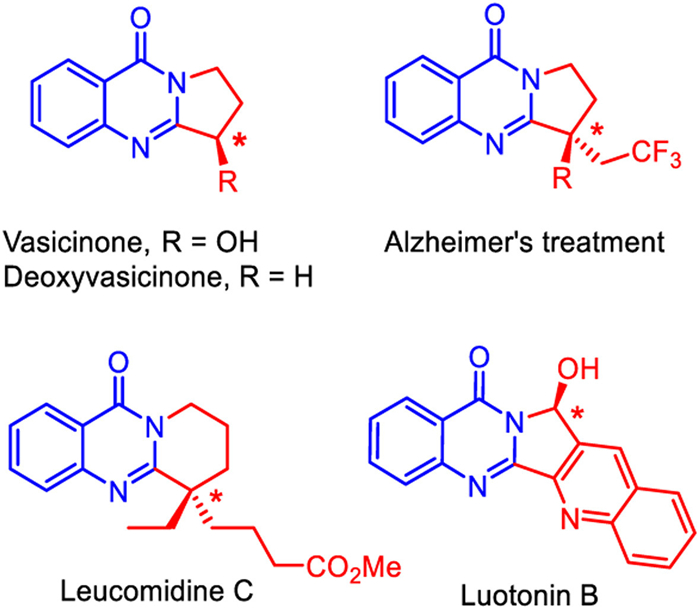
Figure 1.
Natural products and drug molecules of 4-oxoquinazoline skeleton.
Development of sterically hindered SPOs and enantioselective Ni−Al bimetallic catalyzed C−H cyclization of 4-oxoquinazolines with tethered alkenes
Yaqi Zhang , Qiang Ni , Bendu Pan , Long Jiang , Liqin Qiu

Quinazolinone ring skeleton widely exists in natural products and drug molecules. Quinazolinones shows good biological activities in anti-tumor, antibacterial, anti-inflammatory and antihypertension. Similarly, 4-oxoquinazoline natural products are also used as privileged motifs in drug research due to their diverse pharmacological activities (Fig. 1) [1-5]. The synthetic methods of quinazoline have been reported by many researchers [6-10]. However, there are few reports on the simple synthesis of polycyclic quinazolines, especially chiral ones [11-13]. Recently, some research groups attempted to develop methods for the synthesis of tricyclic pyroquinazolone compounds using 4-oxoquinazoline-tethered alkenes or functional group-substituted alkyl chain [14-20]. Notably, in 2021, Dong's group [14] reported a deacylation-aided C–H alkylative annulation of arenes or heteroarenes, in which tricyclic pyroquinazolones were provided in moderate yields (Scheme 1a). Procter [15] disclosed an enantioselective copper-catalyzed borylative cyclization for the assembly of privileged pyroquinazolone motifs (Scheme 1b). Very recently, Wu [16], Jin [17,18], He [19] reported a similar photocatalytic synthesis for polycyclic pyroquinazolones (Scheme 1c). Though there are many ways to prepare achiral or racemic products, few enantioselective synthetic methods could be extended to multifunctionalization of pyroquinazolone scaffolds [21-23]. Enantiomers often exhibit different bioactivities [24], and it is undoubtedly important to prepare these compounds with enantiomeric purity. Therefore, the efficient synthesis of these known and unknown bioactive compounds urgently requires a new asymmetric protocol to build cyclic pyroquinazolone blocks.

Secondary phosphine oxides (SPOs) refer to phosphine oxides containing one hydrogen on the phosphorus atom [25-31]. Compared with most complex phosphine ligands, they are more stable in air and moisture and easy to prepare and preserve. In the presence of alkali, Lewis acid or transition metal, phosphorus can be P(Ⅴ)–P(Ⅲ) tautomerized, in which the stereochemical information around the phosphorus can be retained. For being economical, less toxic and having various coordination modes, Ni [32-35] and Al [36,37] are widely used in their respective catalytic reactions. Nakao [38-40] and colleagues reported direct C-H activation of pyridine compounds catalyzed by Ni-Al bimetallic catalysts with monophosphine ligands or NHC. In the following time, there are also related reports from other groups [41-45]. Their research showed that the Ni-Al catalytic system has a good activation effect on the C-H activation at specific potential electron-deficient sites such as heterocyclic compounds and formamide groups, which provides a possible way for the asymmetric catalytic synthesis of chiral heterocyclic and cyclized compounds. In 2013, Cramer [46] and co-workers discovered that a kind of chiral SPO can provide good activity and enantioselectivity in Ni-Al bimetallic catalyzed hydrocarbamoylation of alkenes. Recently, Ye's group [47-50] also conducted corresponding research in this field. Intramolecular C-H alkylation is undoubtedly a simple and straightforward way for the synthesis of chiral fused 4-oxoquinazoline derivatives. However, suitable catalytic systems have hardly been reported so far. Herein, we disclose the development of two new types of diamine-derived chiral SPO ligands with sterically bulky groups and their application in Ni-Al bimetallic catalytic cyclization of 4-oxoquinazolines with tethered alkenes (Scheme 1d).
Initially, we began to explore the model catalytic reaction of substrates 1ub with traditional SPOs as ligands (Table S1 in Supporting information and Scheme 2, SPO1-5). But the reaction activity or enantioselectivity was poor. Therefore, we realized that modification of the five-membered P-heterocyclic secondary phosphine oxides is necessary and the introduction of large steric substituents into the framework might finely adjust the stereostructure and electrical properties of the ligands and corresponding catalysts. Following this concept, two types of SPOs were designed and successfully synthesized from readily available chiral amines or amino acids (Scheme 2, SPO6-11) [51-54].

With the developed ligands in hand, we continued to conduct ligand screening for substrate 1a (Table 1). Taddol-derived SPO1-2 were found to give the target product 2a in poor yield and ee value (Table 1, entries 1 and 2). In contrast, with traditional N-P-N SPO3-4 instead, the yield of 2a was greatly improved, but the ee value was still not good (Table 1, entries 3-5). Encouragingly, SPO6 provided product 2a with 72% ee, although the yield was only 33%. An interesting phenomenon was also observed, employing SPO7 as the ligand, 3a was obtained as the main product in 87% yield. This implies regioselection can be tuned by appropriate steric effect of the ligand. Further investigations were performed for the prepared SPOs bearing large steric hindrance groups on the N-branched chain (Table 1, entries 8-11). Luckily enough, product 2a was given in 86% yield with 86% ee when SPO10 was used. Through optimization experiments, the optimal reaction conditions of the template reaction were finally determined. The desired product 2a was afforded in 94% yield with 97% ee (Table 1, entry 19).
Under the above optimized reaction conditions, the reaction was further extended to a series of substrates with functional groups at different sites to test the generality (Scheme 3). Substituent effect of monoalkene-tethered 4-oxoquinazoline was first investigated. When the substituents were electron donor groups (2b-2h, 2o), there is less influence on the yield and ee value of the product. The reactions provided the corresponding products in 71%–95% yields and 86%-99% ee. For the substrates with electron-withdrawing substituents, such as CF3, F and ester groups (2i-2m), the reaction activity became poor under the standard conditions. After further optimizing the conditions, acceptable yields were obtained, and the ee values also remained at a high level. For some substrates with large π conjugated groups, reaction results were still good enough under the corresponding conditions to provide 2n, 2p and 2q in 77%–93% yields and 90%–96% ee. Notably, when an additional 6-alkenyloxy substituent was introduced into the skeleton, the enantioselectivity remained excellent (2o) despite a decrease in reactivity. It is speculated that the additional alkenyl group might participate in the coordination competition of Ni, resulting in the reduction of catalytic activity.
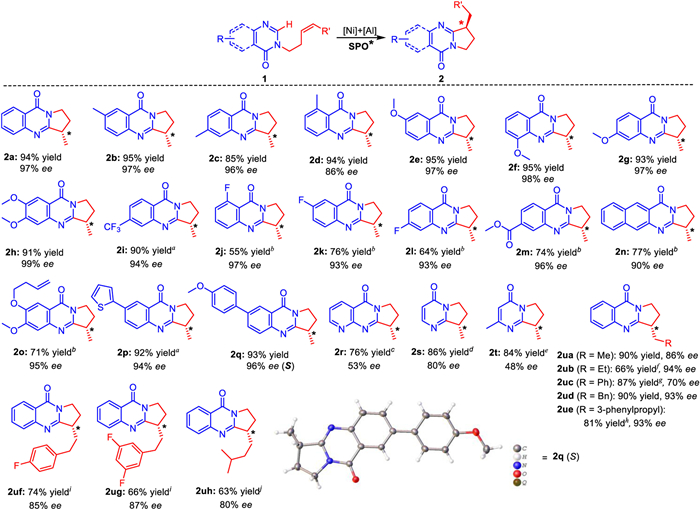
Next, N-heterocyclic substrate 1r was tested. Unfortunately, the ee value of the resulting product 2r was only 51%. Further ligand screening indicated that SPO6 was slightly better than SPO10, and the ee value of the product increased to 53%. Using SPO10 as ligand, the reaction of 4-pyrimidinone substrates 1s and 1t yielded corresponding 2s with 70% ee and racemic 2t. After changing to SPO11, the enantioselectivity of the reactions was improved to 80% and 48%, respectively. Subsequently the suitability of substrates tethered with different N-substituted alkene chains (1ua-1ue) were also investigated. With SPO10 as the ligand, all the catalytic reactions except that of substrate 1uc achieved satisfactory yields and enantioselectivities. These results demonstrate that the alkyl group attached to the end of the olefin chain has less influence on the reaction. However, when phenyl group was directly linked to the end of olefin, product 2uc was only obtained with 25% yield and 47% ee under the standard conditions. In the case of optimized ligand SPO6, 2uc was finally prepared in 87% yield and 70% ee. Changing the linked phenyl group into benzyl or phenylpropyl, interestingly, SPO10 in turn afforded product 2ud or 2ue very well. For substrates 1uf and 1ug with added F atom on the benzene ring of the alkyl chain, no reaction occurred under the standard reaction conditions. After heating to 80 ℃ and adding 15 mol% PPh3, the reaction went well, providing 2uf and 2ug with satisfactory results. Similarly, the activity also decreased when isobutyl was assembled to the olefin chain. The final ee value of 2uh obtained at 80 ℃ was 80%. The configuration of C3 was successfully identified as S by single-crystal X-ray diffraction of 2q.
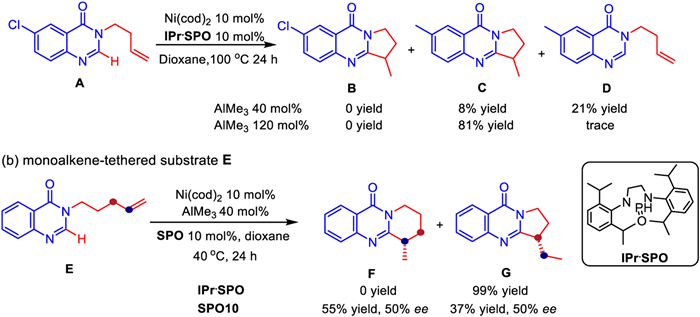
It is also worth noting that the reaction did not proceed as expected when the benzene ring of the substrate contains halogen groups such as Cl and Br. After further exploration (Scheme 4a), it was revealed that the exact reason is that the Cl group became active and was preferentially replaced by methyl group from AlMe3 under the catalysis of IPr. So the corresponding products are C and D, not B. In order to obtain a product with a larger ring, substrate E with a longer N-substituted olefin chain was also tried, but the experimental result were not consistent with our design (Scheme 4b). An interesting phenomenon was noted that the hydrogen of olefin migrated and a five-membered ring product G was finally generated with almost 100% conversion when achiral ligand IPr was used for the catalysis. Replacing IPr with chiral SPO10, however, the five-membered ring product G and the six-membered ring product F coexisted with moderate ee values. This shows that the structure of the ligand affect the regioselectivity of the reaction significantly.
To understand the mechanism, the deuterated substrate D-1aa (88% D) of 1a was prepared from deuterated material (Scheme 5a). In the complete reaction of D-1aa, 54% of deuterium was transferred to the terminal carbon of the alkene moiety, and 36% of deuterium was added to the intermediate carbon atom of the ene. A competitive experiment between D-1aa and 1h revealed that there was no deuterium scrambling distribution. In addition, no significant kinetic isotope effect was observed in the competitive experiment (KIE = 1.0). In order to verify whether SPO has valence change, 31P NMR of SPO10 was identified (Scheme 5b). It was found that the chemical shift of 31P NMR changed from 10.59 ppm to 13.05 ppm after adding AlMe3. This shows that SPO and AlMe3 indeed interact to change the valence state of phosphine, and then SPO-AlMe2 coordinate with nickel [46]. On the basis of our experimental exploration and references [46,50,55], a possibly plausible catalytic cycle is presented (Scheme 5c). First, phosphine oxide is converted into trivalent phosphine under the activation of AlMe3, which then forms a complex with Ni. Al coordinates with the sp2-N atom of the substrate to help the location, then Ni carries out oxidative addition and reductive elimination through the conventional C-H activation steps to generate products. Chiral SPO acts as a ligand and plays a role in chiral induction.

In summary, we have developed two new types of chiral secondary phosphine oxides (SPOs) with bulky and adaptable structures, and their application in the nickel-catalyzed intramolecular enantioselective C−H cyclization of 4-oxoquinazolines with tethered alkenes. For the first time, a new class of chiral tricyclic pyrroloquinazolinone compounds were successfully prepared in up to 95% yield and 99% ee under the mild conditions via the simple Ni-Al bimetallic catalysis. The introduction of sterically hindered chiral amines prepared from readily available chiral raw materials makes the developed ligands significantly superior to the traditional SPOs in this asymmetric catalytic reaction. The effective regulation of enantio- and regioselectivity of the reaction by ligand structure was also demonstrated. The development of Ni-SPO-Al catalytic system and better substrate applicability research are ongoing.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the National Natural Science Foundation of China (No. 22071270), Science and Technology Program of Guangzhou (20220602JBGS02), Guangdong Province Zhu Jiang Talents Plan (No. 2016ZT06C090) and Guangzhou City Talents Plan (No. CYLJTD-201609) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
A. Al-Shamma, S. Drake, D.L. Flynn, et al., J. Nat. Prod. 44 (1981) 745–747. doi: 10.1021/np50018a025
A.G. Sams, G.K. Mikkelsen, R.M. Brodbeck, X.S. Pu, A. Ritzén, Bioorg. Med. Chem. Lett. 21 (2011) 3407–3410. doi: 10.1016/j.bmcl.2011.03.103
I. Khan, A. Ibrar, N. Abbas, A. Saeed, Eur. J. Med. Chem. 76 (2014) 193–244. doi: 10.1016/j.ejmech.2014.02.005
U.A. Kshirsagar, Org. Biomol. Chem. 13 (2015) 9336–9352. doi: 10.1039/C5OB01379H
K. Hemalatha, G. Madhumitha, Eur. J. Med. Chem. 123 (2016) 596–630. doi: 10.1016/j.ejmech.2016.08.001
W.R. Bowman, M.R.J. Elsegood, T. Stein, G.W. Weaver, Org. Biomol. Chem. 5 (2007) 103–113. doi: 10.1039/B614075K
L. He, H. Li, J. Chen, X.F. Wu, RSC Adv. 4 (2014) 12065–12077. doi: 10.1039/C4RA00351A
T.M.M. Maiden, J.P.A. Harrity, Org. Biomol. Chem. 14 (2016) 8014–8025. doi: 10.1039/C6OB01402J
R.S. Rohokale, U.A. Kshirsagar, Synthesis 48 (2016) 1253–1268. doi: 10.1055/s-0035-1560413
X. Zhang, Y. Zhu, Y. Zhu, Z. Li, G. Zhang, Chin. J. Org. Chem. 39 (2019) 2392–2402. doi: 10.6023/cjoc201903025
C. Kaneko, K. Kasai, N. Katagiri, T. Chiba, Chem. Pharm. Bull. 34 (1986) 3672–3681. doi: 10.1248/cpb.34.3672
C. Tsukano, M. Okuno, H. Nishiguchi, Y. Takemoto, Adv. Synth. Catal. 356 (2014) 1533–1538. doi: 10.1002/adsc.201400078
G.R. Jazmín, D.M. Luis, Synthesis 53 (2021) 1471–1477. doi: 10.1055/s-0040-1705975
X. Zhou, Y. Xu, G. Dong, Nature Catal. 4 (2021) 703–710. doi: 10.1038/s41929-021-00661-7
Q. Dherbassy, S. Manna, C. Shi, et al., Angew. Chem. Int. Ed. 60 (2021) 14355–14359. doi: 10.1002/anie.202103259
X. Chen, B. Liu, C. Pei, et al., Org. Lett. 23 (2021) 7787–7791. doi: 10.1021/acs.orglett.1c02819
B. Sun, P. Huang, Z. Yan, et al., Org. Lett. 23 (2021) 1026–1031. doi: 10.1021/acs.orglett.0c04225
B. Sun, R. Shi, K. Zhang, et al., Chem. Commun. 57 (2021) 6050–6053. doi: 10.1039/D1CC02415A
Q.W. Gui, F. Teng, H. Yang, et al., Chem Asian J. 17 (2022) e202101139.
Z. Yang, Y. Shan, J.T. Yu, C. Pan, Eur. J. Org. Chem. 38 (2021) 5382–5385.
S. Eguchi, T. Suzuki, T. Okawa, et al., J. Org. Chem. 61 (1996) 7316–7319. doi: 10.1021/jo9609283
A. Kamal, K.V. Ramana, M.V. Rao, J. Org. Chem. 66 (2001) 997–1001. doi: 10.1021/jo0011484
O.I. Afanasyev, E. Podyacheva, A. Rudenko, et al., J. Org. Chem. 85 (2020) 9347–9360. doi: 10.1021/acs.joc.0c00794
P. Y. Bruice, Organic Chemistry, 4th ed., Pearson Prentice Hall, 2004.
G.Y. Li, Angew. Chem. Int. Ed. 40 (2001) 1513–1516. doi: 10.1002/1521-3773(20010417)40:8<1513::AID-ANIE1513>3.0.CO;2-C
N.V. Dubrovina, A. Borner, Angew. Chem. Int. Ed. 43 (2004) 5883–5886. doi: 10.1002/anie.200460848
T. Nemoto, T. Matsumoto, T. Masuda, et al., J. Am. Chem. Soc. 126 (2004) 3690–3691. doi: 10.1021/ja031792a
L. Ackermann, R. Born, Angew. Chem. Int. Ed. 44 (2005) 2444–2447. doi: 10.1002/anie.200462371
K. Dong, Z. Wang, K. Ding, J. Am. Chem. Soc. 134 (2012) 12474–12477. doi: 10.1021/ja305780z
K. Dong, Y. Li, Z. Wang, K. Ding, Angew. Chem. Int. Ed. 52 (2013) 14191–14195. doi: 10.1002/anie.201307903
C. Chen, Z. Zhang, S. Jin, et al., Angew. Chem. Int. Ed. 56 (2017) 6808–6812. doi: 10.1002/anie.201701394
R.J. Somerville, R. Martin, Angew. Chem. Int. Ed. 56 (2017) 6708–6710. doi: 10.1002/anie.201702188
S.Z. Tasker, E.A. Standley, T.F. Jamison, Nature 509 (2014) 299–309. doi: 10.1038/nature13274
A.L. Clevenger, R.M. Stolley, J. Aderibigbe, J. Louie, Chem. Rev. 120 (2020) 6124–6196. doi: 10.1021/acs.chemrev.9b00682
L. Süsse, B.M. Stoltz, Chem. Rev. 121 (2021) 4084–4099. doi: 10.1021/acs.chemrev.0c01115
H. Mori, K. Hasebe, M. Terano, Polymer 40 (1999) 1389–1394. doi: 10.1016/S0032-3861(98)00379-6
H.R. Yang, L.T. Zhang, D.D. Zang, Z.S. Fu, Z. Q. Fan, Catal. Commun. 62 (2015) 104–106. doi: 10.1016/j.catcom.2015.01.023
Y. Nakao, K.S. Kanyiva, T. Hiyama, J. Am. Chem. Soc. 130 (2008) 2448–2449. doi: 10.1021/ja710766j
Y. Nakao, H. Idei, K.S. Kanyiva, T. Hiyama, J. Am. Chem. Soc. 131 (2009) 5070–5071. doi: 10.1021/ja901153s
Y. Nakao, Y. Yamada, N. Kashihara, T. Hiyama, J. Am. Chem. Soc. 132 (2010) 13666–13668. doi: 10.1021/ja106514b
C.C. Tsai, W.C. Shih, C.H. Fang, et al., J. Am. Chem. Soc. 132 (2010) 11887–11889. doi: 10.1021/ja1061246
W.C. Lee, C.H. Chen, C.Y. Liu, et al., Chem. Commun. 51 (2015) 17104–17107. doi: 10.1039/C5CC07455J
S. Liu, J. Sawicki, T.G. Driver, Org. Lett. 14 (2012) 3744–3747. doi: 10.1021/ol301606y
W.B. Zhang, X.T. Yang, J.B. Ma, Z.M. Su, S.L. Shi, J. Am. Chem. Soc. 141 (2019) 5628–5634. doi: 10.1021/jacs.9b00931
C.S. Wang, S.D. Monaco, A.N. Thai, et al., J. Am. Chem. Soc. 142 (2020) 12878–12889. doi: 10.1021/jacs.0c06412
P.A. Donets, N. Cramer, J. Am. Chem. Soc. 135 (2013) 11772–11775. doi: 10.1021/ja406730t
Q.S. Liu, D.Y. Wang, Z.J. Yang, et al., J. Am. Chem. Soc. 139 (2017) 18150–18153. doi: 10.1021/jacs.7b09947
Y.X. Wang, S.L. Qi, Y.X. Luan, et al., J. Am. Chem. Soc. 140 (2018) 5360–5364. doi: 10.1021/jacs.8b02547
Y.X. Wang, F.P. Zhang, Y.X. Luan, M. Ye, Org. Lett. 22 (2020) 2230–2234. doi: 10.1021/acs.orglett.0c00432
R.H. Wang, J.F. Li, Y. Li, et al., ACS Catal. 11 (2021) 858–864. doi: 10.1021/acscatal.0c04585
K.B. Selim, Y. Matsumoto, Y. Tomioka, Angew. Chem. Int. Ed. 48 (2009) 8733–8735. doi: 10.1002/anie.200904676
O'H. David, T. Mustafa, Tetrahedron: Asymmetry 10 (1999) 1189–1192. doi: 10.1016/S0957-4166(99)00095-6
C. Narendra, M. Sonali, K.G. Sunil, Tetrahedron: Asymmetry 19 (2008) 2721–2730. doi: 10.1016/j.tetasy.2008.12.010
D.A. Klumpp, S.L. Aguirre, G.V. Sanchez, S.J. Leon, Org. Lett. 3 (2001) 2781–2784. doi: 10.1021/ol016408y
J.S. Bair, Y. Schramm, A.G. Sergeev, et al., J. Am. Chem. Soc. 136 (2014) 13098–13101. doi: 10.1021/ja505579f

Scheme 1 Selective cyclization of 4-oxoquinazolines with tethered alkenes or functional group-substituted alkyl chain.

Scheme 3 Scope of substrates. Standard reaction conditions: 1 (0.2 mmol), dioxane (2.0 mL), 5 mol% Ni(cod)2, 40 mol% AlMe3, 10 mol% SPO10, room temperature, under N2 for 24 h; a 10 mol% Ni(COD)2, 15 mol% PPh3, 48 h; b 10 mol% Ni(cod)2, 60 h; c 10 mol% Ni(cod)2, 10 mol% SPO6, 15 mol% PPh3, 80 ℃, 48 h; d 10 mol% Ni(cod)2, 10 mol% SPO11, 40 ℃, 72 h; e 10 mol% Ni(cod)2, 10 mol% SPO11, 60 ℃, 60 h; f 10 mol% Ni(cod)2; g 10 mol% Ni(cod)2, 10 mol% SPO6, 15 mol% PPh3, 60 ℃, 72 h; h 10 mol% Ni(cod)2, 40 ℃, 80 h; i 10 mol% Ni(cod)2, 15 mol% PPh3, 80 ℃, 48 h; j 10 mol% Ni(cod)2, 80 ℃, 48 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



 下载:
下载: