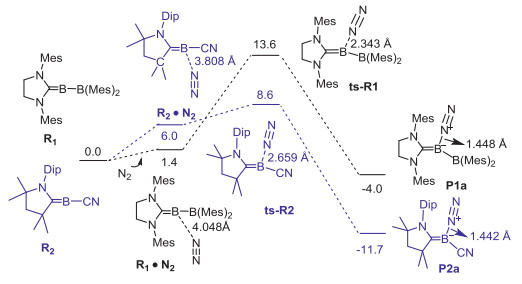
Figure 1.
The Gibbs energy (kcal/mol) profile for the N2 activation reactions by R1 and R2. The B-N bond lengths are given in Å.
Understanding reaction mechanisms of metal-free dinitrogen activation by methyleneboranes
Jie Zeng , Jiaying Su , Feiying You , Jun Zhu

Activating inert dinitrogen (N2), the major component in the air, has attracted considerable attention from both synthetic and theoretical scientists due to its significant challenge [1]. In nature, nitrogenase could be used to activate N2 under ambient temperature and pressure [2]. In 1905, the Haber–Bosch process for dinitrogen fixation has been developed into a feasible fashion on an industrial scale. However, its shortcomings are obvious, including its high energy consumption, environmental pollution, etc. [3]. This calls for the urgent search for alternative methods to activate N2. Due to the donation and backdonation character, transition metals and their complexes are commonly used for dinitrogen activation [4-8].
Unlike transition metals, it was assumed to be difficult for main-group species to activate N2 due to the lack of d orbital. In 2018, Légaré et al. reported that the transient borylene could be successfully used to activate N2 [9]. Subsequently, the dimerization of two nitrene-like radicals led to the formation of a N4 chain via a reductive coupling reaction [10]. These works represented an important breakthrough for N2 activation by p-block elements, bringing new light on N2 reduction [11, 12]. Later, a one-pot reactions of N2 to ammonia was achieved [13, 14]. Moreover, beryllium or boron atom could help to mediate the N2 activation supported by infrared spectra [15, 16]. Very recently, low-valent calcium species was serendipitously discovered for the activation of N2 [17]. In addition, fluoroborylene and phenylborylene were reported to successfully break the N≡N triple bond [18, 19]. Theoretically, two push–pull interactions were found in end-on bridging borylene-N2 complexes which could enhance the N2 activation [20]. Our group have systematically designed a series of carbon-boron frustrated Lewis pairs (FLPs) [21-25] as well as borenium and borinium cations [26] to activate N2 in a thermodynamically and kinetically favorable fashion. In addition, a phosphorus pincer complex was predicted for dinitrogen coupling with a series of small molecules by DFT calculations [27]. The potential of base-stabilized borylenes in dinitrogen activation was probed by DFT calculations in comparison with the intramolecular C—H bond activation [28, 29]. Interestingly, boron radicals [30] have been also reported to be a potential reagent for the activation of N2. Moreover, Xi and co-workers have summarized N2 fixation by carbene species [31].
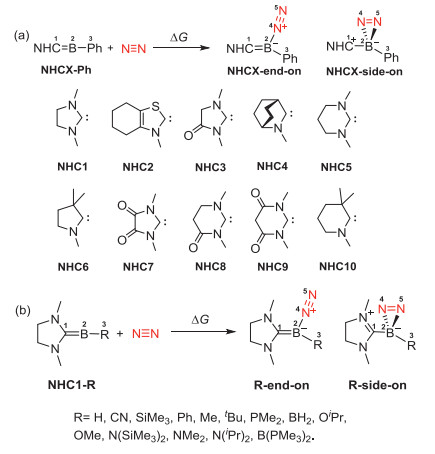
Although much work has been reported, some questions on the reaction mechanisms for N2 activation by methyleneboranes (previously called base-stabilized borylenes) remain unclear. Could other experimentally reported [32-37] methyleneboranes be used to activate dinitrogen? If so, could the moiety of N-heterocyclic carbenes (NHCs) on the methyleneboranes affect the reactions significantly? What is the substituent effect on these reactions? Which factor determines the reaction energy of the dinitrogen activation? Here we perform density functional theory (DFT) calculations to address these issues.
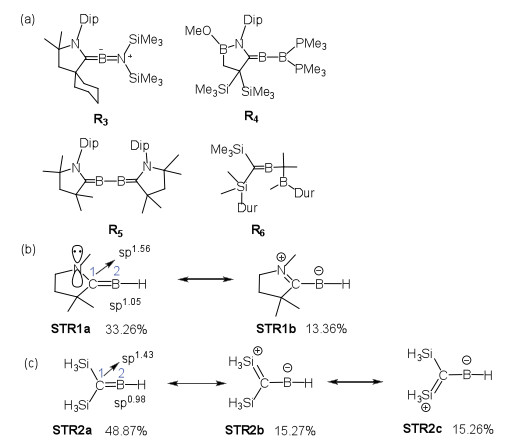
As shown in Fig. 1, R1 and R2 were proposed as intermediates in reduction reactions [32, 33]. Interestingly, the Gibbs energies of reactions to form P1a (−4.0 kcal/mol) and P2a (−11.7 kcal/mol) are exoergic. The corresponding reaction barriers are 13.6 and 8.6 kcal/mol, respectively, suggesting great potential of these methyleneboranes to activate N2. Other experimentally isolated compounds [34-37] failed to activate N2 (Fig. S1b in Supporting information). The structures of R3-R6 are shown in Fig. 2a. Fuzzy bond order (FBO) [38] and natural resonance theory (NRT) [39] were used to analyze the simplified compound (STR1) from R2-R5 (Fig. 2b). The bond length and FBO value of the C1-B2 bond of STR1 are 1.399 Å and 1.71, respectively, indicating a double bond between these two atoms. The major resonance structure (STR1a, 33.26%) of the simplified compound has a hybridization of sp1.56 and sp1.05 for the C1 and B2 atoms, respectively. In addition, R6 was labeled as methyleneborane by Pilz et al. in 1989 [37]. The simplified compound (STR2) from R6 is shown in Fig. 2c, and the bond length and FBO value of the C1-B2 bond of STR2 are 1.364 Å and 1.70, similar to STR1 in Fig. 2b. Again, its major resonance structure (STR2a, 48.87%) has a formal C=B double bond. As the definition of borylene by International Union of Pure and Applied Chemistry (IUPAC) was stated as "the species RB: containing an electrically neutral univalent boron atom with two formally non-bonding electrons", the compounds R2-R5 should be more suitably termed as methyleneboranes rather than borylenes due to the lack of two formally non-bonding electrons in these species.
The influence of NHCs in methyleneboranes for N2 activation was explored. As shown in Fig. 3a and Table 1, from NHC1-Ph to NHC10-Ph, the ΔG values for the formation of the end-on products are gradually increased. Only NHC1-Ph, NHC2-Ph and NHC5-Ph could be used for N2 activation to form a side-on product in a thermodynamically favorable fashion. The cyclic diamino carbenes (CDACs) based methyleneboranes (−12.1 kcal/mol of NHC1-Ph and −4.7 kcal/mol of NHC5-Ph) display higher reactivity than that of cyclic alkyl amino carbenes (CAACs, −2.5 kcal/mol of NHC6-Ph and 8.1 kcal/mol of NHC10-Ph) in activating N2. The two nitrogen atoms in CDAC could donate their lone pair electrons to the empty p orbital of carbene carbon atom, which competes the donation from the boron atom and thus might reduce the strength of the C=B bond. Indeed, the FBO values of the C=B bond of NHC1-Ph and NHC5-Ph are both 1.58, smaller than that of NHC6-Ph (1.67) and NHC10-Ph (1.65).
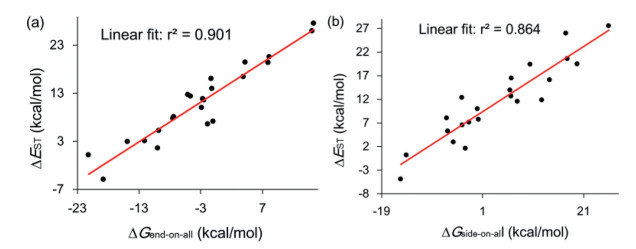
 DownLoad:
CSV
DownLoad:
CSV

|
We choose the NHC1 based methyleneborane which has the best thermodynamic performance in N2 activation (Table 1) to explore the substituent effects. As shown in Fig. 3b and Table 2, the ΔG values for the formation of the end-on products are gradually increased from the substituent H to B(PMe3)2. More geometric details are shown in Tables S1 and S2 (Supporting information). Strong correlations for the singlet–triplet energy gap (ΔEST) values against all end-on products (ΔGend-on-all, r2 = 0.901) and side-on products (ΔGside-on-all, r2 = 0.864) could be identified (Fig. 4). The smaller ΔEST values suggest higher reactivity of the reactants, resulting in a thermodynamically favorable activation of N2 by methyleneboranes. NHC1-H displays the lowest ΔG value, indicating great potential to activate N2 in this work. When the methyleneboranes are substituted at the boron atom by PMe2, OMe, OiPr, NMe2, N(SiMe3)2, N(iPr)2 and B(PMe3)2 groups, the reactions become less favorable or even unfavorable thermodynamically. The principal interacting orbital (PIO) analysis [40, 41] were performed to explain (Fig. S2 in Supporting information). PIO analysis can be used to identify and quantify the main orbital interactions between two fragments. For NHC1-H, the only one PIO pair was located for a B-H σ bond. For NHC1-OiPr and NHC1-B(PMe3)2 besides σ bond interactions of 1st PIO pair, the 2nd PIO pair corresponds to electron donation from the O/B2 atom to an empty p orbital of B1 atom, with a PBI value of 0.39 and 0.66, respectively, which could help to stabilize the reactants, leading to the reduction on the reaction energies in their N2 activation. For NHC1-BH2, the 2nd PIO pair corresponds to electron donation from the B=C double bond to the p orbital of the B2 atom with a charge transfer of 0.39e, which also helps to stabilize the reactant.
DownLoad:
CSV

|
Then, we examined the kinetics of reactions when methyleneboranes with H and OiPr groups at the boron atom, because these reactions have the lowest and highest ΔGR-end-on values in exergonic reactions (Fig. 5). The reaction barriers via ts-OiPr-side-on, ts-OiPr-end-on, ts-H-side-on and ts-H-end-on are 20.9, 15.6, 13.1 and 8.0 kcal/mol, respectively, indicating that these reactions are favorable both thermodynamically and kinetically. Note that the intramolecular C−H bond activation could compete with N2 activation (Fig. S3 in Supporting information).
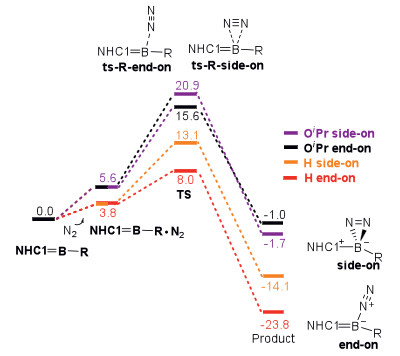
To probe the origin of the difference on reaction barriers of N2 activation, the energy decomposition analysis (EDA) [42, 43] based on the distortion/interaction and activation strain models is performed (Fig. S4 and Table S3 in Supporting information). In general, the reactants deformations (ΔEDeform (N2) and ΔEDeform (methyleneborane)) are positive and destabilize the transition states. On the contrary, the binding energy (ΔEb) is negative and stabilizes transition states. The expression of reaction barrier can be shown as:
|
|
As shown in Table S3, the deformation of methyleneborane and the binding energy are the major factors for the difference of the reaction barriers.
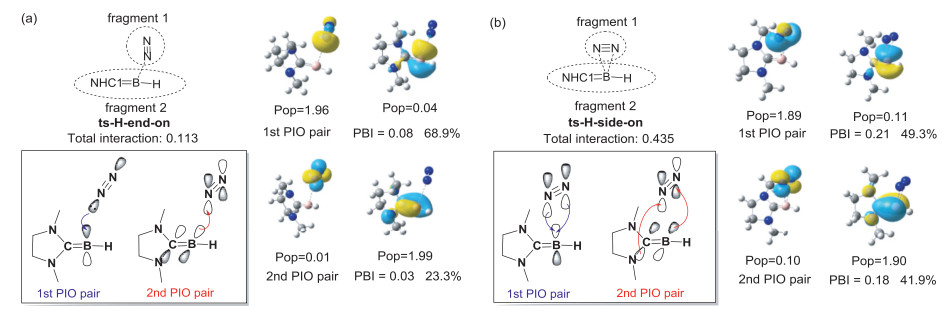
We performed PIO analysis on transition states to further understand the reaction mechanisms, and the diagrams for orbital interaction are shown in black box (Fig. 6). For ts-H-end-on, the first PIO pair shows that N2 donates the lone pair electrons to the empty p orbitals of B with a PBI value of 0.08 only. For ts-H-side-on, the first PIO pair corresponds to electron donation from the N2 to an empty p orbital of the B with a PBI value of 0.21. The dominant two PIO pairs contribute 91.2% to the overall fragment interactions. The second PIO pair of ts-H-end-on and ts-H-side-on corresponds to electron donation from the B=C double bond to the π* orbital of N2 with charge transfer of 0.01e and 0.10e, respectively. The total interaction of ts-H-side-on (0.435) is larger than that of ts-H-end-on (0.113), suggesting the stronger interaction between two fragments of ts-H-side-on. Mimicking transition metals, the B atom in methyleneborane uses filled and vacant p orbitals to break the N≡N triple bond.
The reductive coupling reactions of dinitrogen were reported experimentally [9, 10]. As shown in Fig. S5a (Supporting information), NHC1-H dimerizes to form a cis product D1 (reaction 1, −86.3 kcal/mol), which is less favorable than that of the trans product D2 (reaction 2, −89.3 kcal/mol). The ΔG value of reaction 1 is slightly negative than that of reaction 3 (−72.1 kcal/mol). The reactions shown in Fig. S5b (Supporting information) are the second step of reactions 3 and 4. The first step to form H-end-on is favorable in terms of thermodynamics and kinetics (Fig. 5). The ΔG values of reactions 6 and 7 are −50.9 and 31.5 kcal/mol, respectively, indicating D3 can be formed thermodynamically. Note that a N4 compound was reported previously [10]. The ΔG value of reaction 8 is −15.5 kcal/mol, much lower than that of reaction 7. Note that the multiplicity of H-end-on-K is 2, and the spin density is mainly localized on nitrogen atom (0.746). As shown in Fig. S6 (Supporting information), the FBO values of N1-N2, N2-N3 and N3-N4 bonds in D4 are 1.42, 1.90 and 1.42, respectively. And the FBO values of N1-N2, N2-N3 and N3-N4 bonds in D4-K are 1.50, 1.61 and 1.48, respectively, indicating that the electrons are more delocalized in N4 chain of D4-K than that of D4. When a larger substituent tBu is considered (Fig. S5c in Supporting information), the dimerization reaction of NHC1-tBu (reaction 9) become less exothermic than that of NHC1-H (reaction 2), but it still competes with N2 activation.
The kinetics of reactions 2 and 6 are examined. The scan plots of reaction 2 were carried out by fixing B-B bond lengths from 1.586 Å to 3.586 Å (Fig. S7 in Supporting information). Reaction 2 could be considered as a barrierless process due to the lack of a potential energy maximum at the saddle point. As shown in Fig. S8 (Supporting information), the reaction barrier via ts-D3 is 5.1 kcal/mol, indicating that the reaction to form D3 is favorable kinetically. The FBO values of the N1-N2 bond in free N2, H-end-on and D3 are 3.11, 2.63 and 1.64, respectively, indicating that the triple bond of N2 is gradually broken.
In summary, NRT calculations and FBO analysis suggest that the previously called NHC-stabilized borylene could be more suitably termed as methyleneboranes, which are found to have great potential for N2 activation. The results show that CDAC-based methyleneboranes display higher reactivity than that of CAAC-based ones in activating N2. NHC1-H has the best performance in N2 activation, which could contribute to the small singlet-triplet gap. When the methyleneboranes were substituted by π-donor groups, the reactions become less favourable because the lone pair electrons could be donated to the deficient boron to stabilize the reactants. In addition, due to strong correlations of the ΔEST values against ΔG values, the singlet–triplet gap could play a significant role in N2 activation. The PIO analysis suggests that methyleneboranes can mimic transition metals to break N≡N triple bond. All these findings could be helpful for experimental chemists to activate dinitrogen by main group species.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support by the National Science Foundation of China (No. 22073079) and the Top-Notch Young Talents Program of China is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
M. Falcone, L. Chatelain, R. Scopelliti, I. Živković, M. Mazzanti, Nature 547 (2017) 332–335. doi: 10.1038/nature23279
Z. Lv, J. Wei, W. Zhang, et al., Natl. Sci. Rev. 7 (2020) 1564–1583. doi: 10.1093/nsr/nwaa142
J.B. Howard, D.C. Rees, Chem. Rev. 96 (1996) 2965–2982. doi: 10.1021/cr9500545
Z. Mo, S. Takanori, Z. Hou, Angew. Chem. Int. Ed. 59 (2020) 8635–8644. doi: 10.1002/anie.201916171
G. Wang, J. Yin, J. Li, et al., CCS Chem. 3 (2021) 308–316. doi: 10.31635/ccschem.021.202000712
S. Dong, J. Zhu, Chem. Asian J. 16 (2021) 1626–1633. doi: 10.1002/asia.202100394
M. Zhong, X. Cui, B. Wu, et al., CCS Chem. 4 (2022), 532–539. doi: 10.31635/ccschem.021.202100945
J. Li, J. Yin, C. Yu, W. Zhang, Z. Xi, Acta Chim. Sin. 75 (2017) 733–743. doi: 10.6023/A17040170
M. -. A. Légaré, G. Belanger-Chabot, R.D. Dewhurst, et al., Science 359 (2018) 896–900. doi: 10.1126/science.aaq1684
M.A. Légaré, M. Rang, B.G. langer-Chabot, M.C. Holthausen, H. Brauschweig, Science 363 (2019) 1329–1332. doi: 10.1126/science.aav9593
Q. Wang, S. Pan, S. Lei, et al., Nat. Commun. 10 (2019) 1–8. doi: 10.1038/s41467-018-07882-8
M.A. Légaré, C. Pranckevicius, H. Braunschweig, Chem. Rev. 119 (2019) 8231–8261. doi: 10.1021/acs.chemrev.8b00561
M.A. Légaré, G. Belanger-Chabot, M. Rang, et al., Nat. Chem. 12 (2020) 1076–1080. doi: 10.1038/s41557-020-0520-6
M.G. Walawalkar, Dalton Trans. 50 (2021) 460–465. doi: 10.1039/d0dt03599h
G. Deng, S. Pan, G. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 18201–18207. doi: 10.1002/anie.202007241
G. Deng, S. Pan, X. Dong, et al., Chem. Eur. J. 27 (2021) 2131–2137. doi: 10.1002/chem.202004357
B. Rösch, T.X. Gentner, J. Langer, et al., Science 12 (2021) 1125–1128. doi: 10.1126/science.abf2374
S. Hasenstab-Riedel, B. Xu, H. Beckers, Angew. Chem. Int. Ed. 60 (2021) 17205–17210. doi: 10.1002/anie.202106984
K. Edel, M. Krieg, D. Grote, H.F. Bettinger, J. Am. Chem. Soc. 139 (2017) 15151–15159. doi: 10.1021/jacs.7b08497
H. Zhang, R. Yuan, W. Wu, Y. Mo, Chem. Eur. J. 26 (2020) 2619–2625. doi: 10.1002/chem.201904724
J. Zhu, Chem. Asian J. 14 (2019) 1413–1417. doi: 10.1002/asia.201900010
A.M. Rouf, C. Dai, F. Xu, J. Zhu, Adv. Theory Simul. 3 (2020) 1900205. doi: 10.1002/adts.201900205
A.M. Rouf, C. Dai, S. Dong, J. Zhu, Inorg. Chem. 59 (2020) 11770–11781. doi: 10.1021/acs.inorgchem.0c01754
A.M. Rouf, Y. Huang, S. Dong, J. Zhu, Inorg. Chem. 60 (2021) 5598–5606. doi: 10.1021/acs.inorgchem.0c03520
C. Dai, Y. Huang, J. Zhu, Organometallics 41 (2022) 1480–1487. doi: 10.1021/acs.organomet.2c00069
C. Dai, J. Zhu, Phys. Chem. Chem. Phys. 24 (2022) 14651–14657. doi: 10.1039/d2cp01233b
Q. Zhu, R. Qiu, S. Dong, G. Zeng, J. Zhu, Chem. Asian J. 16 (2021) 2063–2067. doi: 10.1002/asia.202100427
F. Fantuzzi, R. Moral, R.D. Dewhurst, H. Braunschweig, A.K. Phukan, Chem. Eur. J. 28 (2022) e202104123.
F. You, J. Zeng, A.M. Rouf, S. Dong, J. Zhu, Chem. Asian J. 17 (2022) e202200232.
J. Zeng, S. Dong, C. Dai, J. Zhu, Inorg. Chem. 61 (2022) 2234–2241. doi: 10.1021/acs.inorgchem.1c03546
C. Wang, Z. Yin, J. Wei, et al., Tetrahedron 76 (2020) 131703. doi: 10.1016/j.tet.2020.131703
P. Bissinger, H. Braunschweig, A. Damme, et al., J. Am. Chem. Soc. 133 (2011) 19044–19047. doi: 10.1021/ja208372k
M. Arrowsmith, D. Auerhammer, R. Bertermann, et al., Angew. Chem. Int. Ed. 55 (2016) 14464–14468. doi: 10.1002/anie.201608429
F. Dahcheh, D. Martin, D.W. Stephan, G. Bertrand, Angew. Chem. Int. Ed. 53 (2014) 13159–13163. doi: 10.1002/anie.201408371
W. Lu, Y. Li, R. Ganguly, R. Kinjo, Angew. Chem. Int. Ed. 56 (2017) 9829–9832. doi: 10.1002/anie.201704887
J. Böhnke, H. Braunschweig, W.C. Ewing, et al., Angew. Chem. Int. Ed. 53 (2014) 9082–9085. doi: 10.1002/anie.201403888
M. Pilz, M. Stadler, R. Hunold, et al., Angew. Chem. Int. Ed. 28 (1989) 784–786. doi: 10.1002/anie.198907841
I. Mayer, P. Salvador, Chem. Phys. Lett. 383 (2004) 368–375. doi: 10.1016/j.cplett.2003.11.048
E.D. Glendening, F. Weinhold, J. Comput. Chem. 19 (1998) 593–609. doi: 10.1002/(SICI)1096-987X(19980430)19:6<593::AID-JCC3>3.0.CO;2-M
J. Zhang, F. Sheong, Z. Lin, Chem. Eur. J. 24 (2018) 9639–9650. doi: 10.1002/chem.201801220
J. Zhang, F. Sheong, Z. Lin, WIREs Comput. Mol. Sci. 10 (2020) e1469.
M. Phipps, T. Fox, C. Tautermann, C. Skylaris, Chem. Soc. Rev. 44 (2015) 3177–3211. doi: 10.1039/C4CS00375F
F. Bickelhaupt, K. Houk, Angew. Chem. Int. Ed. 56 (2017) 10070–10086. doi: 10.1002/anie.201701486

Figure 1 The Gibbs energy (kcal/mol) profile for the N2 activation reactions by R1 and R2. The B-N bond lengths are given in Å.

Figure 2 The structures of R3-R6 (a), the leading resonance structures of the simplified R2-R5 (b) and R6 (c).

Figure 3 Dinitrogen activation by methyleneboranes containing different NHCs and groups.

Figure 4 Correlations for ΔEST values of reactants against ΔGend-on-all (a) and ΔGside-on-all (b).

Figure 6 The principal interacting orbital (PIO) analyzes of ts-H-end-on (a) and ts-H-side-on (b). The isovalue for the surface is 0.05 a.u.
Table 1. The reaction energy (ΔG) and ΔEST values for dinitrogen activation by methyleneboranes. The energies are given in kcal/mol.
|
|
 下载: 导出CSV
下载: 导出CSV
Table 2. The reaction energy (ΔG) and ΔEST values for dinitrogen activation by methyleneboranes. The energies are given in kcal/mol.
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: