
Scheme 1.
The molecular structures of caffeine and theophylline (THP).
The molecular mechanisms of plasticity in crystal forms of theophylline
Hongji Liu , H.C. Stephen Chan , Li Zhang , Yang Lu , Jiaquan Li , Jianting Li , Liang Li , Zhengzheng Zhou

In recent years, the elastic and plastic deformations of organic crystals are brought into the spotlight of both material science and crystal engineering communities [1-5]. Elastic crystal is thought to require weak isotropic interactions, such as π–π interaction and many others [3,6,7], but more importantly, an interlocking packing architecture that hinders the slippage of molecular layers as lacked in plastic crystals [3,7-9]. Hirshfeld surface calculation is often applied to locate important molecular contacts for maintaining the packing architecture [10,11]. In pharmaceutical industry, plastic crystals with lower stiffness may be pursued for better powder compaction and tabletability [12-14]. Crystal slip plane is a characteristic feature observed in plastic crystal [7,15,16]. Energy framework, parameterized from quantum mechanical calculations, is capable of identifying potential slip planes within a crystal, when the interaction within a certain molecular layer significantly outweighs that between the layers [17-20]. However, the underlying mechanism that restricts these layers from restoring their original positions has not been fully addressed. In this work, we focus on a naturally occurring caffeine derivative, theophylline (THP, Scheme 1), and discovered the plasticity behavior in its anhydrous crystal form Ⅱ. The molecular layers formed via π-π stacking become excellent targets for theoretical investigations. During our characterization experiments, the monohydrate of THP presents excellent elastic property, where π-π stacking also exists. The role of water in tuning the mechanical property therefore becomes another intriguing subject for investigation. DFT level calculations were performed to predict the molecular events in these two crystal forms, in response to various anisotropic deformations [21-24]. The resulting energy landscapes may shed light on their disparate mechanical behaviors.
Our experimental data demonstrate the distinct plastic and elastic behaviors in anhydrous theophylline (THPa) and the monohydrate (THPm) crystals respectively. THPa crystal exhibits super plasticity and undergoes irreversible bending on (200) face (Fig. 1a). By applying the load with a metallic needle on this face, three independent THPa crystals would be molded in the letters “SMU” respectively (Fig. 1b). Meanwhile, THPm crystal undergoes elastic deformation and shows excellent bending resilience when the load is repeatedly applied on its (002) face (Fig. 1c). The maximum tolerable elastic strain was measured to be approximately 1.22% for a 20 µm thick THPm crystal (Fig. S4 in Supporting information), lower than the exceptionally elastic celecoxib crystal (3.56%) [25] but 2–3 times higher than most crystalline alloys (0.50%) [26,27]. Moreover, the fractured THPm crystallites formed beyond its elastic limit do not remain in an irreversible plastic deformation, but restore their linear shapes (Fig. S3 in Supporting information).

Nanoindentation [17,28] coupled with atomic force microscopy (AFM) was used to investigate further the differences in mechanical behaviors between THPa and THPm (Fig. 2a). The load-displacement (P-h) curve of THPa shows a stronger linear relationship and a consistently higher resistance to deformation. In contrast, the P-h curve of THPm significantly deviates from linearity during loading and unloading. THPm is highly compliant to deformation at a small load and develops resistance gradually with the increasing penetration depth. Another key difference is that discrete “pop-in” events are observed in the P-h curve of THPa, but not of THPm. The differences in penetration depth between any two subsequent events are similar, suggesting that the plastic transformation in THPa could be related to certain periodically stacked layers. The images from the scanning electron microscope (SEM) [1,29,30] confirm that the THPa crystals indeed consist of a multilayer topology (Fig. S5a in Supporting information). Meanwhile, surface pile-up phenomenon is observed from the AFM results of THPa, indicating that the molecular slippage direction would be perpendicular to the applied stress [17,28,31]. More importantly, the pile-up occurs predominantly on one particular edge of the indenter, suggesting that the plastic transformation of THPa may also be highly directional on the surface (Fig. 2b). For THPm, the absence of “pop-in” event on P-h curve implies an elastic deformation throughout the whole course of the more profound indentation. At a peak load of 1000 µN, the maximum penetration of the probe in THPm (1318.86 nm) is 4.22 times deeper than that in THPa (312.36 nm). The AFM results of THPm show no pile-up of material, indicating that displacement of molecules proceeds along the same direction of the applied stress [28]. More interestingly, the surface of THPm remains smooth with no sign of previous indentation, after the stress has been released (Fig. 2b). Therefore, THPm demonstrates a more superior ability in recovering its crystal shape.

X-ray crystallography reveals that the inclusion of water molecule significantly alters the packing architecture (Table 1 and Fig. S6 in Supporting information). THPa crystallizes in an orthorhombic space group Pna21 with only one THP molecule in the asymmetric unit. The THP molecules form a 1-D hydrogen bond chain along the [011] direction. Via the screw symmetry, these THP molecules result in wavy molecular sheets that stack on one another by π-π interaction along the b-axis. (Fig. 3a). Meanwhile, THPm crystallizes in a monoclinic space group P1121/b instead, with one THP and one water molecule in the asymmetric unit. Thermal analysis also supports equimolar of water and THP in the lattice (Fig. S7 in Supporting information) [32]. Water molecules presumably form a 1-D hydrogen bond chain along the a-axis. THP molecules no longer adopt a 1-D hydrogen bond chain but 0-D dimers, which alternatingly interact with water along the c-axis and result in a 3-D hydrogen bond network. Noticeably, these THP dimers also stack on one another by π–π interaction, and the putative slip plane appears along [120] direction (Fig. 3b). More plausible slip planes can be identified by further visual inspection, including the (200) plane in THPa at the interface of two 1-D hydrogen bond chains of THP and the (002) plane in THPm along the water channel (Figs. 3c and d). One may conceptualize plastic deformation as a consequence of applied strain along a slip plane, and elastic deformation as a process during which a material undergoes simultaneously compression and dilation along the direction of applied strain (Figs. 3e and f) [2]. However, how exactly water harnesses elasticity in THPm and the determining factor for plasticity of THPa remain unclear. Atomistic modeling was therefore performed to elucidate the underlying reasons.

When subject to an anisotropic strain, atoms inside the deformed lattice are displaced and attain a new configuration that corresponds to a local energy minimum. By probing the structural changes and the relating potential energy surface, the comprehensive interplay between macroscopic mechanical properties and the corresponding atomistic events could be better understood. For a given crystal lattice R matrix, its deformed lattice R' can be expressed as a transformation via a distortion matrix D [33-35]:
|
|
(1) |
where δi corresponds to an anisotropic strain. The internal energy of a crystal E under a set of general strain δi can be expanded in form of a Taylor's series [36]:
|
|
(2) |
where E0 and V are the ground state energy and the volume of the unstrained crystal respectively, σi is the element of stress tensor with respect to δi, and cij is the anisotropic elastic modulus. c11, c22 and c33 account for the axial compression (or dilation) along the a-, b- and c-axis respectively, whereas c44, c55 and c66 account for the shear across the bc-, ca-, and ab-plane respectively [33-35,37]. Note that some shear directions may break the symmetry of the original space group [34]. Other remaining elastic moduli are symmetrical, i.e., cij = cji, and correspond to a combination of any two of the abovementioned deformations [37]. At small strains, the internal energy varies almost like a harmonic function, and each cij can be obtained from the second-order coefficient of the Taylor's series with respect to the deforming strain component (Table 1). Regarding the ground state energy of THPm, the water protons are under the inversion symmetry constraint of the P1121/b space group and result in a suboptimal “head-to-head” contact with one another after DFT-XDM optimization (Fig. 4a) [38-42]. The removal of inversion constraint effectively lowers the space group to P11b and the water protons instead form a “zig-zag” hydrogen bond chain with neighboring water molecules (Fig. 4b). This alternative proton configuration yields an enthalpy excess of nearly 2.4 kcal/mol and inflicts a minimal structural change with a RMSD30 of 0.176 Å only (Table 1). Therefore, the P11b structure was taken as the unstrained state of THPm for calculating the anisotropic elastic moduli.

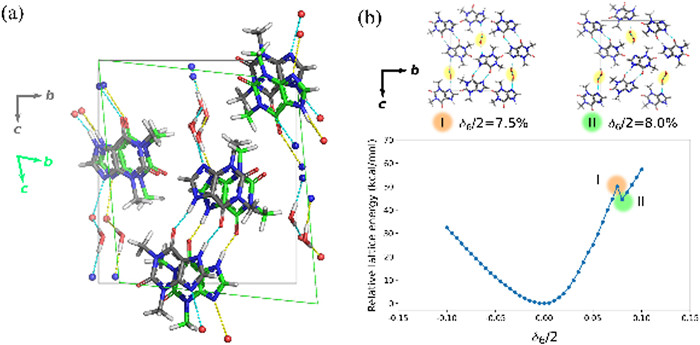
The calculated anisotropic stiffness predominantly depends on the impact of deformation direction on hydrogen bonding. The high axial stiffness in THPm, for example, is attributed to the distortion of the water hydrogen bond chain (for c11), the THP dimer (for c22) or both (for c33) caused by each axial strain (Table 1). Similarly, in THPa, the strains along the a- or c-axis, particularly the former, directly disrupt the hydrogen bond distance and angle, whereas the strain along the b-axis does not significantly alter the relative position of THPs involved in the hydrogen bond. Thus, c11 and c33 are nearly 2 times larger than c22 (Table 1). Nevertheless, THPm in general has larger axial moduli than THPa does. Specifically, c22 and c33 of THPm are around 1.5–2.0 times greater than the largest axial modulus c11 of THPa. The 3-D hydrogen bond network in THPm offers a more robust architecture against axial strains. This prediction result appears paradoxical to the nanoindentation experiment, where THPm shows good compliance to an axial stress. However, its shear moduli are noticeably smaller than those of THPa (Table 1). Its two smallest moduli, c44 and c55, are even less than 50% of the second largest shear modulus c55 of THPa, because these shear directions preserve the quasi-inversion relationship between the THP dimer molecules and inflict minimal effect to the hydrogen bonding (Fig. 5a). Consequently, the generally low shear stiffness of THPm may be the major reason for its high compliance in the nanoindentation experiment, provided that the shear direction would be parallel to the applied stress. It is interesting to note that, when modeling the stiffest shear modulus c66 of THPm, the increasing strain changes the tilting angle of the THP stack, which then squashes against the neighboring water chain and eventually dislodges it slightly along the c-axis. This structural event is reflected from the discontinuity of the energy-strain curve (Fig. 5b). The results from our Hirshfeld surface and energy framework calculations indicate a potential slip plane on the (002) surface here (Figs. S14 and S15b in Supporting information). In the highly strained lattices, the displacement of the water chain occurs exactly on this predicted slip plane, even though no slipping of the THP layer is observed.
Abrupt structural change induced by an applied strain was also predicted in THPa. At a moderate axial strain on a-axis, each strained 1-D hydrogen bond chain is restored to normal at the expense of the weaker dispersion between the chains. This molecular event is also reflected from the discontinuity on the energy-strain curve. Eventually isolated THP chains are observed in the highly strained structure (Fig. 6a). Our Hirshfeld surface and energy framework calculations also indicate this (200) surface as a potential slip plane (Figs. S13 and S15a in Supporting information). Likewise, the simulated shear across the ab-plane unexpectedly results in the largest modulus in THPa, despite its presumably limited impact on hydrogen bonding. One explanation would be the wavy layout of molecular sheet largely hinders its translocation in this particular direction. More strikingly, our simulation shows that, with an increasing strain (δ6/2), the original wavy architecture (up-down-down-up) undergoes rearrangements twice: (1) Beyond 2.5% strain, the architecture gradually turns into an up-up-up-up layout; (2) At 7.0% strain, there is a slight slippage on the (020) plane and the THP in the same hydrogen bond chain becomes almost coplanar (Fig. 6b). Each rearrangement is associated with a huge energy barrier, followed by a gradual convergence to a local energy minimum. This energy landscape may well explain the plastic behavior of THPa: Firstly, each metastable strained state is now restricted from returning to its preceding state, and eventually to the unstrained equilibrium state. Secondly, the multiple “pop-in” events during nanoindentation could be related to the stepwise plastic transformations induced by this shear strain. Thirdly, the highly directional pile-up in AFM may be caused by this directional plastic transformation. Finally, the average intermolecular HO distance between the C—H on the heterocylic ring and the O═C group decreases with an increasing strain (δ6/2), i.e.2.21 Å at 0.0%, 2.18 Å at 6.5% and 2.16 Å at 10.0%. This could have weakened the interactions between these two functional groups. Micro-Raman spectroscopy (Fig. S8 in Supporting information, top) indeed shows a stronger blue shift of the C—H stretch in the inner arc of the bent THPa crystal (2966.19 cm–1), when compared to the outer arc (2964.53 cm–1). This experimental finding may further support this shear direction would be highly related to the plasticity of THPa.

To investigate whether the irreversible deformation in plastic crystal is linked with a significant energy barrier during structural rearrangement, similar calculations were extended to two more well-known examples of plastic crystals. Caffeine monohydrate is noteworthy for its plastic behavior [12], but shares similar packing features with theophylline monohydrate, including space group, 1-D water chain and the xanthine dimer. In our extended calculations, shearing across each of the three faces in caffeine monohydrate crystal does not result in a significant discontinuity on the energy-strain curve as well as rearrangements of the caffeine stacking. However, a compression on the b-axis alongside a dilation on the a-axis does (See supporting information). The weak hydrogen bond between caffeine dimer, instead of the strong hydrogen bond between theophylline dimer, offers more flexibility for rearranging the caffeine stacks. Similarly, in the P21/c setting of hexachlorobenzene [45], another notable example of plastic crystal, our extended calculations also show that molecules overcome an energy barrier and adopt a different tilting angle in response to a simultaneous dilation on the c-axis and an increasing β angle (see Supporting information). Our results possibly suggest the energy barrier hampering the strain-induced π-stacking rearrangement could be a general mechanism for plasticity in crystals of small aromatic molecules.
In this study, water molecule abolishes completely the plastic property of anhydrous theophylline and converts it into a flexible material with excellent resilience against deformation. Under a small applied strain, our predicted anisotropic stiffness of the materials shows strong connections to the directional hydrogen bond network inside the crystal lattice. Axial strain along a 1-D hydrogen bond chain or a 0-D dimer faces huge resistance, whereas shearing about the latter tends to experience less resistance. When a higher strain is further applied, molecular slip planes or related structural events predicted from Hirshfeld surface analysis and energy framework calculations are also captured in our anisotropic stiffness predictions, reflected by a discontinuity on the energy-strain curve. This outcome indicates that all these methods can be complimentary to one another. Both our experimental and predicted results suggest aromatic stacking and molecular slip plane are prerequisites but may not be the only factors for the plasticity of an organic crystal. Additional mechanisms may be required for hindering the recovery of the material back to its unstrained state – here the energy penalty for rearranging the wavy molecular sheets in anhydrous theophylline is a good example. Meanwhile, in the monohydrate form, water molecules form a 1-D chain, break down the wavy sheets of theophylline into isolated columns of theophylline dimers, and offers a robust 3-D hydrogen bonded architecture to undermine the molecular slippage for plasticity. Moreover, the internal energy surface remains as a continuous concave function for a wide range of strain, indicating that a deformed THPm would be capable of returning to its unstrained equilibrium state. Since these calculations investigate the impact of more extreme strains on the molecular packing, there may be a potential application for tracking the pathway of mechanically induced phase transformation [46]. To conclude, searching for a stable crystal form with optimal pharmaceutical properties is the ultimate objective in experiment. The ability to predict further the material properties would definitely add values to crystal structure prediction [47-49].
The authors declare no competing financial interest.
This study was supported by the Young Scientists Promotion Fund of Natural Science Foundation of Guangdong Province, National Natural Science Foundation of China (No. 81703438), the Key R&D Program of Shandong Province (No. 2021ZDSYS26) National Science and Technology Major Project of China (No. 2018ZX09711001-001-013), CAMS Innovation Fund for Medical Sciences (No. 2017-I2M-3-010) and Student Innovation Training Program (No. 201812121093). Authors would like to thank Dr Grahame R. Woollam (Novartis Pharma AG, Switzerland), Dr Hafsa S. Javed (University of Malta) and Dr Sten Nilsson (AstraZeneca, Sweden) for their valuable discussions.
Supplementary material associated with this article can be found, in the online version, at doi:
E. Ahmed, D.P. Karothu, M. Warren, P. Naumov, Nat. Commun. 10 (2019) 3723. doi: 10.1038/s41467-019-11612-z
S. Ghosh, M.K. Mishra, S.B. Kadambi, et al., Angew. Chem. Int. Ed. 54 (2015) 2674–2678. doi: 10.1002/anie.201410730
S. Saha, M.K. Mishra, C.M. Reddy, G.R. Desiraju, Acc. Chem. Res. 51 (2018) 2957–2967. doi: 10.1021/acs.accounts.8b00425
B. Tang, B. Liu, H. Liu, H. Zhang, Adv. Funct. Mater. 30 (2020) 2004116. doi: 10.1002/adfm.202004116
J.R. Wang, M. Li, Q. Yu, et al., Chem. Commun. 55 (2019) 8532–8535. doi: 10.1039/c9cc03542g
H. Liu, Z. Lu, B. Tang, et al., Angew. Chem. Int. Ed. 59 (2020) 12944–12950. doi: 10.1002/anie.202002492
J. Li, J. Li, H. Liu, et al., Chin. Chem. Lett. 33 (2022) 4069–4073. doi: 10.1016/j.cclet.2022.03.090
E. Ahmed, D.P. Karothu, P. Naumov, Angew. Chem. Int. Ed. 57 (2018) 8837–8846. doi: 10.1002/anie.201800137
S. Saha, G.R. Desiraju, J. Am. Chem. Soc. 139 (2017) 1975–1983. doi: 10.1021/jacs.6b11835
S.P. Thomas, M.W. Shi, G.A. Koutsantonis, et al., Angew. Chem. Int. Ed. 56 (2017) 8468–8472. doi: 10.1002/anie.201701972
P.R. Spackman, M.J. Turner, J.J. McKinnon, et al., J. Appl. Crystallogr. 54 (2021) 1006–1011. doi: 10.1107/s1600576721002910
S. Hu, M.K. Mishra, C.C. Sun, Chem. Mater. 31 (2019) 3818–3822. doi: 10.1021/acs.chemmater.9b00441
Z. Zhou, W. Li, W.J. Sun, et al., Int. J. Pharm. 509 (2016) 391–399. doi: 10.1016/j.ijpharm.2016.06.006
D. Guan, B. Xuan, C. Wang, et al., Pharmaceutics, 13 (2021) 2160. doi: 10.3390/pharmaceutics13122160
L. Dezerald, D. Rodney, E. Clouet, et al., Nat. Commun. 7 (2016) 11695. doi: 10.1038/ncomms11695
C. Wang, C.C. Sun. Mol. Pharm. 16 (2019) 1732–1741. doi: 10.1021/acs.molpharmaceut.9b00082
K. Zhang, C.C. Sun, Y. Liu, et al., Chem. Mater. 33 (2021) 1053–1060. doi: 10.1021/acs.chemmater.0c04560
C.F. Mackenzie, P.R. Spackman, D. Jayatilaka, M.A. Spackman, IUCrJ 4 (2017) 575–587. doi: 10.1107/S205225251700848X
M.J. Turner, S.P. Thomas, M.W. Shi, et al., Chem. Commun. 51 (2015) 3735–3738. doi: 10.1039/C4CC09074H
C. Wei, J. Wang, Y. Zhang, et al., Chin. Chem. Lett. (2022) 107896. doi: 10.1016/j.cclet.2022.107896
H. Liu, J. Nie, H.C. Stephen Chan, et al., Int. J. Pharm. 601 (2021) 120537. doi: 10.1016/j.ijpharm.2021.120537
D.D. Huang, H.C.S. Chan, Y.S. Wu, et al., J. Mol. Liq. 329 (2021) 115604. doi: 10.1016/j.molliq.2021.115604
H.M. Yu, B.X. Zhang, W.H. Xing, et al., Chin. Chem. Lett. 34 (2023) 107668. doi: 10.1016/j.cclet.2022.07.011
H.C. Stephen Chan, J. Kendrick, M.A. Neumann, F.J.J. Leusen, CrystEngComm 15 (2013) 3799–3807. doi: 10.1039/c3ce40107c
K. Wang, M.K. Mishra, C.C. Sun, Chem. Mater. 31 (2019) 1794–1799. doi: 10.1021/acs.chemmater.9b00040
M. Telford, Mater. Today 7 (2004) 36–43.
S. Ghosh, M.K. Mishra, S.B. Kadambi, et al., Angew. Chem. Int. Ed. 54 (2015) 2674–2678. doi: 10.1002/anie.201410730
S. Varughese, M.S. Kiran, U. Ramamurty, G.R. Desiraju, Angew. Chem. Int. Ed. 52 (2013) 2701–2712. doi: 10.1002/anie.201205002
Y. Chen, Z. Chang, J. Zhang, J. Gong, Angew. Chem. Int. Ed. 60 (2021) 22424–22431. doi: 10.1002/anie.202108441
P. Commins, A.B. Dippenaar, L. Li, et al., Chem. Sci. 12 (2021) 6188–6193. doi: 10.1039/d1sc01246k
P. Sanphui, M.K. Mishra, U. Ramamurty, G.R. Desiraju, Mol. Pharm. 12 (2015) 889–897. doi: 10.1021/mp500719t
H. Liu, H.H.Y. Tong, Z. Zhou, J. Therm. Anal. Calorim. 147 (2022) 12947–12963. doi: 10.1007/s10973-022-11762-1
J.H. Jang, S.J. Park, T.H. Lee, H.K.D.H. Bhadeshia, Mater. Sci. Tech. 36 (2020) 615–622. doi: 10.1080/02670836.2020.1724402
K.B. Panda, K.S.R. Chandran, Acta. Mater. 54 (2006) 1641–1657. doi: 10.1016/j.actamat.2005.12.003
G. Ghosh, Aip. Adv. 5 (2015), 087102. doi: 10.1063/1.4928208
D.C. Wallace, Am. J. Phys. 40 (1972) 1718–1719. doi: 10.1119/1.1987046
A.U. Ortiz, A. Boutin, A.H. Fuchs, F.X. Coudert, Phys. Rev. Lett. 109 (2012) 195502. doi: 10.1103/PhysRevLett.109.195502
P. Giannozzi, S. Baroni, N. Bonini, et al., J. Phys. Condens. Matter 21 (2009) 395502. doi: 10.1088/0953-8984/21/39/395502
P. Giannozzi, O. Andreussi, T. Brumme, et al., J. Phys. Condens. Matter 29 (2017) 465901. doi: 10.1088/1361-648X/aa8f79
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865–3868. doi: 10.1103/PhysRevLett.77.3865
A. Dal Corso, Comp. Mater. Sci. 95 (2014) 337–350. doi: 10.1016/j.commatsci.2014.07.043
A. Otero-de-la-Roza, E.R. Johnson, J. Chem. Phys. 136 (2012) 174109. doi: 10.1063/1.4705760
J.A. Chisholm, S. Motherwell, J. Appl. Crystallogr. 38 (2005) 228–231. doi: 10.1107/S0021889804027074
C.F. Macrae, I. Sovago, S.J. Cottrell, et al. J. Appl. Crystallogr. 53 (2020) 226–235. doi: 10.1107/s1600576719014092
M.K. Panda, S. Ghosh, N. Yasuda, et al., Nat. Chem. 7 (2015) 65–72. doi: 10.1038/nchem.2123
D. Hasa, W. Jones, Adv. Drug. Deliver. Rev. 117 (2017) 147–161. doi: 10.1016/j.addr.2017.05.001
D.D. Huang, D.Z. Yang, Y. Lv, et al., J. Mol. Struct. 1250 (2022) 131891. doi: 10.1016/j.molstruc.2021.131891
J. Du, J. Guo, D. Kang, et al., Chin. Chem. Lett. 31 (2020) 1695–1708. doi: 10.1016/j.cclet.2020.03.028
A. Halme, M.J. Quayle, S.O. Nilsson Lill, et al., Cryst. Growth. Des. 19 (2019) 3670–3680. doi: 10.1021/acs.cgd.8b01867

Figure 1 (a) THPa crystal shows plastic deformation, when applying a load with forceps and metallic needle. (b) THPa crystals with super plasticity were able to be molded into letters “SMU” respectively. (c) THPm crystal exhibits elastic deformation, where the bent crystal restores its linear shape when the applied force is withdrawn.

Figure 2 (a) Representative load–displacement (P-h) curves obtained from nanoindentation on THPa and THPm crystals. “Pop-in” events (three black arrows) happened when loading THPa. (b) Atomic force microscopy images for the nanoindentation on THPa with pile-up predominantly located on the upper side of the indent. (c) In contrast, a relatively smooth surface after nanoindentation on THPm crystals.

Figure 3 (a) The wavy stacking of THPa along the b-axis. (b) Stacking of THP dimers in THPm along [120] direction. Other putative slip planes (c) (020) in THPa and (d) (002) THPm; Possible molecular arrangement during plastic or elastic deformation in (e) THPa and (f) THPm crystals.

Figure 4 (a) The inversion symmetry in P1121/b space group constraints the water proton positions and interrupts hydrogen bonding between neighboring water molecules; (b) Without inversion symmetry in P11b space group, water protons are free to attain an optimal hydrogen bond geometry.

Figure 5 (a) Shear strain δ4 in THPm has less significant effect on the hydrogen bond of THP dimers. The hydrogen bonds in the unstrained and the sheared structure (δ4/2 = 10.0%) are colored in cyan and yellow respectively. (b) Energy-strain curve of THPm under shear strain δ6 which eventually causes a displacement of water chain in the c-axis direction (highlighted in yellow circles) and a discontinuity on the curve (zones Ⅰ and Ⅱ).

Figure 6 (a) Energy-strain curve of THPa with axial strain δ1. Discontinuity is due to restoration of the 1-D hydrgeon bond chain (zone Ⅰ). Isolated chains are seen in highly strained structure (zone Ⅱ), and (b) with shear strain δ6. The wavy molecular sheet undergoes significant rearrangements and reaches different metastable strained states (zones Ⅱ and Ⅲ respectively).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
 下载:
下载: