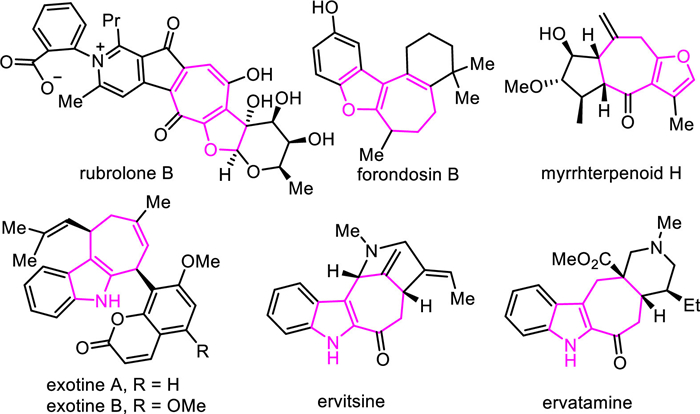
Figure 1.
Cycloheptafuran and cycloheptapyrrole scaffold alkaloids.
[4 + 3] Cycloaddition of ketenimines with furocarbenoids: Divergent and efficient synthesis of fused cycloheptatriene and tropone scaffolds
Jin Wang , Zhaoyang Li , Shengxin Bai , Qinghua Zhou , Tengteng Wu , Zhongyan Hu , Xianxiu Xu

Cycloheptafuran and cycloheptapyrrole scaffolds are widely found in many natural products with significant biological activities (Fig. 1). For example, rubrolone B has cardioprotective activity [1,2], frondosin B is served as an interleukin-8 receptor antagonist [3], myrrhterpenoid H possesses neuroprotective effects, and exotines A and B show inhibition of nitric oxide production [4,5]. While a number of synthetic methods for the construction of these fused frameworks have been documented [6-13], the development of highly efficient synthetic approaches is still desirable.
Ketenimines, a type of unique heterocumulenes, have been explored as versatile synthons for the synthesis of carbocycles and heterocycles via cycloaddition reactions (Scheme 1) [14,15]. In most cases, ketenimines served as a 2π component to undergo [2 + 2] [16-19], [3 + 2] [20-23] and [4 + 2] [24,25] cycloaddition reactions (Scheme 1a). Occasionally, vinyl, phenyl or furyl substituted ketenimines can act as 4π component in [4 + 2] [26-29] and [4 + 1] [30] cycloadditions, respectively (Scheme 1b). Additionally, difluoroketenimines acting as 1,3-dipoles was disclosed by Wang and coworkers [31,32]. Despite these well-developed reaction activity the higher-order cycloaddition of ketenimines remain unexplored.
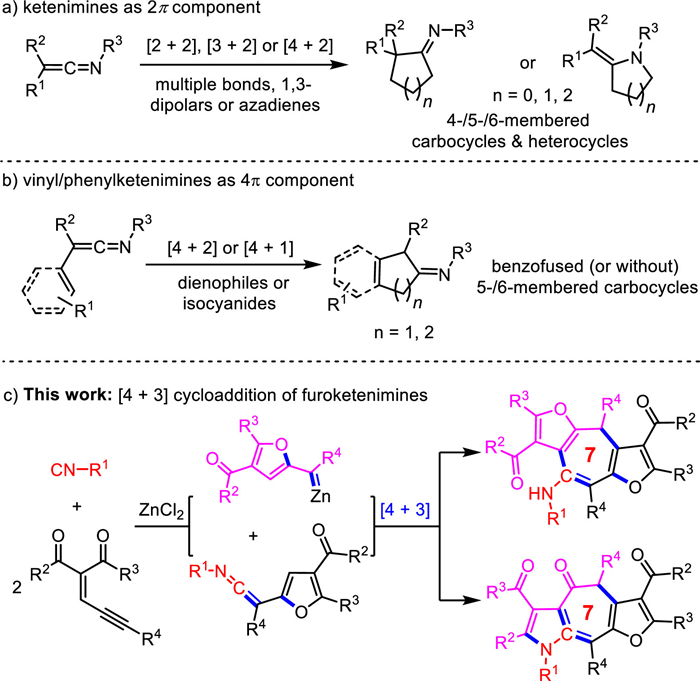
Isocyanide has been proven to be a useful precursor for the synthesis of ketenimines [14,15,33,34] via coupling reactions with carbenes [14,29,31,32,35] and allyl- or propargyl carbonates [36,37], as well as isocyanide-based multicomponent reactions [14,38,39]. Recently, the research groups of Li [30] and us [28] independently developed a convenient method to generate furyl-ketenimines in situ following with [4 + 1] and [4 + 2] cycloadditions by treatment of ene-yne-ketones with isocyanides. With this background and in continuation to our interest in isocyanide-based annulations [40-45], we report herein a [4 + 3] cycloaddition between the in situ generated furyl-ketenimines with zinc furyl-carbenoids, both of which are formed by a zinc promoted reaction of isocyanides with ene-yne-ketones (Scheme 1c). This transformation not only provides a novel protocol for the synthesis of fused cycloheptatriene and tropone scaffolds with highly efficiency, but also represents the first example of higher-order [4 + 3] cycloaddition of ketenimines with perfect atom economy.
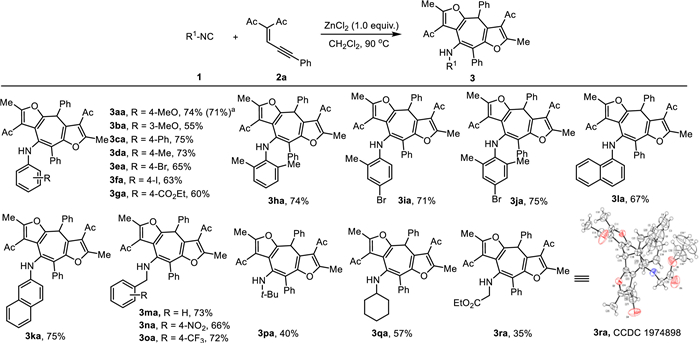
We began our investigation by treating aryl isocyanide 1a with ene-yne-ketone 2a (for more details, see Supporting information). To our delight, under ZnCl2 promoted conditions cycloheptafuran product 3aa was formed in 46% yield, while the cycloheptapyrrole 4aa was obtained in 24% yield (Table 1, entry 1). The structures of 3aa and 4aa were unambiguously assigned by X-ray crystallography (Fig. 2, CCDC Nos. 1974901 and 2018184). Fortunately, rising the reaction temperature to 90 ℃ resulted in improved 74% yield for 3aa along with only 8% of 4aa obtained (entry 2), while higher temperature showed adverse effect on 3aa formation (entry 3). Lowering the ZnCl2 loading from 1.0 equiv. to 0.8 equiv. dramatically affected the reaction efficiency (entry 4). Other zinc salts such as ZnBr2, ZnI2, Zn(OTf)2 and Zn(OAc)2 provided inferior results regarding the yield of 3aa (entries 5–8). This reaction only took place in halogenated solvents (entries 9 and 10); minimal amount of 3aa was formed in other reaction media (entries 11 and 12).

Next, we turned our attention to optimizing the formation of 4aa. Initial effort to convert 3aa directly to 4aa under previously optimized conditions in prolonged reaction time only provided 30% yield (entry 13). When oxidant m-CPBA (or HOAc, see Supporting information for details) was added the conversion and decomposition of 3aa was promoted (entry 14). A more detailed investigation into the reaction procedure revealed when 1.0 equiv. of m-CPBA was added after the reaction mixture was stirred for 10 min at 25 ℃, the desired product 4aa was obtained in 74% yield (entry 15). Finally, 82% yield of 4aa was obtained when 2.5 equiv. of 2a was used at 25 ℃ (entry 16). Herein, the optimal reaction conditions for 3aa and 4aa were determined as shown in entry 2 and entry 16, respectively.
With the optimized conditions in hand (Table 1, entry 2), we began to investigate the generality of this strategy for synthesis of cycloheptafuran 3 (Scheme 2). In general, isocyanide substrates 1 with both electron-donating and electron-withdrawing substituents on the phenyl ring are compatible with this reaction, providing the desired products 3aa−3ga with good yields. Gratifyingly, phenyl isocyanides with sterically hindered 2-methyl or 2,6-dimethyl substitution provided satisfactory yields (3ha−3ja). Naphthyl isocyanides could also be participated in this reaction to afford 3ka and 3la with good yields. In the case of alkyl isocyanides, benzyl isocyanides were converted into corresponding cycloheptafurans in good yields (3ma−3oa). The tert-butyl, cyclohexyl and active methylene isocyanide substrates could also be compatible in this method to afford 3pa−3ra with slightly lower yields. Notably, the structure of product 3ra was further confirmed by X-ray crystallography (CCDC: 1974898). It is worth mentioning that bromo, iodo and ester groups were tolerated in this reaction, thus providing a useful handle for further modification. Particularly, a gram-scale synthesis of 3aa (0.989 g, 71% yield) was carried out, demonstrating the practicability of this protocol.
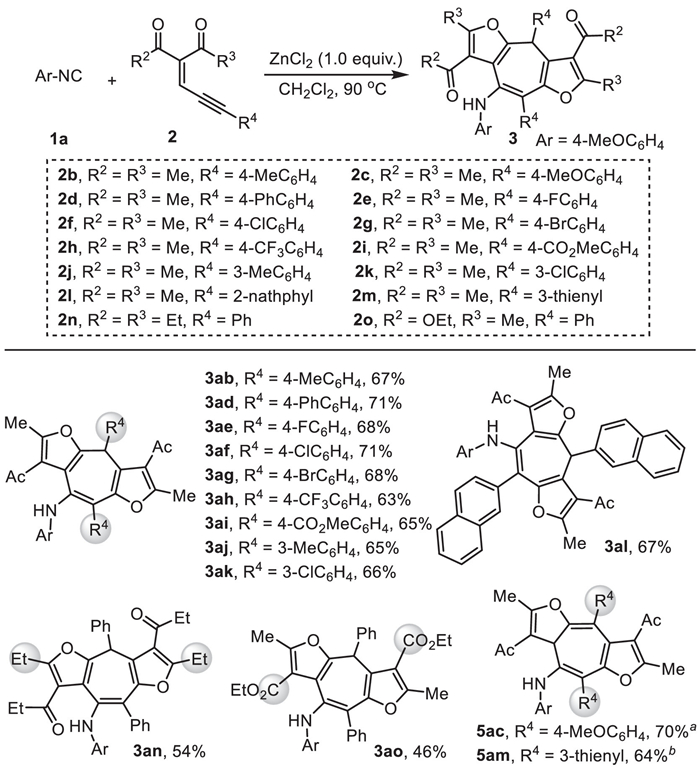
Next, the scope of the ene-yne-ketones 2 was tested (Scheme 3). For acetylacetone derived substrates 2a−2m (R2 = R3 = Me), when aryl groups were adopted at R4 position, including electron-rich (2b and 2j), electron-neutral (2a and 2d), and electron-deficient phenyl groups (2e−2i and 2k), as well as naphthyl group (2l), those yield furo-fused cycloheptatriene products 3ab, 3ad−3al in good result. However, substrates 2c with a 4-methoxylphenyl R4 group and 2m with a 3-thienyl R4 group led to a complex mixture, respectively. Notably, when the reaction temperature was decreased to 0 ℃ for a few minutes, compounds 5ac and 5am, the isomers of 3, were obtained in good yields. In addition, ene-yne-ketone substrates 2n and 2o derived from heptane-3,5-dione and ethyl acetoacetate underwent this transformation smoothly, giving the corresponding products 3an and 3ao in moderate yields. Disappointingly, ene-yne-ketone with alkyl group at R4 position failed to yield desired product 3. It is probably because the alkyl substituted ene-yne-ketone led to a much unstable alkyl substituted zinc furo-carbenoids.
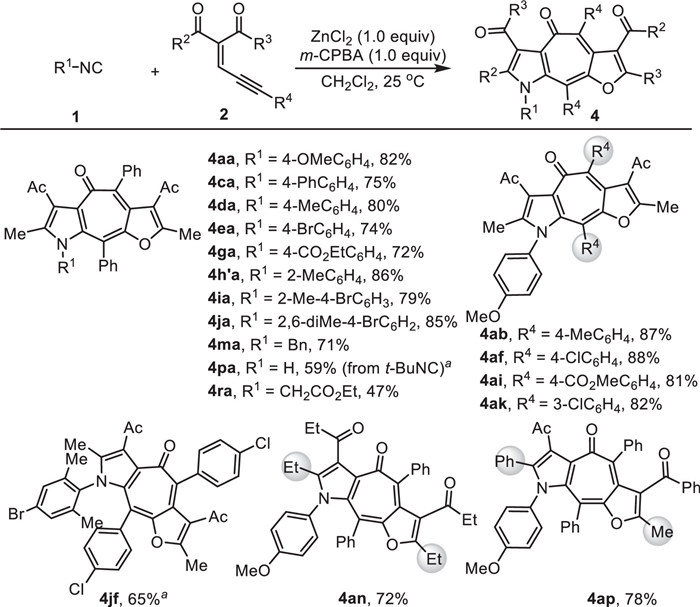
To explore the applicability of this methodology for synthesis of cycloheptapyrrole 4, the scope of both isocyanides 1 and ene-yne-ketones 2 was tested and summarized in Scheme 4. In terms of phenyl isocyanides, various substituent groups on the aromatic ring were tolerated to afford the furo-fused cycloheptapyrrole products 4aa−4ja, 4jf with good yields. The cycloheptapyrrole products 4ma (71%) and 4ra (47%) could also be obtained by using active methylene isocyanide 1m and 1r, respectively. However, when treated with tert-butyl isocyanide 1p, the corresponding product 4pa was obtained with eliminating the tert-butyl group. In general, sterically hindered isocyanide required longer reaction time and excess of ZnCl2 were necessary, suggesting that the formation of the pyrrole ring in product 4 may be the rate-determining step (4ja, 4pa and 4jf). The ene-yne-ketones 2 containing both electron-donating and electron-withdrawing phenyl group on R4 position were all suitable for this reaction, providing 4ab−4ak in high yields. Ene-yne-ketone substrates 2n and 2p derived from other 1,3-diketones underwent this reaction smoothly, and desired products (4an and 4ap) were obtained in good yields. However, ene-yne-ketone 2o derived from β-ketoester was not viable in this reaction.
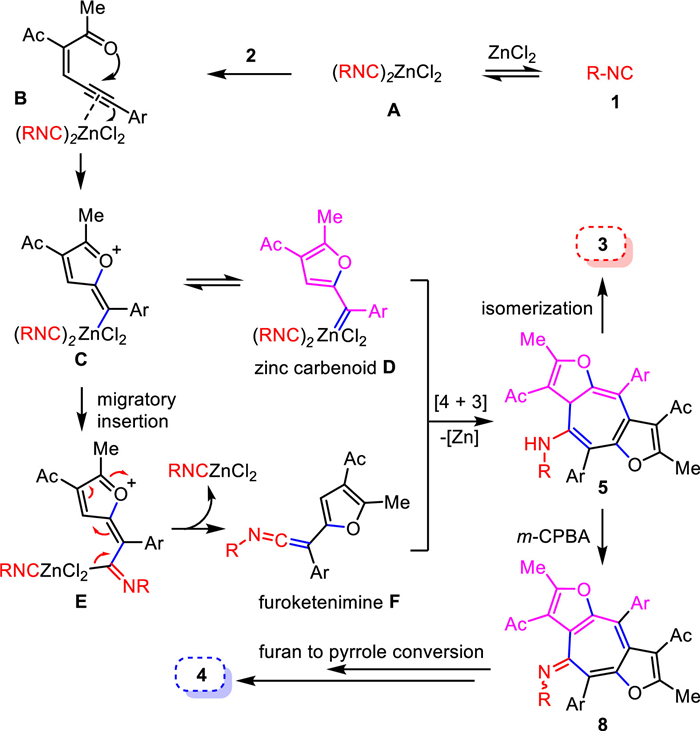
To elucidate the mechanism, several control experiments were carried out as shown in Scheme 5. Cyclopropane derivative 6 was isolated in 85% yield when treating ene-yne-ketone 2a with 4-methoxystyrene under the zinc-promoted reaction conditions, suggesting the formation of a zinc furyl-carbenoid intermediate during the reaction process (Scheme 5a) [46-51]. Presumably, zinc carbenoid can react with isocyanide to form ketenimine [30]. Thus, we synthesized furyl-ketenimine 7 under the known Pd-catalysis conditions [30] and employed it to react with ene-yne-ketone 2a under our zinc-promoted conditions. Gratefully, the desired cycloheptafuran 3ja was obtained in good yield (Scheme 5b). This evidence supports the furyl-ketenimine intermediate is involved in the mechanism. Interestingly, when treating 1a and 2a in the presence of ZnCl2 at 0 ℃, a cyclized product 5aa was obtained in excellent yield. This thermodynamically labile intermediate 5aa was readily isomerized to 3aa upon heating (Scheme 5c). These results suggested that the [4 + 3] cycloaddition likely gave rise to compound 5 firstly, which served as a key intermediate to provide cycloheptafuran 3 by isomerization.

In addition, we found a 4-bromo-2-methylphenyl isocyanide derived 5ia intermediate, formed under similar zinc-promoted reaction conditions, can be oxidized rapidly by m-CPBA at 0 ℃ to provide imine 8ia, which could be isolated and further converted to cycloheptapyrrole 4ia after reacting at 25 ℃ for 24 h. Moreover, imine 8ia could be isolated in excellent yield when reacting 1i and 2a under zinc-promoted oxidative conditions for a shorter time (Scheme 5d). Notably, both structures of the key intermediate 5aa (CCDC: 1974903) and 8ia (CCDC: 2047884) have been confirmed by X-ray crystallography. These results suggested that the [4 + 3] cycloaddition likely gave rise to compound 5 first, which served as a common intermediate to either provide cycloheptafuran 3 by isomerization or form cycloheptapyrrole 4 by an m-CPBA oxidation-rearrangement sequence (see Supporting information for more details). ZnCl2 played multiple roles in this reaction. First, it initiated the intramolecular cyclization to form the zinc carbenoid; second, it promoted the [4 + 3] cycloaddition and subsequent isomerization to form furan product 3; third, it accelerated m-CPBA oxidation to yield imine 8 and triggered rearrangement to form pyrrole product 4. Thus, the synergistic and benign effect of a single zinc accelerator is essential for this methodology. Lastly, product 3ma can undergo hydrogenolysis to provide cycloheptatriene amine 9, which can serve as a useful intermediate to access diverse substitutions on the nitrogen (Scheme 5e).
Based on these preliminary studies and literatures [31,32,46,52], we proposed a tentative mechanism (Scheme 6). First, Coordination of isonitrile 1 with zinc formed complex A [46], which upon activation of alkyne moiety of 2 afforded furyl-carbenoid intermediate C or D [47-52]. Isocyanide migratory insertion from intermediate C or D occurred to form furyl-ketenimine F [30]. Then, ketenimine intermediate F participated in a formal [4 + 3] cycloaddition with another zinc furyl-carbenoid D to yield the furo-fused cycloheptatriene 5 [53]. This process was rather fast and could be completed at 0 ℃. At last, isomerization of 5 provided the thermally stable furo-fused cycloheptatriene product 3. While addition of m-CPBA in the presence of ZnCl2 caused the formation of imine 8, which eventually led to the cycloheptapyrrole product 4 via a ZnCl2 promoted rearrangement (for more details, see Supporting information).
In summary, we have developed an efficient and divergent approach to access cycloheptafuran and cycloheptapyrrole scaffolds, respectively. A novel [4 + 3] cycloaddition process between the in situ generated furyl-ketenimine and zinc furyl-carbenoid is supported by mechanistic investigation. This reaction utilizes readily available starting materials and inexpensive metal additive. Three rings and five bonds are successively constructed, which exemplified the concise and green aspect of this multicomponent domino reaction. Further investigation of this [4 + 3] cycloaddition in the synthesis of structurally relevant natural products and pharmaceuticals is currently under investigation in our group.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors gratefully acknowledge financial support from National Natural Science Foundation of China (Nos. 22171168, 22001152 and 22101159), Taishan Scholar Program of Shandong Province and the Natural Science Foundation of Shandong Province of China (No. ZR2020QB019).
Y. Yan, Y.T. Ma, J. Yan, et al., Org. Lett. 18 (2016) 1254–1257. doi: 10.1021/acs.orglett.6b00074
Y. Yan, J. Yang, Z. Yu, et al., Nat. Commun. 7 (2016) 13083. doi: 10.1038/ncomms13083
A.D. Patil, A.J. Freyer, L. Killmer, et al., Tetrahedron 53 (1997) 5047–5060. doi: 10.1016/S0040-4020(97)00205-6
J. Xu, Y. Guo, Y. Li, et al., Planta Med. 77 (2011) 2023–2028. doi: 10.1055/s-0031-1280087
B.Y. Liu, C. Zhang, K.W. Zeng, et al., Org. Lett. 17 (2015) 4380–4383. doi: 10.1021/acs.orglett.5b02230
M. Inoue, M.W. Carson, A.J. Frontier, S.J. Danishefsky, J. Am. Chem. Soc. 123 (2001) 1878–1889. doi: 10.1021/ja0021060
Z. Dong, C.H. Liu, Y. Wang, M. Lin, Z.X. Yu, Angew. Chem. Int. Ed. 52 (2013) 14157–14161. doi: 10.1002/anie.201306965
X. Han, H. Li, R.P. Hughes, J. Wu, Angew. Chem. Int. Ed. 51 (2012) 10390–10393. doi: 10.1002/anie.201205238
C. Gelis, G. Levitre, J. Merad, et al., Angew. Chem. Int. Ed. 57 (2018) 12121–12125. doi: 10.1002/anie.201807069
H. Zhang, T. Cao, H. Luo, L. Chen, S. Zhu, Org. Chem. Front. 6 (2019) 1118–1122. doi: 10.1039/C9QO00045C
X. Xu, J. Chen, J. Ke, et al., Chin. J. Org. Chem. 41 (2021) 206–228. doi: 10.6023/cjoc202005018
H. Luo, T. Cao, S. Zhu, Chin. J. Org. Chem. 41 (2021) 3521–3531. doi: 10.6023/cjoc202105021
Z. Qu, T. Tian, G.J. Deng, H. Huang, Chin. Chem. Lett. 34 (2023) 107565. doi: 10.1016/j.cclet.2022.05.079
P. Lu, Y. Wang, Chem. Soc. Rev. 41 (2012) 5687–5705. doi: 10.1039/c2cs35159e
M. Alajarin, M. Marin-Luna, A. Vidal, Eur. J. Org. Chem. 2012 (2012) 5637–5653. doi: 10.1002/ejoc.201200383
W. Yao, L. Pan, Y. Zhang, et al., Angew. Chem. Int. Ed. 49 (2010) 9210–9214. doi: 10.1002/anie.201004685
Y. Xing, H. Zhao, Q. Shang, et al., Org. Lett. 15 (2013) 2668–2671. doi: 10.1021/ol4010323
E. Romero, C. Minard, M. Benchekroun, et al., Chem. Eur. J. 23 (2017) 12991–12994. doi: 10.1002/chem.201702545
S. Guo, P. Dong, Y. Chen, X. Feng, X. Liu, Angew. Chem. Int. Ed. 57 (2018) 16852–16856. doi: 10.1002/anie.201810679
M. Alajarin, B. Bonillo, M.M. Ortin, R.A. Orenes, A. Vidal, Org. Biomol. Chem. 9 (2011) 6741–6749. doi: 10.1039/c1ob05745f
S. Li, Y. Luo, J. Wu, Org. Lett. 13 (2011) 4312–4315. doi: 10.1021/ol201653j
K. Namitharan, K. Pitchumani, Org. Lett. 13 (2011) 5728–5731. doi: 10.1021/ol202164x
J. Cai, H. Bai, Y. Wang, et al., Chem. Commun. 55 (2019) 3821–3824. doi: 10.1039/C9CC01257E
W. Lu, W. Song, D. Hong, P. Lu, Y. Wang, Adv. Synth. Catal. 351 (2009) 1768–1772. doi: 10.1002/adsc.200900260
D. Coffinier, L. El Kaim, L. Grimaud, E. Ruijter, R.V.A. Orru, Tetrahedron Lett. 52 (2011) 3023–3025. doi: 10.1016/j.tetlet.2011.04.007
J.L. Alonso-Gómez, Y. Pazos, A. Navarro-Vázquez, J. Lugtenburg, M.M. Cid, Org. Lett. 7 (2005) 3773–3776. doi: 10.1021/ol051433f
M. Alajarin, B. Bonillo, M. Marin-Luna, A. Vidal, R.A. Orenes, J. Org. Chem. 74 (2009) 3558–3561. doi: 10.1021/jo900304a
Z. Hu, J. Dong, Z. Li, et al., Org. Lett. 20 (2018) 6750–6754. doi: 10.1021/acs.orglett.8b02870
L. Zhang, T. Liu, Y.M. Wang, J. Chen, Y.L. Zhao, Org. Lett. 21 (2019) 2973–2977. doi: 10.1021/acs.orglett.9b00307
J. Huang, F. Li, L. Cui, et al., Chem. Commun. 56 (2020) 4555–4558. doi: 10.1039/C9CC09363J
R. Zhang, Z. Zhang, Q. Zhou, L. Yu, J. Wang, Angew. Chem. Int. Ed. 58 (2019) 5744–5748. doi: 10.1002/anie.201901591
R. Zhang, Z. Zhang, K. Wang, J. Wang, J. Org. Chem. 85 (2020) 9791–9800. doi: 10.1021/acs.joc.0c01120
T. Vlaar, B.U.W. Maes, E. Ruijter, R.V.A. Orru, Angew. Chem. Int. Ed. 52 (2013) 7084–7097. doi: 10.1002/anie.201300942
I. Yavari, S. Arab-Salmanabadi, A. Aminkhani, Chin. Chem. Lett. 23 (2012) 49–52. doi: 10.1016/j.cclet.2011.10.006
X. Yan, J. Liao, Y. Lu, et al., Org. Lett. 15 (2013) 2478–2481. doi: 10.1021/ol4009552
D. Coffinier, L. El Kaim, L. Grimaud, Org. Lett. 11 (2009) 1825–1827. doi: 10.1021/ol9004432
Q. Yang, C. Li, M.X. Cheng, S.D. Yang, ACS Catal. 6 (2016) 4715–4719. doi: 10.1021/acscatal.6b01253
G. Qiu, M. Mamboury, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 55 (2016) 15377–15381. doi: 10.1002/anie.201609034
G. Qiu, Q. Wang, J. Zhu, Org. Lett. 19 (2017) 270–273. doi: 10.1021/acs.orglett.6b03592
J. Tan, X. Xu, L. Zhang, Y. Li, Q. Liu, Angew. Chem. Int. Ed. 48 (2009) 2868–2872. doi: 10.1002/anie.200805703
Y. Li, X. Xu, J. Tan, et al., J. Am. Chem. Soc. 133 (2011) 1775–1777. doi: 10.1021/ja110864t
X. Xu, L. Zhang, X. Liu, L. Pan, Q. Liu, Angew. Chem. Int. Ed. 52 (2013) 9271–9274. doi: 10.1002/anie.201303604
Z. Hu, H. Yuan, Y. Men, et al., Angew. Chem. Int. Ed. 55 (2016) 7077–7080. doi: 10.1002/anie.201600257
Z. Hu, J. Dong, Y. Men, et al., Angew. Chem. Int. Ed. 56 (2017) 1805–1809. doi: 10.1002/anie.201611024
J. Dong, L. Bao, Z. Hu, et al., Org. Lett. 20 (2018) 1244–1247. doi: 10.1021/acs.orglett.8b00186
Y. Odabachian, S. Tong, Q. Wang, M.X. Wang, J. Zhu, Angew. Chem. Int. Ed. 52 (2013) 10878–10882. doi: 10.1002/anie.201305506
R. Vicente, J. Gonzalez, L. Riesgo, J. Gonzalez, L.A. Lopez, Angew. Chem. Int. Ed. 51 (2012) 8063–8067. doi: 10.1002/anie.201203914
J. Gonzalez, J. Gonzalez, C. Perez-Calleja, L.A. Lopez, R. Vicente, Angew. Chem. Int. Ed. 52 (2013) 5853–5857. doi: 10.1002/anie.201301625
D. Zhu, J. Ma, K. Luo, et al., Angew. Chem. Int. Ed. 55 (2016) 8452–8456. doi: 10.1002/anie.201604211
J. Ma, L. Zhang, S. Zhu, Curr. Org. Chem. 20 (2016) 102–118.
W. Chen, D.S. Ji, Y.C. Luo, Z.Y. Wang, P.F. Xu, Org. Chem. Front. 5 (2018) 1768–1771. doi: 10.1039/C8QO00232K
L. Chen, K. Chen, S. Zhu, Chem 4 (2018) 1208–1262. doi: 10.1016/j.chempr.2018.02.001
B. Song, L.H. Li, X.R. Song, et al., Chem. Eur. J. 20 (2014) 5910–5913. doi: 10.1002/chem.201402513

Scheme 2 Scope of isocyanides 1. Reaction conditions: ZnCl2 (1.0 equiv.) was added to a solution of isocyanide 1 (0.3 mmol) and ene-yne-ketone 2 (0.6 mmol) in CH2Cl2 (2 mL) at 90 ℃ in an air atmosphere for 1.5 h. Isolated yields. a Gram scale synthesis, 0.989 g 3aa was obtained.

Scheme 3 Scope of ene-yne-ketones 2. Reaction conditions: ZnCl2 (1.0 equiv.), 1a (0.3 mmol) and ene-yne-ketone 2 (0.6 mmol) in CH2Cl2 (2 mL) at 90 ℃ in an air atmosphere for 1.5 h. Isolated yields. a At 0 ℃ for 10 min. b At 0 ℃ for 15 min.

Scheme 4 Synthesis of cycloheptapyrroles 4. Reaction conditions: unless otherwise noted, ZnCl2 (1.0 equiv.) was added to a solution of isocyanide 1 (0.3 mmol) and ene-yne-ketone 2 (0.75 mmol) in CH2Cl2 (2 mL), and the reaction mixture was kept at 25 ℃ in an air atmosphere for 10 min, then m-CPBA (1.0 equiv.) was added. The reaction mixture was stirred for another 18 h. Isolated yields. a ZnCl2 (1.5 equiv.) was used.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:




 下载:
下载: