
Figure 1.
Schematic diagram of PROTACs-mediated degradation of POI. E2, Ubiquitin-conjugating enzyme; Ub, Ubiquitin.
Current strategies for improving limitations of proteolysis targeting chimeras
Chunlan Pu , Shirui Wang , Lei Liu , Zhonghui Feng , Hongjia Zhang , Qianyuan Gong , Yueshan Sun , Yuanbiao Guo , Rui Li

Proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules consisting of E3 ligase-recruiting ligand, protein of interests (POI) ligand, and a chemical linker to connect the above two moieties. PROTACs serve as bridging molecules to induce POI degradation by specifically recruiting proximity between ubiquitin ligases (E3) and POI to form a ternary complex (Fig. 1) [1-3]. Utilizing PROTACs technology can effectively inhibit and rapidly degrade the key proteins which are related to tumor occurrence and development, and successfully promote the rapid development of targeted protein degradation (TPD) for cancer therapy [4,5]. Till now, research in small-molecule PROTACs is surging. There are more than thousands of reported PROTACs in PROTAC-DB database [6]. Among them, over 29 PROTACs have passed in vivo or pharmacokinetics (PK) studies and at least 15 degraders have entered the clinical trial stage (Table 1) [7,8]. PROTACs technology is becoming increasingly mature with wide developing prospect.
Compared with conventional small-molecule drugs, PROTACs have several advantages in the field of antineoplastic drug design and discovery [9,10].
Firstly, PROTACs can effectively overcome drug resistance. Since PROTACs exhibit the potential to directly remove or deplete of intracellular protein through degradation, it is helpful to conquer cancer resistance caused by target protein mutation or over-expression, thus enhancing the therapeutic effect on tumors [11,12]. PROTACs could also induce the degradation of other components of the complex that the POI ligands can bind to exhibit more extensive antitumor effects [13]. Recently, Homo-PROTACs was reported as an approach to induce the homo-dimerization of E3 ubiquitin ligases and trigger its suicide-type chemical knockdown inside cells by causing proteasome-dependent degradation [14]. Studies have shown that Homo-PROTACs could be used to induce the degradation of different oncogenic targets in cancer, such as glioblastoma [15], or prostate cancer [16].
Secondly, PROTACs can eliminate "undruggable" disease-causing proteins. Studies have shown that PROTACs are indeed capable of degrading these proteins which lack suitable drug binding sites [17], such as transcription factors (TFs) (e.g., STAT3) [18], large multiprotein complexes BAF (e.g., SMARCA2/4) [19], v-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS) [20] and ubiquitin conjugating enzyme E2C (UBE2C) [21], which can transform "undruggable" targets that play a key role in cancer or other diseases into "druggable" [22].
Thirdly, PROTACs could be used to repurpose some natural compounds. Recently, the natural compound Celastrol was linked to JQ1 to induce BRD4 degradation [23], and some natural compounds with potent anticancer effects have shown to interact with E3 ligases, such as the case of the natural compound Obtusaquinone [24], indicating that those natural compounds have the potential to be used in PROTAC drug design to induce selective oncoprotein degradation.
Lastly, PROTACs can drive the knockout of the isomer or subtype of POI in selective ways. PROTACs prefer to degrade proteins that can form a sufficiently stable POI-PROTAC-E3 ternary complex [5,25,26], thus reducing the non-specific effects on other proteins. However, recently, it was observed that different POI ligands, E3 ligase and inherent linker composition could affect the degradation selectivity of closely-related protein families [27,28]. As shown in Fig. 2a, BSJ-02–162 and BSJ-01–187 with different POI ligand show degradation selectivity of CDK6 or CDK4, respectively [29]. BSJ-02–162 consisting of 4-carbon alkyl linker and Palbociclib could degraded CDK6 (DC50 < 100 nmol/L, Dmax < 90%) effectively, while BSJ-01–187 with the same linker but conjugated to Ribociclib preferred to degraded CDK4 in Jurkat cells. Additionally, Palbociclib-based degrader pal-pom and CP-10, with distinct triazole linker composition and lengths, can degrade CDK6 or both CDK4/6 degradation selectively (Fig. 2b). Among them, pal-pom was reported as a CDK4/6 degrader [30], while CP-10 can degrade CDK6 (DC50 = 2.1 nmol/L, Dmax < 90%) in U251 cells selectively [31]. Furthermore, as shown in Fig. 2c, the PROTACs DAS-6–2–2–6-CRBN and DAS-6–2–2–6-VHL, consisting of different E3 ligase recruiters linked to the same POI inhibitor Dasatinib, are capable of selective degradation of c-ABL and BCR-ABL [32].

Despite many advantages for PROTACs in term of tumors and other diseases therapy, due to their various drawbacks or limitations, most of PROTACs drugs remain in vitro research.
First of all, PROTACs drugs usually have poor cellular permeability and oral bioavailability. Drug metabolism and PK are key factors that have to be optimized in drug development [33,34]. Bifunctional PROTACs usually have a higher molecular weight (MW) (0.7~1.1 kDa), with excessive hydrogen bond donors (HBDs), hydrogen bond acceptors (HBAs) and large polar surface area (PSA), which make them out of the "Rule of 5″ chemical space [35,36] and impose hurdles to their biofilm permeability, resulting in low drug bioavailability and ineffective efficacy [37].
Secondly, it is difficult to control the degradation activity of PROTACs. The degradation of POI often shows typical shape of the mountain dependence on PROTACs concentration called "hook effect" [38,39], which is caused by the distinct mechanism of three-component system. The ineffective binary complexes are formed at high protein degrader concentrations for PROTACs binding with POI or E3 ligases, and competing with effective ternary complexes, which result in negative impacts on PROTAC's degradation potency [40,41]. As shown in Fig. 3a, the hook effect is related to binding affinities and relative concentrations of the POI, PROTACs and E3 ligases. Therefore, the binding cooperativity of three elements in PROTACs is a key design point to modulate this hook effect [42,43].

Thirdly, there are many challenges in expanding the E3 ligase toolbox. To date, more than 600 E3 ligases were reported in human cells [44], however, only a few of them have been used in the PROTACs. Among them, CRBN and VHL E3 ligases are commonly used [45,46]. Recent studies reported some PROTACs that harness new E3 ligases such as DCAF15 [47], DCAF16 [48], RNF4 [49] and RNF114 [50]. E3 ligases to degrade POI, but the degradation efficiency is generally low. Compared with the "occupation driven" theory of small molecule inhibitors, PROTACs with low degradation efficiency will lose the advantage of low doses "event driven" pharmacology and fail to meet clinical requirements [51].
The last drawback of PROTAC is the side effects and toxicity. The potential of on-target as well as off-target side effects becomes a bottleneck for PROTACs development [52]. On one hand, PROTACs aims at tackling those pathogenesis-related proteins for fulfilling the disease therapy, but the catalytic nature of PROTACs have the potential to degrade entire proteins completely, which may result in unacceptable toxicity [5,53]. On the other hand, PROTACs molecules do not have the target effect on tumors, while the target proteins usually expressed in normal cells widely [54]. If the E3 ligases are not tumor-specific, it can easily lead to uncontrolled protein degradation and the increase of side effects and toxicity on normal cells [55,56].
The factors mentioned above may contribute to cancer treatment failure of PROTACs drug. Although there have been many previous reviews of recent advances in developing PROTACs [57-59], these strategies will be reinterpreted from a new perspective in this review. In particular, we focus on those allowing to improve cell permeability or reduce toxicity of PROTACs by optimizing structural or favorable modification, and shed light on their merit and demerit (Fig. 3b). These strategies that will give a guide to the reasonable PROTACs design, and are beneficial for PROTACs drug development and safety considerations.
Western blot is generally performed to assess the efficacy of PROTACs degradation. Nevertheless, when the results are undesired, we should consider whether it is connected with low solubility and permeability of PROTACs or a stable ternary complex failure [60,61]. Since most of the POI are intracellular localization, the acceptable cell permeability or solubility is crucial for PROTACs to induce successful POI degradation. Recent progress in the PROTACs discloses several ternary complex crystal structures, which make researchers gradually realize that small structural changes will have a vital impact on the PK characteristics of drugs, especially in the optimization of the linker region [62,63]. There are two key methods which can be used for linker optimization, including shortening the linker length to reduce flexibility and MW of PROTACs and replacement of linker types to enhance PROTACs stability and cell permeability. Degrader optimization may be particularly complex, but the balance between PROTACs activity, PK and selectivity can be achieved by finding the core issues that need to be addressed [34].
To acquire potent target POI degradation, PROTACs require appropriate linker to connect an E3 ligase binding scaffold with POI ligand [64,65]. Therefore, optimizing the geometry and chemical composition of linker i.e., the length and types, is crucial to obtain favorable oral PROTACs [66,67]. Current linker type of PROTACs is shown in Table 2. Among them, the hydrophilic polyethylene glycol (PEG) chains can improve water solubility because of their polarity and flexible nature, which make them the most common type of linkers used in PROTACs [68,69]. Lipophilic alkyl chains are also widely used in design of PROTACs [70,71]. The above-mentioned linkers are usually long and flexible. By change the number of HBDs or HBAs, the exposed polarity and lipophilicity can be modulated. Although the increase of lipophilicity will improve the cell permeability of PROTACs, it might also reduce the solubility and increase toxicity. Therefore, it is very important to balance the permeability, lipophilicity and solubility.
Recent studies have shown that increase of linker rigidity may improve degradation potency, while introducing conformational flexibility to the linker might boost aqueous solubility and cell permeability simultaneously [52,72,73]. Interestingly, by using of cyclization strategy, Alessio Ciulli's group firstly reported a macrocyclic PROTACs (macroPROTAC-1) which exhibits rapid and potent intracellular degradation of BRD4 (Fig. 4a) [73]. Macrocyclization can stabilize the active conformation by locking it to the bound state, which is favorable for not only increase degradation potency and selectivity, but also exhibits optimized PK properties [74,75]. Besides, the introduction of heterocyclic scaffolds (e.g., piperazine/piperidines) into linker structures will improve water solubility and balance lipophilicity by modulating the PROTACs physico-chemical properties [19,42,76]. For instance, by replacing chain-typed linker with polar pyridine/piperidine motifs, ARV-110 and ARV-471 show good oral bioavailability and now are in Phase Ⅱ clinical trials (Figs. 4b and c) [7,58].

The 1,2,3-triazole ring may not only provide additional motifs for novel binding sites on target proteins, but also may improve the physico-chemical properties of PROTACs. Besides, protein ligand and E3 recruiting motif can be readily prepared in the form of azides and alkynes [77,78]. The triazole containing chimera is formed through "click chemistry" [79], which involving the combination of azides and alkynes moiety via a copper-catalyzed Huisgen 1,3-dipolar cycloaddition (Fig. 5a) [80]. Because this reaction is typically high yielding, rapid response and boasts excellent functional group compatibility under mild reaction conditions, this linking strategy has wide applicability in situ bonding and reactions in and out of organisms [81]. Furthermore, compared to linear linkers, the triazole linkers have other advantages. For example, with more structural rigidity, the triazole ring may get a more stable oxidative metabolism in vivo [82]. Besides, the introduction of the triazole ring is more likely to produce additional contact points or surfaces in the spatial structure of protein interaction [83], which is beneficial to form stable ternary complexes and induce effective protein degradation. However, such class of PROTACs showed poor cell permeability, so they cannot pass through the cell membrane to perform good degradation.
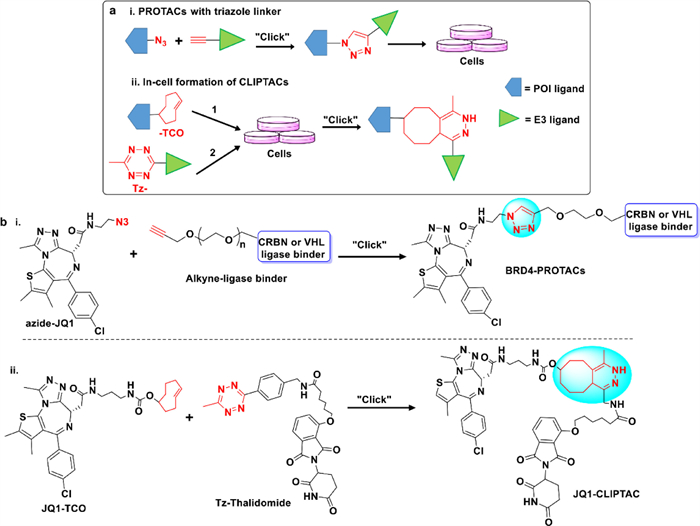
Researchers are committed to finding alternative ways to solve the solubility and permeability problems related to high MW and lipophilicity. Recently, Lebraud et al. reported a novel PROTAC technology, named as in-cell click-formed PROTACs (CLIPTAC), which can combine two smaller chemical moieties to generate PROTACs in cells by using "click chemistry" reactions, that is to say the rapid and high efficiency click reaction between a tetrazine tagged CRBN E3 recruiter (Tz-thalidomide) and a trans-cyclooctene (TCO) tagged ligand of POI by the inverse electron demand Diels-Alder (IEDDA) cycloaddition (Fig. 5a) [84]. In their study, by using the CLIPTAC tactics, JQ1-CLIPTAC (Fig. 5b) and ERK-CLIPTAC can successfully degrade oncogenic proteins, e.g., BRD4 and ERK1/2, respectively [58,85]. Compared to the traditional PROTAC molecules, the in-cell CLIPTACs not only have smaller MW and good cell permeability, but also showed indeed an excellent degradation potency. It provides us an advanced strategy for improving the bioavailability of PROTAC drugs. However, the click reaction of TCO-groups and Tz-groups may also occur outside the cells, so, choosing the appropriate click reaction is worth noting [86].
Clinically, PROTACs are all orally administered. To improve oral bioavailability, in addition to linker structural optimization, it can also be obtained by developing new dosage forms. New nano-drug delivery systems (NDDS), such as nanoparticles [87], polymer micelles and liposomes [88], could effectively overcome the oral absorption barrier and improve the oral bioavailability of drugs, which provides a new research direction for the realization of oral drug delivery of PROTACs [61,89]. Chen et al. proposed a novel two-component PROTAC system, i.e., the pre-fusing E3-ligases linking proteins to PROTACs delivered by lipid nanoparticle (LNP) platform (Fig. 6a) [90]. The results demonstrated that the pre-fused PROTACs could significantly increase POI degradation efficiency compared with free PROTACs (ternary complex: POI-PROTAC-E3). Besides, encapsulated pre-fused PROTACs into LNP for intracellular delivery could enhance the cell permeability of PROTACs by endocytosis. This approach has been successfully used to degrade BRD4 (pre-fused ARV-771 LNPs) and AR (pre-fused SARD279 LNPs) (Fig. 6b) [90]. Nevertheless, despite above advantages, the per-fused PROTACs did not solve the problem of active targeting of tumor sites in vivo.

It is generally accepted that large-in-size compounds have been uptake in cell via passive transmembrane permeation preferentially, which may be affected by multiple factors and transporters, while the exact mechanism of PROTACs cell penetration remains unclear [91]. Wang's group have developed a reversible covalent BTK PROTAC (RC-1) for BTK efficacious degradation [92]. Notably, they have serendipitously discovered that cyano-acrylamide-based moiety could produce a fast and reversible reaction with intracellular glutathione to overcome the nonreusable drawback of irreversible covalent PROTACs, thus significantly enhancing the intracellular accumulation. The specific mechanism of this strategy is showed in Fig. 7. When the PROTAC molecule binds to the active site of POI, the nearby cysteine side chain can react with the α-cyano-acrylamide group to form a covalent and reversible bond [93], thus allowing ternary complex formation and target degradation, and subsequent reversible covalent PROTAC regeneration [92]. In fact, some recent works suggested that the free cysteines and electrophilic groups occur in the covalent reversible binding on the cell surface, which benefits the non-permeable molecules to enhance their transmembrane penetration via a thio-mediated cellular uptake [94]. Therefore, reversible covalent chemistry for PROTAC development provides a new idea for PROTACs' active uptake in cells and gives an efficient tool for ameliorating poor cellular permeability of PROTACs.

Trying different E3 ligases binder is another way to improve physicochemical properties of PROTACs [95]. It should be noted that the expression levels of the corresponding E3 ligases in target cells may determine the tissue/disease specificity of the degrader [54,55,96]. To date, most PROTACs have designed and developed by recruiting CRBN or VHL E3 ligases. Although these two types of PROTACs usually have potent efficacies both in vitro and in vivo, which have been successfully applied in clinical research, the catalytic properties of PROTACs may cause potential toxicities due to the systemic target degradation, and complete elimination of target proteins, thus leading to the unpredictable consequences [97]. For instance, it is tolerated when BET bromodomains is inhibited by its inhibitor, but it is lethal when the BRD2 and BRD4 are completely degraded [98]. On the other hand, CRBN or VHL E3 ligases and target proteins are also spared in healthy cells, so if PROTACs engage these E3 ligases, it will increase cytotoxicity and lead to serious side effects, thus finally restrict them from entering the clinic, such as the PROTAC of CDK9 [99,100] or AURKA [101]. Totally, toxicology is an issue that could not be ignored in clinical application of PROTACs [102]. It is extremely important to achieve a more precise control and pathological localization for degradation of PROTACs. Nowadays, researchers have carried out multi-functional PROTACs from many aspects to ensure their safety.
Light with high spatiotemporal accuracy and fewer off-target side effects is generally used to modulate biological response in neurobiology, chemical biology, disease treatment and other fields [103,104]. Due to the advantages of light, photochemically targeting chimeras (PHOTACs) which fuse the photochemistry moiety with these bifunctional PROTACs provided a novel pathway to control PROTAC activity [62,105,106]. Photocaged protective group or opto-electronic switches which are introduced into the PROTAC structure can be released by artificial light-induced cleavage of protecting groups or conformational switching. This method has the advantages of good release performance and photocleavage and non-toxicity. There are two strategies in design of PHOTACs: incorporation of photo-caged groups to PROTACs or making photoswitchable PROTACs (Fig. 8a).
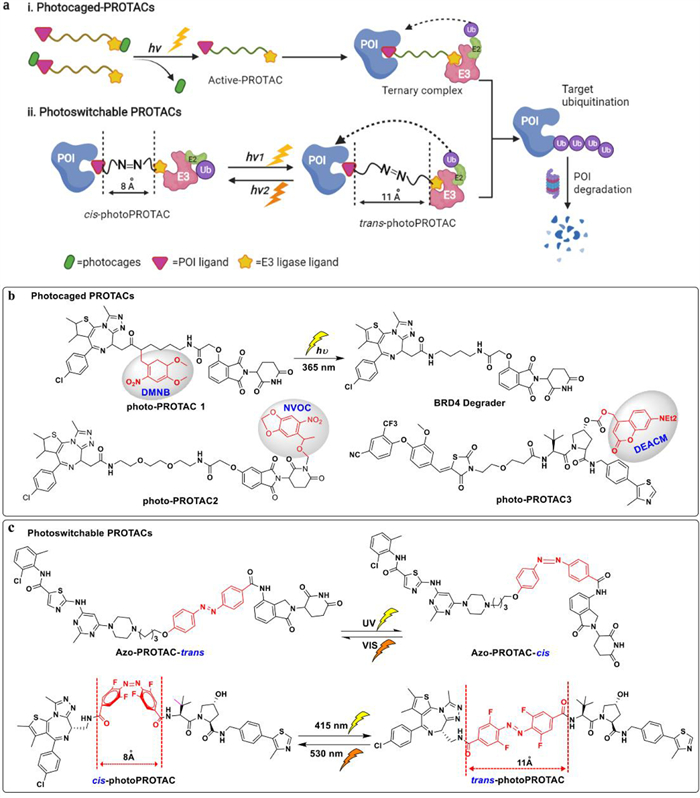
Photocaged PROTACs consist of a photo-removable blocking group that detaches only upon irradiation at a specific wavelength, thus releasing the active PROTAC and inducing POI degradation [107]. A photo labile caging substituents such as dimethoxy-2-nitrobenzyl (DMNB) [108], nitroveratryloxycarbonyl (NVOC) [109] and [7-(diethylamino)coumarin-4-yl]methyl (DEACM) [110] groups were used in photocaging of the POI binder or E3 recruiter (Fig. 8b), which are capable of blocking the formation of the ternary complex in the dark, while it can be efficiently dislodge upon irradiation at specified wavelength, such as at 365 nm. Xue and Kounde's groups reported photo-caged PROTAC for degradation of BRD4 by synthesizing potent CRBN\VHL-based PROTACs, respectively [108,111]. In parallel, Liu et al. also regulated protein degradation by adding a photo-removable group, i.e., DMNB on pomalidomide of ALK-PROTACs and BET-PROTACs to form opto-PROTACs [112]. All of them displayed no activities in the dark, and after light irradiating, the active PROTACs removed the photoprotective group and showed powerful degradation activity in cells, which demonstrated as light-inducible protein degradation.
By using the ability of azobenzene to switch reversibly between cis and trans conformation upon visible light pulses [113], the degradation capability of photoswitchable PROTACs bearing the azobenzene linkages can be reversibly activated and deactivated by using suitable wavelengths of light (Fig. 8a) [107]. Reynders et al. developed PHOTACs by incorporating the azobenzene moiety into the CRBN E3 ligase ligand (lenalidomide) for targeting BRD2\4 and FKBP12 [114]. After irradiation with 390 nm light pulses, the isomerization changes to the active cis form, leading to potent degradation of POI. While the trans isomerization could be achieved by irradiating with wavelengths > 450 nm or in the absence of light. Similarly, Jin et al. obtained azo-cis and azo-trans isomer of CRBN-based PROTACs to target oncogenic BCR-Abl fusion and Abl proteins, and the azo-trans isomer can degrade the POI efficaciously [115]. In addition, Crew and Carreira group discovered a reversible photoPROTAC based on ARV-771 template, which is switchable between irradiation with 530 nm to get inactive cis-photoPROTAC and acquired the active trans-photoPROTAC in irradiation with 450 nm (Fig. 8c) [116]. To shed light on protein degradation mechanism, they analyzed the critical difference in linker length between active and inactive degraders. The inactive isomerization may be ascribed to the linker distance, which is too short to product a stable ternary complex. Specifically, the active trans form is 3 Å longer than that of inactive cis isomer [116].
Compared with photo-caged groups, introducing azobenzene and its derivatives in PROTACs may have many advantages, since they may not dramatically increase the MW of PROTACs, and needless of switchable elements which will leading to more stable and facile modulation of these PROTACs [60]. Despite the above light-induced PROTACs techniques enable protein degradation in a spatiotemporal manner and make great possibility to avoid the damage to normal cells, it has obvious disadvantages because light stimuli usually happened in external control, leading to the fact that it can only treat tumors that can be accessibly irradiated by ultraviolet light like some skin cancers, which limits the application range of PHOTACs.
Targeting receptors that are highly and specifically expressed on tumor cells enables the selective delivery of drugs to cancerous tissue to minimize toxic side effects in the patients [117]. The folate receptor α (FOLR1) has been identified as one of overexpressed proteins in cancer cells and is a well-defined target for drug delivery [118]. FOLR1 is capable of transporting folate (FA) into cells, therefore, folate-conjugating strategy is beneficial for tumor-targeted delivery of PROTACs to achieve controllable target degradation of POI (Fig. 9a) [119,120]. Liu et al. developed a folate-caged PROTACs by linking the folate group with BRD-PROTAC(ARV-771) to synthesis folate-ARV-771, which is capable of degrading BRDs via cleavage by endogenous hydrolase in FOLR1-positive cancer cells (Fig. 9b) [119]. By using similar way, MEK-PROTAC (folate-MS432) and ALK-PROTAC (folate-MS99) have been developed to degrade MEK or ALK in tumor cells. Furthermore, they reported a folate-caged pomalidomide prodrug (FA-S2-POMA) and a folate-caged ALK-PROTAC (FA-S2-MS4048) (Fig. 9b) [120], both of which can be cleaved by intracellular glutathione (GSH) to release the active IMiD-based molecular glues or ALK degrader MS4048 in FOLR1-expressing cancer cells. It is confirmed that the folate-caged PROTACs strategy is successful in circumventing potential toxicity for uncontrolled or incorrect degradation of POIs in normal cells. There is also limitation in this novel method, as after introducing of folate group, the MW of PROTACs increase dramatically (usually more than 1000 Da), which will inevitably decrease their oral bioavailability and PK. To date, the in vivo data on folate-PROTACs have not been described.

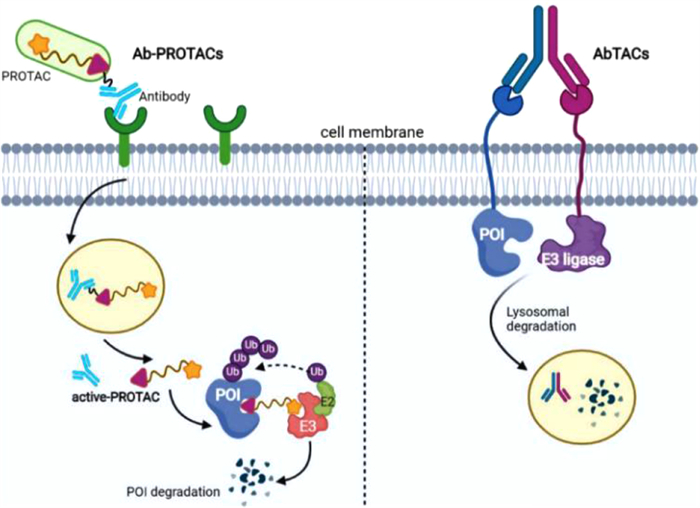
Antibody-drug conjugates (ADCs) can specifically deliver cytotoxic payload to a specific cancer cell type based on high antigen-antibody specificity and affinity [121]. Recently, studies showed that antibody-PROTAC conjugates (Ab-PROTACs) and antibody-based PROTACs (AbTACs) which conjugate PROTACs with antibody could overcome limitations of PROTAC selectivity to some extent (Fig. 10) [122,123]. The difference between the two tools is that they have different protein degradation mechanisms. By linking E3 ligase-directed degrader to a monoclonal antibody (Trastuzumab [124]), Maneiro et al. developed an antibody-PROTAC conjugate (Ab-PROTAC 3) which can specifically degrade BRD4 in HER2 positive breast cancer cell lines through antigen-dependent manner [125]. Similarly, Wells group developed an AbTACs AC-1 by utilizing recombinant bispecific antibodies to recruit a transmembrane E3 ligases RNF43 for inducing degradation of programmed death-ligand 1 (PD-L1) [126]. It should be mentioned that the degradation mechanism of AC-1 is in lysosome-dependent manner, which is different with Ab-PROTAC 3 [58]. Both of these two strategies bear the protein degradation capabilities of PROTACs with the precision selectivity of antibody that can help to further reduce undesired side effects over traditional PROTACs, but due to complex PK properties of antibody, Ab-PROTACs and AbTACs are still need to be administrated by injection as reported.
Cancer immunotherapy can effectively prevent cancer cells from escaping the immune system compared with traditional chemotherapy, thus reducing drug-resistance and increasing the therapeutic index [127]. In 2021, Zhang et al. reported a Semiconducting polymer nano-PROTAC (SPNpro) as a multifunctional nano-PROTACs system bearing photo-therapeutic, immune-related protein degradation and tumor targeting capabilities [128]. SPNpro consists of a semiconducting polymer core coupled with a PROTAC fragment via a cancer-biomarker-cleavable peptide (Fig. 11). On one side, SPNpro was expected to be effectively inhibit cancer cells by producing the singlet oxygen (1O2) under near-infrared light and simultaneously stimulating T cells to promote the apoptosis of cancer cells. On the other hand, Cathepsin B (CatB), a cancer biomarker, can cleave SPNpro and release PROTACs which target immunosuppressive indole-amine-2,3-dioxygenase (IDO) and induce its degradation. SPNpro-mediated in situ immune metabolism intervention combined with immunogenic phototherapy to stimulate the function of T cells in immunity and effectively inhibit tumor growth and metastasis [128]. SPNpro showed high cancer tissue specificity since both CatB and IDO can effectively target cancer cells. As a new mode of multifunctional PROTACs, SPNpro could solve the problems of the uncontrollable protein degradation and off-target side effects, and provide a new idea to boost the antitumor activity of PROTACs. However, SPNpro is a complex nanohybrid system with the high MW and unstable elements (e.g., polypeptide), so the bioavailability and effectivity in vivo should further be evaluated.

In recent years, TPD is a hot field of new drug discovery, which is potential in tackling disease-causing proteins by harnessing the cell's own disposal system [122,129]. Apart from cell permeability and potential toxicology, the repertoire of proteins they can degrade is also a key point that PROTAC needs to break through in the future [52]. There is some novel and exciting TPD approaches to remedy this PROTAC's limitation. Lysosome-based degradation technology has been reported as they can effectively remove certain large proteins, damaged or excess organelles and extracellular proteins that are closely related to cancer and other diseases (Fig. 12) [130,131]. For example, lysosome targeting chimeras (LYTACs), which link a targeted antibody with a glycan to harness the lysosomal degradation pathway can induce degradation of those extracellular proteins effectively [132,133]. Besides, by linking a target ligand to an autophagic degradation tag, autophagy-targeted chimeras (AUTACs) [134,135] and autophagosome-tethering compounds (ATTECs) [136] can hijack the autophagy-lysosome pathway and degrade entire organelles and protein aggregates potentially [130,137]. Otherwise, some new heterobifunctional molecules have been reported to contribute to alternative PROTACs drug design strategies, such as phosphorylation targeting chimeras (PhosTACs) [138], ribonuclease-targeting chimeras (RIBOTACs) [139] and acetyltransferase-targeting chimeras (AceTAGs) [140]. Unfortunately, from the perspective of degradation efficiency, these new TPD strategies typically have low degradation efficiency, which needs to be further optimized. In 2021, Ciulli's group designed trivalent PROTACs SIM1, which showed more sustained and higher degradation efficacy than that of bivalent PROTACs MZ1 (a BRD4 degrader), since SIM1 consist of two BET inhibitor (JQ1) and an E3 ligand (VHL) tethered via a branched linker [141]. This example provides valuable guidance for optimizing PROTAC degradation efficiency.

In this article, firstly, we reviewed some existing strategies to solve low solubility and poor cell permeability of PROTACs, such as the optimization of linker structure or changing linker flexibility or rigidity, low MW intracellular assemble CLIPTAC, thio-mediated cellular uptake, and pre-fused PROTACs for intracellular delivery. However, these strategies are trapped in tumor targeting and tissue selectivity. Besides, we uncovered the relationship between the chemical structure of linker and cell permeability in PROTACs. This is helpful to propose perspectives for future research directions. Secondly, to cope with the off-targeted toxicity of PROTACs, we summarized some solutions including spatiotemporal localization via exogenous light induction, folate-mediated and antibody-binding mediated tumor specificity and SPNpro by combining phototherapy, immunotherapy with protein degradation. However, they usually have high WM and need to be further improved and evaluated.
To date, there is no clear design rules and principles to improve PROTACs oral bioavailability, so optimization of PROTACs is laborious and still experience-based. Although currently most PROTACs are administered orally in clinical trials, their PK profiles, solubility, and cell permeability could be further modified. Hence, improving these properties via structural optimization or multifunctional modification are of great value for the clinical advancement of PROTACs. These new ideas of PROTACs will open up new promising perspectives in accurate and effective degradation of proteins in human diseases and conducive to the future targeted therapy drugs development. Totally, this review gives guidelines for PROTACs reasonable design to address bioavailability or toxicology issues.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank all members for the writing and development of this review. This work was supported by National Natural Science Foundation of China (No. 81773195), Sichuan Science and Technology Program (No. 2021YJ0220), Technology Innovation Research and Development Project of Chengdu (No. 2022-YF05−01982-SN), the Foundation of Science and Technology Department of Sichuan Province (No. 2022YFS0172).
M. Pettersson, C.M. Crews, Drug Discov. Today: Technol. 31 (2019) 15-27. doi: 10.1016/j.ddtec.2019.01.002
X. Sun, H. Gao, Y. Yang, et al., Signal Transduct. Target. Ther. 4 (2019) 64. doi: 10.1038/s41392-019-0101-6
J. Lu, Y. Qian, M. Altieri, et al., Chem. Biol. 22 (2015) 755-763. doi: 10.1016/j.chembiol.2015.05.009
M. Lv, W. Hu, S. Zhang, et al., Cancer Lett. 539 (2022) 215716. doi: 10.1016/j.canlet.2022.215716
B. Dale, M. Cheng, K.-.S. Park, et al., Nat. Rev. Cancer 21 (2021) 638-654. doi: 10.1038/s41568-021-00365-x
G. Weng, C. Shen, D. Cao, et al., Nucleic. Acids. Res. 49 (2020) D1381-D1387.
M. Békés, D.R. Langley, C.M. Crews, Nat. Rev. Drug Discov. 21 (2022) 181-200. doi: 10.1038/s41573-021-00371-6
A. Mullard, Nat. Rev. Drug. Discov. 20 (2021) 247-250. doi: 10.1038/d41573-021-00052-4
W. Li, M.R. Elhassan, X. Hou, H. Fang, Curr. Med. Chem. 28 (2021) 4893-4909. doi: 10.2174/0929867327666201117141611
M. Ao, J. Wu, Y. Cao, et al., Chin. Chem. Lett. 34 (2023) 107741. https://doi.org/10.1016/j.cclet.2022.107741 doi: 10.1016/j.cclet.2022.107741
F. Yu, M. Cai, L. Shao, J. Zhang, Front. Chem. 9 (2021) 679120. doi: 10.3389/fchem.2021.679120
Y. Wu, J. Zhang, X. Zhu, Y. Zhang, Cancer Lett. 544 (2022) 215808. doi: 10.1016/j.canlet.2022.215808
G.M. Burslem, B.E. Smith, A.C. Lai, et al., Cell. Chem. Biol. 25 (2018) 67-77. doi: 10.1016/j.chembiol.2017.09.009
C. Maniaci, S.J. Hughes, A. Testa, et al., Nat. Commun. 8 (2017) 830. doi: 10.1038/s41467-017-00954-1
S. Maksoud, Mol. Neurobiol. 58 (2021) 3252-3269. doi: 10.1007/s12035-021-02339-4
J. Qi, L. Fan, A. Hussain, Cur. Opin. Oncol. 27 (2015) 172-176. doi: 10.1097/CCO.0000000000000178
Z. Liu, X. Hu, Q. Wang, et al., J. Med. Chem. 64 (2021) 2829-2848. doi: 10.1021/acs.jmedchem.0c02234
L. Bai, H. Zhou, R. Xu, et al., Cancer Cell 36 (2019) 498-511. doi: 10.1016/j.ccell.2019.10.002
W. Farnaby, M. Koegl, M.J. Roy, et al., Nat. Chem. Biol. 15 (2019) 672-680. doi: 10.1038/s41589-019-0294-6
M.J. Bond, L. Chu, D.A. Nalawansha, K. Li, C.M. Crews, ACS Central Sci. 6 (2020) 1367-1375. doi: 10.1021/acscentsci.0c00411
J. Wang, M. Zhang, S. Liu, et al., Chin. Chem. Lett. 34 (2023) 107732. https://doi.org/10.1016/j.cclet.2022.08.012 doi: 10.1016/j.cclet.2022.08.012
A. Poso, J. Med. Chem. 64 (2021) 10680-10681. doi: 10.1021/acs.jmedchem.1c01126
N.C. Payne, S. Maksoud, B.A. Tannous, R. Mazitschek, Cell. Chem. Biol. 29 (2022) 1333-1340. doi: 10.1016/j.chembiol.2022.05.003
C.E. Badr, C.C. da Hora, A.B. Kirov, et al., ACS Chem. Biol. 15 (2020) 1445-1454. doi: 10.1021/acschembio.0c00104
K.G. Coleman, C.M. Crews, Annual Rev. Cancer Biol. 2 (2018) 41-58. doi: 10.1146/annurev-cancerbio-030617-050430
K.A. Donovan, F.M. Ferguson, J.W. Bushman, et al., Cell 183 (2020) 1714-1731. doi: 10.1016/j.cell.2020.10.038
B.E. Smith, S.L. Wang, S. Jaime-Figueroa, et al., Nat. Commun. 10 (2019) 131. doi: 10.26506/pharmhist.61.3-4.0131
C. Pu, Y. Tong, Y. Liu, et al., Eur. J. Med. Chem. 236 (2022) 114321. doi: 10.1016/j.ejmech.2022.114321
B. Jiang, E.S. Wang, K.A. Donovan, et al., Angew. Chem. Int. Ed. 58 (2019) 6321-6326. doi: 10.1002/anie.201901336
B. Zhao, K. Burgess, Chem. Commun. 55 (2019) 2704-2707. doi: 10.1039/c9cc00163h
S. Su, Z. Yang, H. Gao, et al., J. Med. Chem. 62 (2019) 7575-7582. doi: 10.1021/acs.jmedchem.9b00871
A.C. Lai, M. Toure, D. Hellerschmied, et al., Angew. Chem. Int. Ed. 55 (2016) 807-810. doi: 10.1002/anie.201507634
Y. Li, Q. Meng, M. Yang, et al., Acta Pharm. Sin. B 9 (2019) 1113-1144. doi: 10.1016/j.apsb.2019.10.001
D.W. Bartlett, A.M. Gilbert, Chem. Soc. Rev. 51 (2022) 3477-3486. doi: 10.1039/d2cs00114d
S.D. Edmondson, B. Yang, C. Fallan, Bioorg. Med. Chem. Lett. 29 (2019) 1555-1564. doi: 10.1016/j.bmcl.2019.04.030
V.G. Klein, A.G. Bond, C. Craigon, R.S. Lokey, A. Ciulli, J. Med. Chem. 64 (2021) 18082-18101. doi: 10.1021/acs.jmedchem.1c01496
C. Cantrill, P. Chaturvedi, C. Rynn, et al., Drug. Discov. Today 25 (2020) 969-982. doi: 10.1016/j.drudis.2020.03.012
E.F. Douglass, C.J. Miller, G. Sparer, H. Shapiro, D.A. Spiegel, J. Am. Chem. Soc. 135 (2013) 6092-6099. doi: 10.1021/ja311795d
F.P. Rodriguez-Rivera, S.M. Levi, ACS Central Sci. 7 (2021) 1117-1125. doi: 10.1021/acscentsci.1c00389
D.W. Bartlett, A.M. Gilbert, J. Pharmacokinetic. Phar. 48 (2021) 149-163. doi: 10.1007/s10928-020-09722-z
K.M. Riching, S. Mahan, C.R. Corona, et al., ACS Chem. Biol. 13 (2018) 2758-2770. doi: 10.1021/acschembio.8b00692
X. Han, L. Zhao, W. Xiang, et al., J. Med. Chem. 62 (2019) 11218-11231. doi: 10.1021/acs.jmedchem.9b01393
H. Zhang, H.-.Y. Zhao, X.-.X. Xi, et al., Eur. J. Med. Chem. 189 (2020) 112061. doi: 10.1016/j.ejmech.2020.112061
B. Medvar, V. Raghuram, T. Pisitkun, A. Sarkar, M.A. Knepper, Physiol. Genomics 48 (2016) 502-512. doi: 10.1152/physiolgenomics.00031.2016
C. Wang, Y. Zhang, Y. Wu, D. Xing, Eur. J. Med. Chem. 225 (2021) 113749. doi: 10.1016/j.ejmech.2021.113749
A.B. Benowitz, K.L. Jones, J.D. Harling, Expert Opin. Ther. Pat. 31 (2021) 1-24. doi: 10.1080/13543776.2021.1840553
L. Li, D. Mi, H. Pei, et al., Signal. Transduct. Target. Ther. 5 (2020) 129. doi: 10.1038/s41392-020-00245-0
X. Zhang, V.M. Crowley, T.G. Wucherpfennig, M.M. Dix, B.F. Cravatt, Nat. Chem. Biol. 15 (2019) 737-746. doi: 10.1038/s41589-019-0279-5
C.C. Ward, J.I. Kleinman, S.M. Brittain, et al., ACS Chem. Biol. 14 (2019) 2430-2440. doi: 10.1021/acschembio.8b01083
J.N. Spradlin, X. Hu, C.C. Ward, et al., Nat. Chem. Biol. 15 (2019) 747-755. doi: 10.1038/s41589-019-0304-8
P.M. Cromm, C.M. Crews, Cell. Chem. Biol. 24 (2017) 1181-1190. doi: 10.1016/j.chembiol.2017.05.024
K. Garber, Nat. Biotechnol. 40 (2022) 12-16. doi: 10.1038/s41587-021-01173-2
A.C. Lai, C.M. Crews, Nat. Rev. Drug Discov. 16 (2017) 101-114. doi: 10.1038/nrd.2016.211
M.J. Bond, C.M. Crews, RSC Chem. Biol. 2 (2021) 725-742. doi: 10.1039/d1cb00011j
R.G. Guenette, S.W. Yang, J. Min, B. Pei, P.R. Potts, Chem. Soc. Rev. 51 (2022) 5740-5756. doi: 10.1039/d2cs00200k
M. Schapira, M.F. Calabrese, A.N. Bullock, C.M. Crews, Nat. Rev. Drug Discov. 18 (2019) 949-963. doi: 10.1038/s41573-019-0047-y
K. Li, C.M. Crews, Chem. Soc. Rev. 51 (2022) 5214-5236. doi: 10.1039/d2cs00193d
C. Wang, C. Zheng, H. Wang, et al., Eur. J. Med. Chem. 235 (2022) 114290. doi: 10.1016/j.ejmech.2022.114290
G.R. Hughes, A.P. Dudey, A.M. Hemmings, A. Chantry, Drug. Discov. Today 26 (2021) 2377-2383. doi: 10.1016/j.drudis.2021.04.010
C. Cecchini, S. Pannilunghi, S. Tardy, L. Scapozza, Front. Chem. 9 (2021) 672267. doi: 10.3389/fchem.2021.672267
A. Juan, M. Del Mar Noblejas-Lopez, M. Arenas-Moreira, C. Alonso-Moreno, A. Ocana, Front. Cell. Dev. Biol. 9 (2021) 805336.
S.-.L. Paiva, C.M. Crews, Cur. Opin. Chem. Biol. 50 (2019) 111-119. doi: 10.1016/j.cbpa.2019.02.022
M. Teng, J. Jiang, Z. He, et al., Angew. Chem. Int. Ed. 59 (2020) 13865-13870. doi: 10.1002/anie.202004087
D. Garcia Jimenez, M. Rossi Sebastiano, M. Vallaro, et al., J. Med. Chem. 65 (2022) 12639-12649. https://doi.org/10.1021/acs.jmedchem.2c00201 doi: 10.1021/acs.jmedchem.2c00201
R.P. Nowak, S.L. DeAngelo, D. Buckley, et al., Nat. Chem. Biol. 14 (2018) 706-714. doi: 10.1038/s41589-018-0055-y
Y. Dai, N. Yue, J. Gong, et al., Eur. J. Med. Chem. 187 (2020) 111967. doi: 10.1016/j.ejmech.2019.111967
R.I. Troup, C. Fallan, M.G.J. Baud, Explor. Targeted Anti-Tumor Ther. 1 (2020) 273-312.
M. Brand, B. Jiang, S. Bauer, et al., Cell. Chem. Biol. 26 (2019) 300-306. doi: 10.1016/j.chembiol.2018.11.006
S. Rana, M. Bendjennat, S. Kour, et al., Bioorg. Med. Chem. Lett. 29 (2019) 1375-1379. doi: 10.1016/j.bmcl.2019.03.035
C. Cao, J. Yang, Y. Chen, et al., J. Med. Chem. 63 (2020) 11012-11033. doi: 10.1021/acs.jmedchem.0c00821
R.R. Shah, J.M. Redmond, A. Mihut, et al., Bioorgan. Med. Chem. 28 (2020) 115326. doi: 10.1016/j.bmc.2020.115326
C.J. Gerry, S.L. Schreiber, Nat. Chem. Biol. 16 (2020) 369-378. doi: 10.1038/s41589-020-0469-1
T.A. Bemis, J.J. La Clair, M.D. Burkart, J. Med. Chem. 64 (2021) 8042-8052. doi: 10.1021/acs.jmedchem.1c00482
A. Testa, S.J. Hughes, X. Lucas, J.E. Wright, A. Ciulli, Angew. Chem. Int. Ed. 59 (2020) 1727-1734. doi: 10.1002/anie.201914396
E. Valeur, S.M. Guéret, H. Adihou, et al., Angew. Chem. Int. Ed. 56 (2017) 10294-10323. doi: 10.1002/anie.201611914
X. Han, C. Wang, C. Qin, et al., J. Med. Chem. 62 (2019) 941-964. doi: 10.1021/acs.jmedchem.8b01631
Y. Chen, X. Yuan, M. Tang, et al., Bioorgan. Chem. 119 (2022) 105508. doi: 10.1016/j.bioorg.2021.105508
L. Zhou, W. Chen, C. Cao, et al., Eur. J. Med. Chem. 189 (2020) 112028. doi: 10.1016/j.ejmech.2019.112028
H.C. Kolb, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 40 (2001) 2004-2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5
R.P. Wurz, K. Dellamaggiore, H. Dou, et al., J. Med. Chem. 61 (2018) 453-461. doi: 10.1021/acs.jmedchem.6b01781
W.C. Gao, K. Feng, J. Tian, et al., Chin. Chem. Lett. 34 (2023) 107587. https://doi.org/10.1016/j.cclet.2022.06.010 doi: 10.1016/j.cclet.2022.06.010
L.W. Xia, M.Y. Ba, W. Liu, et al., Future Med. Chem. 11 (2019) 2919-2973. doi: 10.4155/fmc-2019-0159
H. Yang, W. Lv, M. He, et al., Chem. Commun. 55 (2019) 14848-14851. doi: 10.1039/c9cc08509b
M.L. Blackman, M. Royzen, J.M. Fox, J. Am. Chem. Soc. 130 (2008) 13518-13519. doi: 10.1021/ja8053805
H. Lebraud, D.J. Wright, C.N. Johnson, T.D. Heightman, ACS Cent. Sci. 2 (2016) 927-934. doi: 10.1021/acscentsci.6b00280
H. Lebraud, T.D. Heightman, Essays Biochem. 61 (2017) 517-527. doi: 10.1042/EBC20170030
A. Yau, J. Lee, Y. Chen, Pharmaceutics 13 (2021) 155. doi: 10.3390/pharmaceutics13020155
D. Guimarães, A. Cavaco-Paulo, E. Nogueira, Int. J. Pharmaceut. 601 (2021) 120571. doi: 10.1016/j.ijpharm.2021.120571
M. Germain, F. Caputo, S. Metcalfe, et al., J. Control. Release 326 (2020) 164-171. doi: 10.1016/j.jconrel.2020.07.007
J. Chen, M. Qiu, F. Ma, et al., J. Control. Release 330 (2021) 1244-1249. doi: 10.1016/j.jconrel.2020.11.032
B.C. Doak, J. Zheng, D. Dobritzsch, J. Kihlberg, J. Med. Chem. 59 (2016) 2312-2327. doi: 10.1021/acs.jmedchem.5b01286
W.H. Guo, X. Qi, X. Yu, et al., Nat. Commun. 11 (2020) 4268. doi: 10.1038/s41467-020-17997-6
S. Krishnan, R.M. Miller, B. Tian, et al., J. Am. Chem. Soc. 136 (2014) 12624-12630. doi: 10.1021/ja505194w
Y. Cheng, A.-.T. Pham, T. Kato, et al., Chem. Sci. 12 (2021) 626-631. doi: 10.1039/d0sc05447j
C. Steinebach, Y.L.D. Ng, I. Sosič, et al., Chem. Sci. 11 (2020) 3474-3486. doi: 10.1039/d0sc00167h
D.A. Nalawansha, K. Li, J. Hines, C.M. Crews, J. Am. Chem. Soc. 144 (2022) 5594-5605. doi: 10.1021/jacs.2c00874
L.E. Evans, K. Jones, M.D. Cheeseman, Chem. Commun. 53 (2017) 5167-5170. doi: 10.1039/C7CC01376K
E. Shang, X. Wang, D. Wen, D.A. Greenberg, D.J. Wolgemuth, Dev. Dynam. 238 (2009) 908-917. doi: 10.1002/dvdy.21911
C.M. Robb, J.I. Contreras, S. Kour, et al., Chem. Commun. 53 (2017) 7577-7580. doi: 10.1039/C7CC03879H
X. Qiu, Y. Li, B. Yu, et al., Eur. J. Med. Chem. 211 (2021) 113091. doi: 10.1016/j.ejmech.2020.113091
R. Wang, C. Ascanelli, A. Abdelbaki, et al., Commun. Biol. 4 (2021) 640. doi: 10.1038/s42003-021-02158-2
C. Nieto-Jimenez, E.C. Morafraile, C. Alonso-Moreno, A. Ocana, Mol. Cancer 21 (2022) 67. doi: 10.1186/s12943-022-01535-7
A. Ambrosone, V. Marchesano, S. Carregal-Romero, et al., ACS Nano 10 (2016) 4828-4834. doi: 10.1021/acsnano.5b07817
R. Wen, L. Yang, S. Wu, D. Zhou, B. Jiang, Chin. Chem. Lett. 34 (2023) 107204. https://doi.org/10.1016/j.cclet.2022.02.010 doi: 10.1016/j.cclet.2022.02.010
S. Verma, D. Manna, ChemMedChem. 15 (2020) 1258-1261. doi: 10.1002/cmdc.202000249
J. Liu, J. Ma, Y. Liu, et al., Semin. Cancer Biol. 67 (2020) 171-179.
Z.W. Wang, Y. Liu, X. Zhu, Trends Cell Biol. 30 (2020) 749-751. doi: 10.1016/j.tcb.2020.08.003
G. Xue, K. Wang, D. Zhou, H. Zhong, Z. Pan, J. Am. Chem. Soc. 141 (2019) 18370-18374. doi: 10.1021/jacs.9b06422
L. Baumann, A.G. Beck-Sickinger, Angew. Chem. Int. Ed. 52 (2013) 9550-9553. doi: 10.1002/anie.201302242
Y. Naro, K. Darrah, A. Deiters, J. Am. Chem. Soc. 142 (2020) 2193-2197. doi: 10.1021/jacs.9b12718
C.S. Kounde, M.M. Shchepinova, C.N. Saunders, et al., Chem. Commun. 56 (2020) 5532-5535. doi: 10.1039/d0cc00523a
J. Liu, H. Chen, L. Ma, et al., Sci. Adv. 6 (2020) eaay5154. doi: 10.1126/sciadv.aay5154
D. Bléger, J. Schwarz, A.M. Brouwer, S. Hecht, J. Am. Chem. Soc. 134 (2012) 20597-20600. doi: 10.1021/ja310323y
M. Reynders, S. Matsuura Bryan, M. Bérouti, et al., Sci. Adv. 2365 (2021) 315-329. doi: 10.1007/978-1-0716-1665-9_17
Y.H. Jin, M.C. Lu, Y. Wang, et al., J. Med. Chem. 63 (2020) 4644-4654. doi: 10.1021/acs.jmedchem.9b02058
P. Pfaff, K.T.G. Samarasinghe, C.M. Crews, E.M. Carreira, ACS Central Sci. 5 (2019) 1682-1690. doi: 10.1021/acscentsci.9b00713
M. Fernandez, F. Javaid, V. Chudasama, Chem. Sci. 9 (2018) 790-810. doi: 10.1039/c7sc04004k
J.D. Seitz, J.G. Vineberg, E. Herlihy, et al., Bioorgan. Med. Chem. 23 (2015) 2187-2194. doi: 10.1016/j.bmc.2015.02.057
J. Liu, H. Chen, Y. Liu, et al., J. Am. Chem. Soc. 143 (2021) 7380-7387. doi: 10.1021/jacs.1c00451
H. Chen, J. Liu, H.U. Kaniskan, W. Wei, J. Jin, J. Med. Chem. 64 (2021) 12273-12285. doi: 10.1021/acs.jmedchem.1c00901
P. Hoppenz, S. Els-Heindl, A.G. Beck-Sickinger, Front. Chem. 8 (2020) 571. doi: 10.3389/fchem.2020.00571
L. Zhao, J. Zhao, K. Zhong, A. Tong, D. Jia, Signal Transduct. Target. Ther. 7 (2022) 113. doi: 10.1007/978-981-16-9515-5_9
J.A. Gramespacher, A.D. Cotton, P.W.W. Burroughs, I.B. Seiple, J.A. Wells, ACS Chem. Biol. 17 (2022) 1259-1268. doi: 10.1021/acschembio.2c00185
S.J. Keam, Drugs 80 (2020) 501-508. doi: 10.1007/s40265-020-01281-4
M.A. Maneiro, N. Forte, M.M. Shchepinova, et al., ACS Chem. Biol. 15 (2020) 1306-1312. doi: 10.1021/acschembio.0c00285
A.D. Cotton, D.P. Nguyen, J.A. Gramespacher, I.B. Seiple, J.A. Wells, J. Am. Chem. Soc. 143 (2021) 593-598. doi: 10.1021/jacs.0c10008
F. Martins, L. Sofiya, G.P. Sykiotis, et al., Nat. Rev. Clin. Oncol. 16 (2019) 563-580. doi: 10.1038/s41571-019-0218-0
C. Zhang, Z. Zeng, D. Cui, et al., Nat. Commun. 12 (2021) 2934. doi: 10.1038/s41467-021-23194-w
K.T.G. Samarasinghe, C.M. Crews, Cell. Chem. Biol. 28 (2021) 934-951. doi: 10.1016/j.chembiol.2021.04.011
J. Pei, G. Wang, L. Feng, et al., J. Med. Chem. 64 (2021) 3493-3507. doi: 10.1021/acs.jmedchem.0c01689
Y. Miao, Q. Gao, M. Mao, et al., Angew. Chem. Int. Ed. 60 (2021) 11267-11271. doi: 10.1002/anie.202102170
E.J. Hanan, J. Liang, X. Wang, et al., J. Med. Chem. 63 (2020) 11330-11361. doi: 10.1021/acs.jmedchem.0c00093
G. Ahn, S.M. Banik, C.L. Miller, et al., Nat. Chem. Biol. 17 (2021) 937-946. doi: 10.1038/s41589-021-00770-1
C.H. Ji, H.Y. Kim, M.J. Lee, et al., Nat. Commun. 13 (2022) 904.
J. Pei, X. Pan, A. Wang, et al., Chem. Commun. 57 (2021) 13194-13197. doi: 10.1039/d1cc04661f
G. Dong, Y. Wu, J. Cheng, et al., J. Med. Chem. 65 (2022) 7619-7628. doi: 10.1021/acs.jmedchem.1c02001
Z. Li, C. Wang, Z. Wang, et al., Nature 575 (2019) 203-209. doi: 10.1038/s41586-019-1722-1
P.-.H. Chen, Z. Hu, E. An, et al., ACS Chem. Biol. 16 (2021) 2808-2815. doi: 10.1021/acschembio.1c00693
R.B. Kargbo, ACS Med. Chem. Lett. 12 (2021) 1872-1873. doi: 10.1021/acsmedchemlett.1c00576
W.W. Wang, L.Y. Chen, J.M. Wozniak, et al., J. Am. Chem. Soc. 143 (2021) 16700-16708. doi: 10.1021/jacs.1c07850
S. Imaide, K.M. Riching, N. Makukhin, et al., Nat. Chem. Biol. 17 (2021) 1157-1167. doi: 10.1038/s41589-021-00878-4

Figure 1 Schematic diagram of PROTACs-mediated degradation of POI. E2, Ubiquitin-conjugating enzyme; Ub, Ubiquitin.

Figure 2 The degradation selectivity of the POI, which is affected by warhead (a), linker (b) and E3 ligase (c).

Figure 3 (a) The hook effect is related to PROTAC concentration, and which could influence on PROTAC-mediated ternary complex formation. (b) Schematic summary of PROTAC's advantages and disadvantages.

Figure 4 Successful case of linker optimization strategies. (a) Structure-based design of macrocyclic macroPROTAC-1 according to linker optimization. (b) Optimization evolution of ARV-110. (c) Evolution of ER degrading PROTACs leading to ARV-471.

Figure 5 Applications of "click chemistry" to design PROTACs. (a) Schematic representation of the mode of action of pre-clicked PROTACs (ⅰ) or in-cell formation of CLIPTACs (ⅱ). (b) The CuCAAC and IEDDA click reactions to generate BRD4-PROTACs (ⅰ) and JQ1-CLIPTAC (ⅱ), respectively.

Figure 6 (a) Schematic representation of the pre-fused PROTACs strategy. The pre-fused PROTAC was then encapsulated into LNPs and could escape from the endo/lysosome after the endocytosis, releasing the active PROTACs to degrade POI. (b) The ARV-771 was pre-fused with VHL and SARD279 was pre-fused with HSP70, and then encapsulated into 80-O14B LNPs.

Figure 7 Structures of reversible covalent PROTAC RC-1 and its action mechanism demonstration of catalytic degradation of targeted proteins. α-cyano-acrylamide group as an electrophilic group allows a reversible covalent bond between the PROTACs and a reactive Cys-belonging to the targeted protein. When the targeted protein is degraded, the reversible covalent PROTACs regenerated.

Figure 8 Schematic depiction of the PHOTACs strategy. (a) Under light irradiation: (ⅰ) a photo-removable group (the green circle) is cleaved from pc-PROTAC and release of an active PROTAC to degrade POI; (ⅱ) the molecules toggle between an inactive form (cis-) and an active form (trans-) upon irradiation. (b) Uncaging reaction of pc-PROTAC1and chemical structure of different photo-caged PROTACs. (c) The illustration of the activation of reversible cis-trans isomerization of photoswitchable PROTACs upon different irradiation wavelength.

Figure 9 (a) Schematic representation of the tumor-specific folate-caged PROTACs mode of action. Following the FOLR1-mediated entrance into cancer cells, the folate group (the green ball) is cleaved by endogenous hydrolase or GSH, releasing the active PROTAC to form ternary complex to degrade the POI. (b) Schematic illustration of the activation of folate-ARV-771 by endogenous hydrolase (ⅰ) and FA-S2-MS4048 by intracellular GSH (ⅱ).

Figure 10 Schematic diagram of the action mechanism of Ab-PROTACs (left) and AbTAC (right). Ab-PROTACs via part of antibody binding to a receptor, internalized them and release the active PROTACs, resulting in specific receptor-dependent protein degradation.

Figure 11 Schematic illustration of SPNpro-mediated IDO degradation for photo-immunometabolic cancer therapy. SPNpro provides a new combinational therapeutic modality that is synergizes phototherapeutic function with biomarker-activated protein degradation.

Figure 12 Schematic representation of LYTAC degrade extracellular proteins and transmembrane proteins via the lysosomal pathway (left). AUTAC/ATTEC small molecules can effectively promote the formation of POI-specific autophagosomes (e.g., LC3) to degrade pathogenic proteins by autophagy.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
 下载:
下载: