
Figure 1.
(a) Bioactive compounds comprising DHQ core. (b) Workflow of DEL technology. (c) Antimony trichloride-promoted synthesis of on-DNA DHQ scaffold.
Antimony salt-promoted cyclization facilitating on-DNA syntheses of dihydroquinazolinone derivatives and its applications
Qigui Nie , Jie Sun , Xianfu Fang , Xun He , Feng Xiong , Gong Zhang , Yangfeng Li , Yizhou Li

Nitrogen-containing heterocycles are predominant in many natural products and pharmaceutical compounds. Statistically, over 75% of FDA-approved drugs contain nitrogen-containing heterocycles [1]. Thus, medicinal chemistry research and development of nitrogen-containing heterocycles are overwhelming, attracting a large amount of work toward such skeletons. Among those nitrogen-containing heterocycles, dihydroquinazolinone (DHQ) is an important subtype that is broadly found in a variety of bioactive molecules (Fig. 1a). Its structure contains pharmacophores that forge interactions with specific targets [2]. For instance, compound 1, a Tankyrase 2 inhibitor, forms a bidentate hydrogen bond by the carbonyl and adjacent NH group with glycine residue of the receptor protein in the co-crystal structure [3]. Besides, the sp3 carbon in DHQ contributes to conformational flexibility of the fused ring system and could serve as an important structural diversifying site. The spiro-locked modification at such a site gives compound 2, an antibacterial agent which exhibits activities against two Gram-negative bacterial strains [4]. Consequently, it is of great value to develop synthetic methodologies to generate DHQ scaffold for medicinal use [5]. Furthermore, the diverse activities elicited by the heterocycle convinced us of the significance to construct a DHQ-focused compound library, especially with a combinatorial synthetic strategy gaining high chemical diversities.
DNA-encoded chemical libraries (DELs) technology, emerging as a novel platform for drug discovery, has showcased the great potential for early hit identification [6-9]. Unlike conventional compound libraries, DEL leverages genetic barcoding to record the structural information of each member in the libraries [10-12]. In a typical DEL workflow, for diverse building blocks (BBs) after each synthetic reaction stepwise, an amplifiable unique DNA sequence as the molecular identifier is tethered via enzymatic ligation. By iterative "split-and-pool" strategies, the unprecedented size of the encoded libraries is gained (Fig. 1b). DEL technology enables selecting millions or billions of molecules against protein-of-interest (POI) simultaneously in a single tube instead of one-by-one screening [13-17]. Afterward, the chemical information of selected binders is determined by amplification of PCR and decoded by next-generation sequencing. The merits of cost-effectiveness and time efficiency render DEL an alternative to high-throughput screening, and thus DEL is widely adopted in pharmaceutical companies and academia. Recent years have witnessed the discovery of drug candidates that are derived from corresponding binders by means of DEL technology, exemplified by the sEH inhibitor and RIPK1 inhibitor that have entered clinical trials [18].
Structural diversity and chemical space coverage of a library are critical to the success of selection process since druglike scaffolds such as nitrogen-containing heterocycles play a crucial role in the interaction with targets on the basis of the point from medicinal chemistry. Owing to the existence of DNA tags, synthetic reactions employed in DEL construction must ensure nucleotide integrity. Implementation of DNA-compatible reactions should be under mild and aqueous conditions to provide high-yielding conversions of the desired products [19-25]. Ideally, from the perspective of diversity-oriented synthesis, reactions that utilize abundant BBs are preferred [26-30]. Meanwhile, as the manipulation of DEL construction is on-plate parallel combinatorial synthesis (Fig. 1b), the involved reactions must be well-tolerated for numerous structural analog syntheses instead of the need to optimize reaction conditions for each substrate.
The rapid progress of DNA-compatible reactions developed in recent years has brought deeper chemical space into the DEL field and expanded the on-DNA synthetic toolbox [31-34]. To the best of our knowledge, DNA-compatible synthesis of DHQ has not been achieved yet. Considering the value of dihydroquinazolinone in medicinal chemistry and drug development, we herein present the facile synthesis of on-DNA dihydroquinazolinone from aldehyde and anthranilamide (Fig. 1c). The reaction was promoted by antimony trichloride and demonstrated to be utilized in DNA-compatible parallel synthesis. The resulting on-DNA DHQ derivatives were further easily oxidized into quinazolinones by sodium periodate in situ. Remarkably, DHQ as a privileged scaffold has been efficiently integrated into macrocycles via our developed on-DNA cyclization reaction, to further facilitate macrocycle-focused drug discovery by DEL selection.
Initially, 2-aminobenzoic acid was coupled with DNA head-piece (HP) to form DNA-conjugate a1, which was then used in condition optimization to react with 4-nitrobenzaldehyde b1 (Table 1). A trace of desired product c1 was detected in methanol-water co-solvent system after 3 h (entry 1). Prolongation of time raised the conversion to 90% (entry 2). We sought preliminary exploitation of aryl aldehyde substrate scope, only to find a portion of the defined substrate set gain good conversions under the above condition. However, some substrates beyond benzaldehyde derivatives failed to provide synthetically useful conversions even after 48 h reaction time (Table S1 in Supporting information). The conditions of choice, from the perspective of parallel synthesis and combinatorial chemistry, should be inherently robust and general and tolerate highly functionalized building blocks for DEL constructions [35]. Thus, more efforts were made to accelerate the reaction rate and furnish synthetically useful conversions. We anticipated that some metal salts as mild Lewis acids would promote cyclization [2]. Zirconium tetrakis(dodecyl sulfate), Zr(DS)4, which was reported to catalyze ring opening of on-DNA epoxides in water [36], was adopted. Compared to entry 1, a slight increase to 25% conversion yield was observed with Zr salt (entry 3). We then conducted a screening of various antimony salts as the promoters for the cyclization reaction (entries 4–9). Among them, c1 was obtained with > 95% conversion yield after 3 h, when antimony trichloride was applied (entry 6) [37]. The influence of co-solvent system was further carefully examined (entries 10–16), indicating that the methanol/water solvent system worked best. Reducing antimony salt equivalence from 500 equiv. to 250 equiv. resulted in lower conversion (entry 17). The optimal reaction condition (entry 6) was then confirmed by examining the previous aryl aldehydes (Table S1), and we were delighted to obtain satisfying conversions on some substrates that showed low reactivity before. Given a new chiral center formation, we attempted to differentiate the stereoisomers. UPLC-MS as the sole effective way for analysis was then used, showing only one single peak on achiral columns [38]. With the desired DNA-conjugated dihydroquinazolinone c1 at hand, we attempted further oxidation by the commonly used sodium periodate in the DNA-compatible chemistry [39,40]. Gratifyingly, 4000 equiv. usage of sodium periodate at 25 ℃ yielded the corresponding quinazolinone product with a conversion of 90%, verified by deconvoluted mass. This direct oxidation would provide a DNA-compatible alternative to prepare quinazolinones, while synthesizing such DNA-conjugated quinazolinones by use of other BBs was reported [41,42]. A co-injection experiment was then implemented to validate on-DNA DHQ product and its oxidative state quinazolinone structure. (Figs. S8 and S9 in Supporting information).
Encouraged by these results, we first began the investigation of aldehyde scope by utilizing DNA conjugate a1 (Scheme 1). Aldehydes are commercially available as building blocks, thus contributing to structural diversity in DEL construction [41,43]. Mono-substituted benzaldehydes (b2-b18) were examined to give moderate-to-excellent conversions. Notably, product c11 resembles Tankyrase 2 inhibitor 1 in Fig. 1a, implying that these synthesized molecules could be potentially screened against Tankyrase 2 [3]. Likewise, these DHQ-focused members might also interact with tubulin, as the small-molecule part in the synthesized DNA-conjugated c5 was reported to be a potent tubulin-targeting inhibitor (Fig. 1a) [44]. Pleasing results were also observed in di-substituted and tri-substituted aldehydes. Notably, a variety of functional groups including amine (b4), phenol (b6, b16, b22, b23, b26, b30), halogen (b11, b12, b24-b29), alkyne (b9), alkene (b10), trifluoromethyl (b13), boronic acid (b33) and carboxylic acid (b14, b18, b21, b23, b24, b27) were tolerated. Some of these substrates bearing an additional handle for diversification are ideal since increasing dimensional parameters will boost the library size. And thus, we performed a Suzuki-Miyaura reaction by using c12 and heteroboric acid to obtain the desired coupling product (Fig. S4 in Supporting information). Aldehydes featuring drug-like heteroaryl rings (thiazole, thiophene, pyridine, quinoline) and fused rings were also compatible in the reaction. Moreover, we also tested alkyl aldehydes, since they are more sp3-rich, an important factor that is frequently precluded in the selection of building blocks for DEL construction [45]. To our delight, different chain-length aliphatic aldehydes including natural products like citronellal (b47) and melonal (b48) yielded desired DHQ products. The scope was further expanded to b51-b53. Besides, alicyclic aldehydes (b54-b57) regardless of ring size reacted with a1 to afford the corresponding DHQ scaffolds in high conversions. Attracted by these aldehyde-derived DHQ scaffolds, we performed an oxidation reaction on some of the products at hand to convert them into corresponding quinazolinones and included the results in Scheme 1. Collectively, moderate-to-excellent conversions were received among 22 tested DHQ-containing compounds, demonstrating good tolerance for functional groups like hydroxyl (b16 and b26) and carboxylic acid (b14, b32 and b39).

Isatins as precursors form a unique kind of spiro-locked architecture when reacting with other reagents, and thus they have been widely used in DNA-compatible chemistry [43,46]. While substrates d7, d9, and d15 impeded the reaction to give relatively low conversions, most of the tested isatins afforded acceptable conversions by the prolongation of reaction time to 40 h. Particularly, product e8 was successfully obtained, implying that structurally similar compounds with potential antibacterial activity could be parallelly synthesized [4]. Taken together, our on-DNA synthesis of DHQ core would possibly facilitate ligand-based hit identification in a rational DEL design.
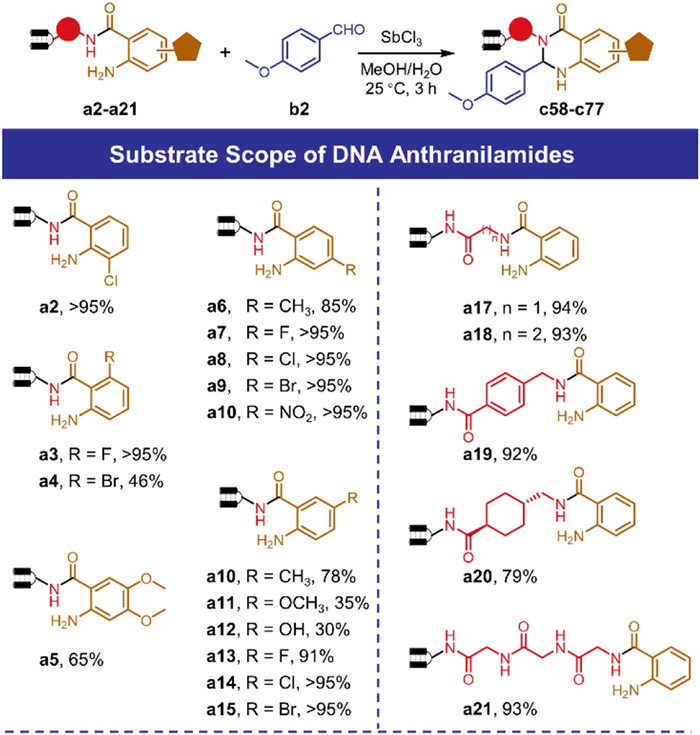
We continued to investigate the scope of DNA-conjugated anthranilamide by using b2 in Scheme 2. DNA-conjugated Anthranilamides bearing different groups were obtained by treatment of corresponding isatoic anhydride with HP. Electron-deficient substrates with halogen (a2-a4, a7-a9, a13-a15) or nitro (a10) furnished well, while compounds equipped with electron-donating groups like methoxy (a11) and phenol (a12) gave inferior conversions. Moreover, we varied the linker between HP and distant anthranilamide and assumed that it was the first-dimensional diversifying site. The DHQ formation reaction was conveniently implemented on these substrates (a17-a20) to provide desired products in great conversions. We further extended the linker length by synthesizing a peptidic molecule a21 to better mimic the real library construction step, since peptide-focused DELs have attracted the interest of researchers [47]. This DNA-conjugated peptide decorated with a distant anthranilamide delivered the corresponding DHQ product in 93% conversion, highlighting the feasibility of our developed reaction in a late-stage modification to install privileged DHQ scaffold into molecules.
DNA-tethered aldehydes were reversely prepared via amide coupling reaction to further evaluate substrate scope. As illustrated in Scheme 3a, benzaldehydes with distinct substituents and heteroaryl aldehydes proceeded smoothly to afford desired products, noting that hindered substrates led to poor conversions. Similarly, subsequent oxidation of DHQ-containing products delivered the corresponding quinazolinone partners in good conversions, indicating that two drug-like moieties could be conveniently constructed. In total, 16 out of 22 DNA-conjugated aldehydes provided conversions over 70% under the optimal reaction condition.

Considering that structurally diverse building blocks are responsible for broadening chemical space in DNA-compatible reactions, we attempted to expand the scope of anthranilamide in Scheme 3b. First, six substrates (a23-a28) reacted readily with f1, giving the cyclized products in satisfactory conversions. We envisioned that N-substituted anthranilamide that increased chemical complexity could be from substituted isatoic anhydrides with abundant amines via ring-opening reaction [48]. To our satisfaction, these N-substituted anthranilamide derivatives achieved good transformations in the DHQ formation reaction except a34. Although it is not convenient to synthesize N-modified products by substitution in a DNA-compatible manner, using previously decorated N-substituted anthranilamide substrates provides synthetic feasibility in obtaining products g29-g35. Besides, carboxylic acid in a31 highlighted its great potential in DEL construction by permitting further diversification via amide coupling. Collectively, anthranilamide as a type of commercially available building block enables structural coverage of DHQ formation reaction.
The feasibility of our developed reaction in DEL construction was then validated in two aspects. First, we tested its DEL-encoding compatibility by performing enzymatic ligation assays. Since the DNA sequence is the sole barcode to record structural information of linked small molecules, it is vital to maintain DNA integrity after each chemical transformation. As shown by the polyacrylamide gel electrophoresis (PAGE) analysis, no signs of DNA damage were observed (Fig. S12 in Supporting information), indicating DNA integrity. Second, we evaluated the DEL-synthesis compatibility of the reaction. Parallel synthesis on a 96-well plate was performed by using 12 small-molecule aldehydes and 8 DNA-conjugated anthranilamides (Fig. S7 in Supporting information). Following the standard protocol, we obtained the satisfying result that 86 of the total 96 wells furnished over 70% conversions. Notably, alkyl and alicyclic aldehydes, together with other functional group decorated aldehydes, were well-tolerated. Aldehydes, which showed inferior conversions under the reaction condition (Table S1), gave better chemical transformations under the antimony trichloride-promoted DHQ formation reaction. This agreed with previous findings in that a general reaction condition should be evenly feasible for a broader substrate scope. Encouraged by the delightful result from 96-well plate data, we further conducted subsequent diversification based on DHQ products bearing an additional aldehyde handle, delivering double-heterocycle products with excellent conversions (Figs. S2 and S3 in Supporting information) [41]. Moreover, a preliminary scalable reaction at 5 nmol underwent smoothly with high conversion maintained (Figs. S10 and S11 in Supporting information). Collectively, antimony trichloride-promoted DHQ formation reaction has been proven to be amenable for DEL synthesis.
Macrocyclic compounds, bridging the gap between small molecules and biomacromolecules, represent a class of promising ligands that are advantageous in interacting with protein targets [49,50]. Among the strategies that have been developed to discover new macrocycles, DEL technology is featured the rapid synthesis of macrocycle library by means of combinatorial chemistry. The crucial step in the construction of a macrocycle library is macrocyclization [51]. Currently, prevalent methods in DNA-compatible macrocyclization are amide bond formation [52], Cu-catalyzed azide/alkyne cycloaddition [53-56], Wittig olefination [57], ring-closing metathesis [58,59], and others [60-64]. Although these approaches demonstrate robustness and efficacy, seeking more chemical diversity during macrocyclization remains unexplored. We envisioned that DHQ formation has several merits. First, it would accomplish a privileged scaffold into the rigid macrocycle to consequently yield druglike properties, advantageous over most used amide bond formation. Second, an increase in molecular weight after the reaction eases analytical monitoring, in contrast to the CuAAC method. As shown in Scheme 4, we implemented our method onto DNA-conjugated linear peptides carrying a free amine and a distant aldehyde, by using isatoic anhydride via a "one-pot" synthesis. To our delight, macrocycles (m1-m5) with different ring sizes and chemical compositions were achieved with moderate-to-good conversions (40%−80%). DNA-tethered oligopeptidic S1-S5, regardless of amino acid replacement influence including alpha-amino acid (m3), beta-turn unit like proline (m4), and adjacent hindrance (m5), all gave desired macrocycles. The product structures, unlike ordinary macrocycles generated by methods discussed above, are featured a medium macrocycle fused with a small DHQ cycle which could serve as a pharmacophore. Moreover, we would gain more structural diversity after the cyclization reaction if more decorations are installed on the ring of isatoic anhydride. Since reductive amination reaction is also feasible in cyclization when there is a free amine group and a free aldehyde group in the substrate structure [65], we conducted macrocyclization of m1 precursor via reductive amination to make a comparison between these two methods. The desired macrocycle was obtained with an inferior 40% conversion, and by deconvoluted mass analysis, the main by-product was probably the product of aldehyde reduction (Fig. S6 in Supporting information) [66]. This implies that to some extent our antimony trichloride-promoted DHQ formation reaction improved macrocyclization efficiency. Overall, antimony trichloride-promoted DHQ formation reaction can be readily employed in DNA-compatible macrocyclization, with merits of high efficiency and simultaneous chemical diversity introduction.

In summary, the present work describes a mild, rapid synthetic protocol to form on-DNA DHQ heterocycle featured with antimony salt-promoted cyclization. The antimony trichloride has proved to shorten the reaction time and improve conversions. The broad substrate scope of small-molecule aldehydes including both commercially available (hetero)aryl and alkyl derivatives was well-explored, allowing the introduction of structural diversity. Moreover, DHQ-containing spirocycles were obtained by employing isatins as the replacement for aldehydes. With the reversely-prepared DNA-conjugated aldehydes, a broad range of anthranilamides were tested to be well-tolerated with moderate-to-excellent conversions, affirming the robustness and generality of the optimal condition in parallel synthesis. Furthermore, quinazolinone moiety was achieved by mild oxidation of DHQ with sodium periodate. Remarkably, we applied the reaction into DNA-compatible macrocyclization, where the incorporation of druglike scaffold DHQ into the cyclic architecture would leverage the macrocycle products in the DEL selection. We expect our developed method can readily expand the coverage of unprecedented chemical structures to fulfill the potential of DEL technology in drug discovery.
The authors declare that this technology is covered by patent applications (Nos. 202211099109.6 and 202211117979.1).
This work was supported by grants from the National Natural Science Foundation of China (Nos. 22222702, 22107016, 22107017 and 21907011); the Fundamental Research Funds for the Central Universities (No. 2022CDJQY-001); Beijing National Laboratory for Molecular Sciences (No. BNLMS202104); the Natural Science Foundation of Chongqing (Nos. cstc2020jcyj-jqX0009, cstc2021jcyj-msxmX0016 and cstc2021jcyj-cxttX0002); High-end Foreign Expert Introduction Program (No. G2022165020L); Shenzhen Innovation Center for Small Molecule Drug Discovery Co. (No. H20220687).
Supplementary material associated with this article can be found, in the online version, at doi:
N. Kerru, L. Gummidi, S. Maddila, K.K. Gangu, S.B. Jonnalagadda, Molecules 25 (2020) 1909. doi: 10.3390/molecules25081909
M. Badolato, F. Aiello, N. Neamati, RSC Adv. 8 (2018) 20894–20921. doi: 10.1039/c8ra02827c
Y. Nkizinkiko, B.V.S. Suneel Kumar, V.U. Jeankumar, et al., Bioorg. Med. Chem. 23 (2015) 4139–4149. doi: 10.1016/j.bmc.2015.06.063
J. Zhang, J.W. Zhao, L.P. Wang, et al., Tetrahedron 72 (2016) 936–943. doi: 10.1016/j.tet.2015.12.055
Z. Xie, J. Lan, H. Zhu, et al., Chin. Chem. Lett. 32 (2021) 1427–1431. doi: 10.1016/j.cclet.2020.09.059
Y. Huang, L. Meng, Q. Nie, et al., Nat. Chem. 13 (2021) 77–88. doi: 10.1038/s41557-020-00605-x
M. Song, G.T. Hwang, J. Med. Chem. 63 (2020) 6578–6599. doi: 10.1021/acs.jmedchem.9b01782
K.D. Hook, J.T. Chambers, R. Hili, Chem. Sci. 8 (2017) 7072–7076. doi: 10.1039/C7SC02779F
S. Brenner, R.A. Lerner, Proc. Natl. Acad. Sci. USA 89 (1992) 5381–5383. doi: 10.1073/pnas.89.12.5381
Y.K. Sunkari, V.K. Siripuram, T.L. Nguyen, M. Flajolet, Trends Pharmacol. Sci. 43 (2022) 4–15. doi: 10.1016/j.tips.2021.10.008
G. Zhao, S. Zhong, G. Zhang, Y. Li, Y. Li, Angew. Chem. Int. Ed. 61 (2022) e202115157. doi: 10.1002/anie.202115157
G. Zhao, Y. Huang, Y. Zhou, Y. Li, X. Li, Expert Opin. Drug Discovery 14 (2019) 735–753. doi: 10.1080/17460441.2019.1614559
B. Cai, D. Kim, S. Akhand, et al., J. Am. Chem. Soc. 141 (2019) 17057–17061. doi: 10.1021/jacs.9b08085
G. Zimmermann, Y. Li, U. Rieder, et al., ChemBioChem 18 (2017) 853–857. doi: 10.1002/cbic.201600637
C. Zambaldo, J.P. Daguer, J. Saarbach, S. Barluenga, N. Winssinger, Med. Chem. Commun. 7 (2016) 1340–1351. doi: 10.1039/C6MD00242K
F.V. Reddavide, W. Lin, S. Lehnert, Y. Zhang, Angew. Chem. Int. Ed. 54 (2015) 7924–7928. doi: 10.1002/anie.201501775
L.M. McGregor, T. Jain, D.R. Liu, J. Am. Chem. Soc. 136 (2014) 3264–3270. doi: 10.1021/ja412934t
P.A. Harris, S.B. Berger, J.U. Jeong, et al., J. Med. Chem. 60 (2017) 1247–1261. doi: 10.1021/acs.jmedchem.6b01751
H.A. Stanway-Gordon, J.S. Graham, M.J. Waring, Angew. Chem. Int. Ed. 61 (2022) e202111927. doi: 10.1002/anie.202111927
M.K. Skopic, F. Losch, A.E. McMillan, et al., Org. Lett. 24 (2022) 1383–1387. doi: 10.1021/acs.orglett.2c00228
F. Ma, J. Li, S.N. Zhang, et al., ACS Catal. 12 (2022) 1639–1649. doi: 10.1021/acscatal.1c05338
X. Fu, J. Tang, R. Hua, et al., Org. Lett. 24 (2022) 2208–2213. doi: 10.1021/acs.orglett.2c00516
B. Lin, W. Lu, Z.Y. Chen, et al., Org. Lett. 23 (2021) 7381–7385. doi: 10.1021/acs.orglett.1c02562
Z. Fan, S. Zhao, T. Liu, et al., Chem. Sci. 11 (2020) 12282–12288. doi: 10.1039/d0sc03935g
S.O. Badir, J. Sim, K. Billings, et al., Org. Lett. 22 (2020) 1046–1051. doi: 10.1021/acs.orglett.9b04568
J. Sun, Q. Nie, X. Fang, et al., Org. Biomol. Chem. 20 (2022) 5045–5049. doi: 10.1039/d2ob00862a
E. Lenci, L. Baldini, A. Trabocchi, Bioorg. Med. Chem. 41 (2021) 116218. doi: 10.1016/j.bmc.2021.116218
R. Wu, S. Gao, T. Du, et al., Chem. Asian J. 15 (2020) 4033–4037. doi: 10.1002/asia.202001105
C.J. Gerry, S.L. Schreiber, Curr. Opin. Chem. Biol. 56 (2020) 1–9.
R.M. Franzini, C. Randolph, J. Med. Chem. 59 (2016) 6629–6644. doi: 10.1021/acs.jmedchem.5b01874
Y. Gao, G. Zhao, P. He, et al., Bioconjugate Chem. 33 (2022) 105–110. doi: 10.1021/acs.bioconjchem.1c00567
P.R. Fitzgerald, B.M. Paegel, Chem. Rev. 121 (2021) 7155–7177. doi: 10.1021/acs.chemrev.0c00789
D.K. Kolmel, A.S. Ratnayake, M.E. Flanagan, et al., Org. Lett. 22 (2020) 2908–2913. doi: 10.1021/acs.orglett.0c00574
Y. Li, R. De Luca, S. Cazzamalli, et al., Nat. Chem., 10 (2018) 441–448. doi: 10.1038/s41557-018-0017-8
A.W. Dombrowski, A.L. Aguirre, A. Shrestha, K.A. Sarris, Y. Wang, J. Org. Chem. 87 (2022) 1880–1897. doi: 10.1021/acs.joc.1c01427
L. Fan, C.P. Davie, ChemBioChem 18 (2017) 843–847. doi: 10.1002/cbic.201600563
S.A. Pourmousavi, A. Kanaani, H.R. Fatahi, F. Ghorbani, D. Ajloo, J. Phys. Chem. Solids 106 (2017) 82–93. doi: 10.1016/j.jpcs.2017.03.008
J. Chai, X. Lu, C.C. Arico-Muendel, Y. Ding, M.P. Pollastri, Bioconjugate Chem. 32 (2021) 1973–1978. doi: 10.1021/acs.bioconjchem.1c00363
S. Yang, G. Zhao, Y. Gao, et al., Chem. Sci. 13 (2022) 2604–2613. doi: 10.1039/d1sc06268a
X. Li, Z.J. Gartner, B.N. Tse, D.R. Liu, J. Am. Chem. Soc. 126 (2004) 5090–5092. doi: 10.1021/ja049666+
X. Fang, Y. Wang, P. He, et al., Org. Lett. 24 (2022) 3291–3296. doi: 10.1021/acs.orglett.2c01187
A.L. Satz, J. Cai, Y. Chen, et al., Bioconjugate Chem. 26 (2015) 1623–1632. doi: 10.1021/acs.bioconjchem.5b00239
Y. Gao, Y. Sun, G. Zhao, et al., Org. Lett. (2022) 6664–6669. doi: 10.1021/acs.orglett.2c02714
B.D. Rupnar, T.R. Kachave, P.D. Jawale, S.U. Shisodia, R.P. Pawar, J. Iran. Chem. Soc. 14 (2017) 1853–1858. doi: 10.1007/s13738-017-1124-y
C.J. Gerry, M.J. Wawer, P.A. Clemons, S.L. Schreiber, J. Am. Chem. Soc. 141 (2019) 10225–10235. doi: 10.1021/jacs.9b01203
S. Zhong, X. Fang, Y. Wang, et al., Org. Lett. 24 (2022) 1022–1026. doi: 10.1021/acs.orglett.1c04169
Q. Liang, J.Y. He, X. Zhao, et al., J. Med. Chem. 64 (2021) 4196–4205. doi: 10.1021/acs.jmedchem.1c00123
Y. Bao, Y. Yan, K. Xu, et al., J. Org. Chem. 80 (2015) 4736–4742. doi: 10.1021/acs.joc.5b00191
A.A. Peterson, A.M. Rangwala, M.K. Thakur, et al., Nat. Chem. Biol. 18 (2022) 1184–1195. doi: 10.1038/s41589-022-01116-1
J.P. Maianti, A. McFedries, Z.H. Foda, et al., Nature 511 (2014) 94–98. doi: 10.1038/nature13297
L. Plais, J. Scheuermann, RSC Chem. Biol. 3 (2022) 7–17. doi: 10.1039/d1cb00161b
C.J. Stress, B. Sauter, L.A. Schneider, T. Sharpe, D. Gillingham, Angew. Chem. Int. Ed. 58 (2019) 9570–9574. doi: 10.1002/anie.201902513
Y. Onda, G. Bassi, A. Elsayed, et al., Chem. Eur. J. 27 (2021) 7160–7167. doi: 10.1002/chem.202005423
M.H. Shin, K.J. Lee, H.S. Lim, Bioconjugate Chem. 30 (2019) 2931–2938. doi: 10.1021/acs.bioconjchem.9b00628
Z. Zhu, A. Shaginian, L.C. Grady, et al., ACS Chem. Biol. 13 (2018) 53–59. doi: 10.1021/acschembio.7b00852
W.H. Connors, S.P. Hale, N.K. Terrett, Curr. Opin. Chem. Biol. 26 (2015) 42–47. doi: 10.1016/j.cbpa.2015.02.004
Z.J. Gartner, B.N. Tse, R. Grubina, et al., Science 305 (2004) 1601–1605. doi: 10.1126/science.1102629
O.B.C. Monty, P. Nyshadham, K.M. Bohren, et al., ACS Comb. Sci. 22 (2020) 80–88. doi: 10.1021/acscombsci.9b00199
X. Lu, L. Fan, C.B. Phelps, C.P. Davie, C.P. Donahue, Bioconjugate Chem. 28 (2017) 1625–1629. doi: 10.1021/acs.bioconjchem.7b00292
Q.G. Nie, S. Zhong, Y. Li, G. Zhang, Y. Li, Chin. Chem. Lett. 33 (2022) 2559–2563. doi: 10.1016/j.cclet.2021.09.041
Q. Nie, X. Fang, C. Liu, et al., J. Org. Chem., 87 (2022) 2551–2558. doi: 10.1021/acs.joc.1c02496
E. Koesema, A. Roy, N.G. Paciaroni, et al., Angew. Chem. Int. Ed., 61 (2022) e202116999. doi: 10.1002/anie.202116999
P. Yang, X. Wang, B. Li, et al., Chem. Sci. 12 (2021) 5804–5810. doi: 10.1039/d1sc00789k
M.V. Pham, M. Bergeron-Brlek, C. Heinis, ChemBioChem 21 (2020) 543–549. doi: 10.1002/cbic.201900390
L.R. Malins, J.N. deGruyter, K.J. Robbins, et al., J. Am. Chem. Soc., 139 (2017) 5233–5241. doi: 10.1021/jacs.7b01624
J. Shan, X. Ling, J. Liu, X. Wang, X. Lu, Bioorg. Med. Chem. 42 (2021) 116234. doi: 10.1016/j.bmc.2021.116234

Figure 1 (a) Bioactive compounds comprising DHQ core. (b) Workflow of DEL technology. (c) Antimony trichloride-promoted synthesis of on-DNA DHQ scaffold.

Scheme 3 (a) Synthesis of DHQ scaffolds using diversified DNA-conjugated aldehydes. (b) Synthesis of DHQ scaffolds using diversified anthranilamides.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



 下载:
下载: