi: 10.1016/j.cej.2021.130803
-
[44]
Y. Li, S. Ma, S. Xu, et al., Chem. Eng. J. 387 (2020) 124094. doi: 10.1016/j.cej.2020.124094
-
[45]
E. Yun, S. Park, H. Shin, et al., Appl. Catal. B Environ. 279 (2020) 119360. doi: 10.1016/j.apcatb.2020.119360
-
[46]
L. Chen, S. Wang, Z. Yang, J. Qian, B. Pan, Appl. Catal. B Environ. 292 (2021) 120193. doi: 10.1016/j.apcatb.2021.120193
-
[47]
C.A. Akinremi, S. Rashid, P.D. Upreti, G.T. Chi, K. Huddersman, RSC Adv. 10 (2020) 12941–12952. doi: 10.1039/d0ra00520g
-
[48]
A.A. Oladipo, A.O. Ifebajo, M. Gazi, Appl. Catal. B Environ. 243 (2019) 243–252. doi: 10.1016/j.apcatb.2018.10.050
-
[49]
Z. Yang, J. Qian, A. Yu, B. Pan, P. Natl Acad. Sci. U. S. A. 116 (2019) 6659–6664. doi: 10.1073/pnas.1819382116
-
[50]
X. Wang, J. Jing, M. Zhou, R. Dewil, Chin. Chem. Lett. 34 (2023) 107621. doi: 10.1016/j.cclet.2022.06.044
-
[51]
X. Hu, D. Zhou, H. Wang, et al., Chin. Chem. Lett. 34 (2023) 108050. doi: 10.1016/j.cclet.2022.108050
-
[52]
M. Cheng, C. Lai, Y. Liu, et al., Coord. Chem. Rev. 368 (2018) 80–92. doi: 10.1016/j.ccr.2018.04.012
-
[1]
-
Figure 2 Comparison of the catalytic stability of metal NPs catalysts and SACs by (a) H2O2 activation and (b) PMS/PDS activation.
Figure 3 (a) Stability of SA Co-N/C and NPs Co/C. (b) N 1s of original and used SA Co-N/C. Reprinted with permission [1], Copyright 2020, Elsevier.
-

 DownLoad:
DownLoad:



 下载:
下载:




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 2
- 文章访问数: 290
- HTML全文浏览量: 9
 下载:
下载:
