Figure 1.
Each step of CAR-T immunotherapy.
Application of nanotechnology in CAR-T-cell immunotherapy
Qiang Zeng , Zhigang Liu , Ting Niu , Chuan He , Ying Qu , Zhiyong Qian

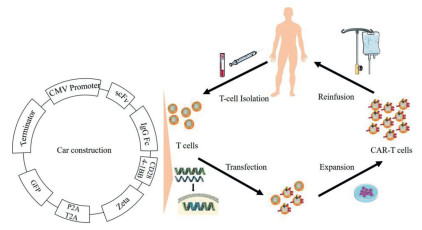
Tumor cells are deceitful and easily resistant to chemotherapy through mutations and adaptations to their surroundings [1, 2]. Chimeric antigen receptor T-cell (CAR-T) immunotherapy is an advanced approach for patients with tumors. Precisely targeted immunotherapy, CAR-T therapy is characterized by a single chain variable fragment (scFv) that recognizes neoantigen epitopes independent of the major histocompatibility complex (MHC) [3, 4]. CAR-T immunotherapy involves the following procedures: isolation of T cells from donors, transfection of CAR genes into isolated T cells, functional validation and expansion of CAR-T cells in vitro, and reinfusion of CAR-T cells into patients (Fig. 1) [5, 6].
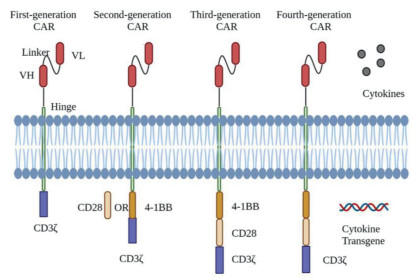
The CAR comprises the extracellular domain (scFv and hinge domain), transmembrane domain, and intracellular domain (costimulatory and signaling domain) [7, 8]. As soon as the extracellular domain of the CAR recognizes the tumor-associated antigen (TAA), the intracellular domain is activated and triggers downstream signaling pathways that exert tumor-killing action [9, 10]. To date, the construction of CAR-T cells has undergone tremendous progress. Four generations of CAR-T cells have been designed and manufactured. The first-generation CAR-T cells were constructed with scFv to recognize tumor antigens fused to the CD3ζ signaling domain and exert specific antitumor effects [11-13]. However, first-generation CAR-T cells cannot expand efficiently, which results in limited sustained antitumor effects due to the costimulatory pathway not being activated. Thus, second-generation CAR-T cells were born. Based on the first-generation CAR-T, the costimulated domain (4–1BB/CD28) was added to the second-generation CAR-T cells to present more durable and efficient antitumor effects [14]. Third-generation CAR-T cells are second-generation CAR-T cells with another costimulated domain [15]. As a result, third-generation CAR-T cells are further boosted in proliferation and antitumor effects. Fourth-generation CAR-T cells are designed to achieve fine-grained control of the whole treatment via a constitutive or inducible expression cassette integrated for suicide switches or immunomodulation, including interleukin (IL)-12 [16], IL-15 [17], IL-18 [18] and suicide genes (Fig. 2) [17].
All CAR-T cells currently on the market are second generation. Since Kymriah was the first CAR-T product approved by the Food and Drug Administration (FDA) in 2017, another four CAR-T-cell productions targeting CD19 or B-cell maturation antigen (BCMA) have been launched in succession for relapsed/refractory hematological tumors [19-21]. The milestone event that advanced CAR-T immunotherapy into clinical practice is the story of a young girl named Emily with relapsed acute lymphoblastic leukemia (ALL). CAR-T immunotherapy saved the girl's life and drew the world's attention. Emily has so far survived for 9 years without relapse.
However, CAR-T immunotherapy currently has the following shortcomings. First, CAR-T therapy is not as effective for solid tumors as it is for hematological tumors. Numerous studies have demonstrated that CAR-T-cell effectiveness is weakened due to poor infiltration of CAR-T cells into solid tumor cells caused by the tumor microenvironment (TME) and the inadequate trafficking of CAR-T cells. In addition, the efficacy of CAR-T cells is limited by the diversity of antigens on solid tumor cells [22-24]. Therein, TME involves various complicated conditions, including hypoxia, chronic inflammation and immunosuppression. The overproduction of hypoxia inducible factor (HIF) was shown to play roles in many signaling pathways of immunosuppression and tumorigenesis, including upregulating PD-L1 and enhancing CXCR4/CXCL12 [25]. Inflammatory cytokines, such as IL-4, IL-6, and transforming growth factor-β (TGF-β), appear in large numbers in the TME. In addition to causing chronic inflammation, these cytokines derived from tumor-associated macrophages (TAMs) help to produce M2 macrophages and T helper 2 (Th2) cells and in turn promote cancer [26, 27]. Immunosuppression of the TME interferes with the antitumor response of CAR-T cells via overactivation of checkpoints (CTLA-4 and PD-1) [28] and overproduction of immune suppressor cells (regulatory T cells, Tregs; myeloid-derived suppressor cells, MDSCs; TAMs) [29-32]. The second shortcoming is the severe CAR-T therapy-associated toxicities. Of these toxicities, cytokine release syndrome (CRS) and CAR-T-related encephalopathy syndrome (CRES) occur most frequently [33-36]. It has been demonstrated that CRS is caused by a large number of proinflammatory cytokines secreted by activated T cells [37]. Patients suffering from CRS usually present with constant fever, myalgia, hypotension and respiratory disorders [38]. Based on the rationale of CAR-T immunotherapy, CRS is almost unavoidable and manifests in various forms and degrees in different patients. Compared to that of predictable CRS, the mechanism of CRES remains unclear. We need to be vigilant about CRES when headache, delirium, or epilepsy symptoms present in patients [38]. CRES can occur either secondary to CRS or independently. Studies have reported that cytokines and CAR-T cells are found in cerebrospinal fluid [39, 40]. Parker et al. found that compared with treatments targeting other proteins on the surface of B cells, neurotoxicity occurred more frequently in immunotherapy with anti-CD19 [41]. Their findings further inferred that, in addition to B cells, other cells expressing CD19, such as blood–brain barrier cells, cause neurotoxicity. Despite updated guidelines regarding CAR-T therapy toxicities [42], severe toxicities bring patients sequelae, such as B-cell aplasia, hypogammaglobulinemia [43, 44], or even death. For these reasons, toxicity has always been a key concern in CAR-T immunotherapy.
Considering these findings, a new approach should be introduced into CAR-T-cell manufacturing. Nanotechnology has developed very rapidly in recent years. Compared with being used alone, many traditional drugs and treatments combined with nanotechnology have emerged with better efficacy and lower toxicity. Importantly, among the many nanotechnologies, nanoparticles possess unique qualities, including a large surface area, high deformability, good biocompatibility, photothermal effect and so on. Compared with traditional drugs and treatments, nanotechnology can significantly improve the properties of drugs to increase the drug concentration in target organs and decrease the biodistribution in healthy tissues, prolong drug retention at the tumor site via the enhanced permeability and retention effect (EPR) [45-47], mimic conditions in vivo, and so on. These benefits have contributed to the combination of nanotechnology and CAR-T immunotherapy increasingly appearing in various studies. Herein, based on the literature review and recent results in this field, we focus on the following areas: (1) construction of CAR; (2) transfection of T cells; (3) expansion of CAR-T cells in vitro; (4) modification of CAR-T cells; and (5) visual monitoring of CAR-T cells in vivo.
In the construction of CAR, the most commonly used nanotechnology is nanosized scFv, and replacing scFv with nanobody is one of the most studied methods. Nanobody, also called the variable domain of heavy chain of the heavy-chain antibody (VHH), is a camelid-derived heavy-chain antibody fragment without the CH1 domain and light chain (Fig. 3A) [48, 49]. Compared with traditional scFvs, nanobodies are smaller, more stable and less immunogenic and comparable in affinity and specificity [50-52]. In the construction of CAR, nanobodies can avoid misfolding and aggregation of scFvs that will cause overexpression of cytotoxic signaling, which in turn leads to early T-cell exhaustion [53, 54]. Furthermore, nanobodies bring enhanced tissue penetration to CAR-T cells, which is beneficial to CAR-T therapy for solid tumors (Fig. 3B) [55-57]. Because of these advantages, nanobodies are easy to mass produce, such as generation from Escherichia coli or yeast [58]. To date, multiple studies referring to nanobody-based CAR-T cells have been listed in Table 1.

Research on nanobody-based CAR-T cells had already begun before CAR-T therapy was launched [59-61]. In 2008, Bakhtiari and colleagues first reported the application of nanobodies in the construction of CARs [62]. In their study, the encoding sequence of the nanobody targeting MUC1 was integrated into chimeric receptor genes instead of scFv and successfully expressed on the surface of Jurkat cells. In the in vitro experiment, lysis of MCF7 cells was observed in the coculture with receptor-grafted Jurkat cells, whereas no similar result was found in the coculture with non-transfected and control vector-transfected Jurkat cells. In addition, compared with non-transfected and control vector-transfected Jurkat cells, receptor-grafted Jurkat cells were shown to secrete more IL-2. However, there were no in vivo experiments and no comparison between nanobody-based CAR and scFv-based CAR on efficacy and safety in this study.
Li et al. demonstrated that nanobody-based CAR-T cells could inhibit tumor growth in mice with neuroblastoma in a study targeting glypican-2 [63]. They isolated anti-glypican-2 single-domain antibody fragments to link to the transmembrane region and generated glypican-2-specific CAR. In an in vivo experiment, they built a neuroblastoma mouse model with disseminated tumor lesions surrounding the spine and bones, and after injection of glypican-2-specific CAR-T cells, they found that metastatic tumors were effectively suppressed, and half of the mice were tumor-free. The excellent results of the in vivo experiments indicated that nanobody-based CARs are very competitive candidates for clinical use. Additionally, many conventional CAR-T therapy targets, including CD38, CD30, CD20, and HER2, have been successfully nanomodified, and have shown good efficacy in the in vivo and in vitro experiments [64-76].
Furthermore, nanobody-based CAR-T therapy has already been applied to clinical trials. Han and coworkers published the results of a clinical trial regarding the safety and efficacy of anti-BCMA nanobody-based CAR-T therapy in patients with multiple myeloma (MM) in 2020 (NCT03664661) [77]. A total of nine patients with relapsed/refractory MM were included in the trial. During a median follow-up of 9.8 months, six patients achieved stringent complete response (CR), two achieved very good partial response (VGPR) and one showed a minimal response. Regarding toxicity, no CRES was observed, and grade 1 CRS was observed in four patients, grade 2 CRS was observed in two patients, grade 3 CRS was observed in only one patient, and no CRS was observed in two patients. It seems that anti-BCMA nanobody-based CAR-T therapy has good efficacy and safety in patients. Another registered clinical trial of CAR-T therapy for relapsed/refractory MM has also been launched in China (NCT03661554). This BCMA CAR comprises a BCMA nanoantibody, a CD8 strand region, a transmembrane region and 4–1BB costimulatory domain, and a CD3− T-cell activation domain, aiming to overcome the disadvantages of conventional CAR-T therapy for hematological tumors. The latest clinical data from the trial are not currently available, however. There are currently no studies comparing scFv-based CAR-T cells with nanobody-based CAR-T cells. Despite the theoretical advantages of nanobody-based CAR-T cells compared to traditional CAR-T cells, we did not know which one was better in experiments or used in patients. Thus, further study on the advantages and disadvantages of these two CAR-Ts is needed in the future.
In addition to the single-target CARs mentioned above, some advanced CARs based on nanobodies have been developed and studied, such as bispecific CARs and universal CARs. Compared with traditional CARs, bispecific CARs are more specific and less prone to the on-target, off-tumor effect. However, because of the large size and ease of aggregation, scFv is not applicable for the construction of bispecific CAR. The advent of nanobodies can very well improve the situation. Munter et al. utilized nanobodies to design a bispecific CAR targeting CD20 and HER2 [78]. They substituted a tandem of two nanobodies for scFv and successfully expressed it on the surface of Jurkat cells. However, in the in vitro experiments, the advantages of bispecific nanobody-based CAR cells were not shown. Compared with anti-CD20 or anti-HER2 CAR-T cells, bispecific CAR-T cells did not exhibit higher efficacy and affinity in either cytokine secretion or cytotoxicity assays. Additionally, as a result of only in vitro experiments, the safety and efficacy of the bispecific CAR-T cells in vivo were not assessed. Wang et al. subsequently designed another bispecific nanobody-based CAR-T targeting CD19 and CD20 [79]. In this study, anti-CD19 and anti-CD20 nanobodies were incorporated into basic CARs in a tandem fashion. Compared to mock cells, bispecific CAR-T cells exerted better tumor-killing effects on K562, Daudi, and human primary ALL cells in the cytotoxicity assay. However, as the study did not involve at comparison with CD19 or CD20 CAR-T cells and in vivo experiments, we did not know about the superiority and safety of the bispecific nanobody-based CAR-T cells. According to the limited studies available, bispecific nanobody-based CAR-T therapy still has a long way to go for clinical use. Nevertheless, bispecific nanobody-based CAR-T therapy remains a promising method for enhancing efficacy and reducing toxicity, and the authors also believe that optimally designed bispecific nanobody-based CAR-T cells could perform well in the treatment of tumors.
Universal CAR is a platform for retargeting T cells to TAAs. Unlike traditional CARs, the extracellular domain of a universal CAR does not recognize a certain antigen of the tumor but the short peptide epitope used as the target module tag, which could reduce side effects and simplify the preparation process of CAR-T cells [80]. Albert and colleagues developed a nanobody targeting epidermal growth factor receptor (EGFR) based on a universal CAR platform [81, 82]. The EGFR-targeted nanobody was linked to E5B9, an epitope of universal CAR-armed T cells, and successfully redirected to EGFR+ tumors. In the cytotoxicity and cytokine release experiments, the universal CAR performed better than the α-EGFR target module. In the in vivo experiment, the universal CAR-T cells exerted an excellent antitumor effect. Eight days after administration, the luciferase activity of the mice receiving the universal CAR-T therapy was not detected via an in vivo imaging system (IVIS), while the luciferase activity of the mice in the control group was easily detected. The study demonstrated that apart from scFvs, nanobodies were used for the generation of target modules to redirect universal CAR-T cells, which opens up new ideas for novel CAR-T development based on a universal CAR-T-cell platform. In summary, the advantages of nanobodies for CAR-T have been shown in animal experiments, but further studies are needed to clarify their effects in humans.
Transferring cargo into T cells is the first major step of CAR-T-cell manufacturing. The efficiency of transfection affects the effectiveness of the subsequent CAR-T therapy. Transfection with viral vectors and electroporation are the two transfer methods currently approved by the FDA [83]. Therein, the viral vector is the most commonly used transfection technology, including lentivirus and retrovirus [84]. Lentivirus and retrovirus vectors that have inherently low immunogenicity enable CAR genes to integrate into the T-cell genome and sustain expression [85, 86]. Overall, statistics showed that approximately 54% of CAR-T cells were produced with lentiviruses and 41% with retroviruses [87]. Nevertheless, transfection with viral vectors has weaknesses. There are, for instance, reports of increasing risk of oncogenesis and lower cell viability during viral vector transfection [88-90]. Multiple genes are also not codelivered by viral vectors. Additionally, delivery of the large CAR gene is reluctant for viral vectors due to their packaging size having an upper limit [91]. Usually, a base pair longer than 7–15 kbp is not allowed [92, 93].
Electroporation is the second FDA-approved method that has been in development for over 30 years. Electroporation can transport cargo, mainly mRNA and plasmids, through pores on the membrane transiently formed by high voltages, except for nucleotides [94, 95]. Moreover, different species of cargo can be simultaneously transported by electroporation [83, 96]. Similar to viral vectors, however, electroporation cannot deliver large CARs [83]. Electroporation can also decrease cell viability and the efficiency of transfection and impair the antitumor effect of CAR-T cells [83, 97, 98]. Importantly, the expression of CAR via electroporation is transient, which requires the prepared CAR-T cells to be reinfused as soon as possible.
In recent years, the transposon system has also been explored for T-cell transfection in CAR-T-cell manufacturing [99-101]. In the presence of transposase, terminal inverted repeats (TIRs) are bound and mobilize DNA flanked by TIRs [102]. Compared to viral vectors and electroporation, the transposon system can generate stable transgenes with less toxicity but is not as efficient [103, 104]. As a result of these issues, nanotechnology is starting to be used for T-cell transfection.
Nanoparticles, as vectors enveloped with cargo, are one of the most common nanoforms for penetrating cell membranes. It has been demonstrated that nanoparticles can deliver genes or proteins into human T cells to increase the efficiency of transfection, avoid immunogenicity, and enhance cancer-killing activity [105-107]. While nanoparticles deliver cargo across the cell membrane via endocytosis or membrane fusion [108], the exact mechanism remains unclear. Generally, cationic nanoparticles are attached to sulfated proteoglycans on the T-cell membrane and enter the cell [109-111]. Then, the nucleic acid cargo is released after endosomal escape and enters the host nuclear membrane via nuclear uptake [112].
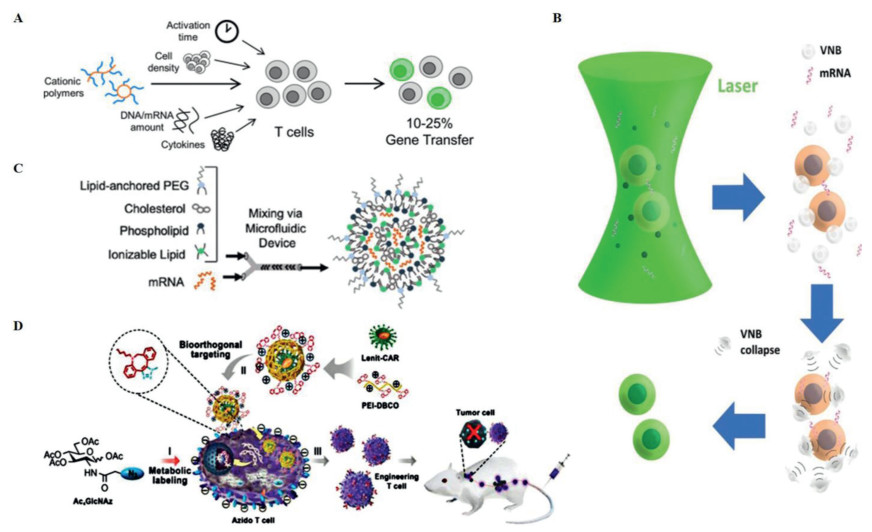
Due to the characteristics of high customizability [113], low cost [114], and single route [115, 116], research on nanoparticles in T-cell transfection is now very prevalent (Table 2). The polymer is one of the most commonly used nanoparticles. Tyrel et al. reported that CAR genes could be imported into the nuclei of T cells with the help of biodegradable poly(β-amino ester) nanoparticles. These nanoparticles were functionalized with peptides containing microtubule-associated sequences (MTAS) and nuclear localization signals (NLS) via the microtubule transport machinery [105]. Despite the comparable treatment results of traditional viral vectors in the in vivo experiments, the nanoparticles simplified storage and reduced cost. Researchers further found that the cationic polyethyleneimine (PEI) polymer interacts with the phosphate backbone of DNA to form a PEI-DNA complex that crosses the cell membrane and delivers loaded DNA into the cell [117, 118]. Using the property of PEI, Fan and colleagues designed a three-segment amphiphilic copolymer, methoxy poly(ethylene glycol)-branched polyethyleneimine-poly(2-ethylbutyl phospholane) (mPEG-bPEI-PEBP) with PEI [119]. This polymer could transport CARs into Jurkat cells with approximately 25% transfection efficiency. Yu and coworkers took the approach a step further. They used adamantane-grafted polyamidoamine (Ad-PAMAM) dendrimers and cyclodextrin-grafted branched polyethylenimine (CD-PEI) to construct a self-assembled nanoparticle, which exhibited efficient gene transfection and low cytotoxicity in Jurkat cells and a relatively simple manufacturing process [120]. Poly(2-dimethylaminoethyl methacrylate) (pDMAEMA) is another common cationic polymer similar to PEI. pDMAEMA promotes electrostatic binding to the negative cell membrane and is taken up into cells and then internalized [121, 122]. Olden and coworkers have researched a series of pDMAEMA. They synthesized a panel of polymers that can carry large plasmids and found that comb- and sunflower-shaped poly(hydroxyethyl methacrylate)-g-poly(2-dimethylaminoethyl methacrylate) (pHEMA-g-pDMAEMA) polymers could significantly increase gene transfer efficiency by up to 50% in Jurkat cells and 18%−25% in primary human T cells, respectively, with high viability (Fig. 4A) [109]. Additionally, nanoparticle-sensitized photoporation is an efficient method to open pores on the cell surface, allowing external cargo to then enter the cell. Recently, Harizaj and colleagues successfully transfected mRNA into HeLa, Jurkat and human T cells by exploiting the photosensitive properties of biodegradable polydopamine nanoparticles, which could transiently permeabilize cells, and achieved 2.5 times more living transfected T cells than electroporation (Fig. 4B) [123]. This technology, which exploits the photothermal properties of polymers, can efficiently produce therapeutic engineered T-cells at a throughput easily exceeding 105 cells per second.
Apart from polymers, lipid nanoparticles are another commonly used type of nanovector. Lipid nanoparticles are often used for the delivery of nucleic acids due to their excellent abilities to concentrate large nucleic acid molecules and block the degradation of nucleic acids caused by widely available nucleases [124-126]. There have been reports that lipid nanoparticles enveloped with siRNA have efficiently entered dendritic, Jurkat, and natural killer (NK) cells [127, 128]. Based on established construction protocols, lipidic vectors for gene transfection are determined by screening libraries of lipids, modifying current existing lipid materials, and developing new materials [129]. With the advancement of nanomaterials, lipid nanoparticles have evolved to the point where there is no need to balance the charges of constituent compounds for effective and efficient delivery into cells. Lipid nanoparticles can also present a shell without a lipid bilayer [130].
The lipidoid, a subtype of lipid nanoparticle that can be easily prepared, shares many of the physicochemical properties with the lipid. Thus, lipidoids can also independently deliver nucleic acids. One study reported that a lipoid containing imidazole group delivered mRNA into mouse T cells, resulting in 8.2% of gene-recombined cells being harvested [131]. This lipidoid was synthesized based on lipid screening libraries. Researchers thought the low efficacy was the result of the nanoparticle structure, especially the amine head part. Nonetheless, the authors believed that the nanoparticle has great potential for cell transfection after process optimization. Ionizable lipids are another subtype of lipid nanoparticles that can change the cell surface charge according to the environmental pH and have a certain ability to self-assemble. Billingsley and colleagues screened out an ionizable lipid nanoparticle, C14–4, with the B-10 formulation, and the mRNA-coated C14–4 was delivered to primary human T cells with an equal level of transfection to electroporation and lower cytotoxicity (Fig. 4C) [132, 133]. Furthermore, the CAR-T-cell engineering method elicited similar antitumor activity against ALL cells in vitro as electroporation or lentiviral vectors. Given that the nanoparticle was prepared as a clinically approved precursor, the manufacturing of CAR-T cells can be utilized soon. Nanotechnology, especially the nanoparticles, is now very mature, and it also develops very rapidly in the field of T-cell transfection. Currently, a nanoparticle platform from Tidal Therapeutics is used for preclinical development [134]. Furthermore, Alnylam's Onpattro, a lipid nanoparticle platform, has been approved by the FDA for mRNA delivery [135].
Nanotechnology combined with conventional transfection methods, including viral vector or electroporation, could also improve CAR-T-cell manufacturing. It was demonstrated that bioorthogonal chemistry can selectively link two biological molecules without interfering with other components [136]. After complementary bioorthogonal motifs are anchored on the surface of viruses and T cells, bioorthogonal chemistry can facilitate the binding of viruses and T cells, which significantly improves the success rate of viral transfection into T cells. Pan et al. reported that bioorthogonal chemistry was utilized to achieve high-efficiency viral transduction into human primary T cells [137]. The researchers anchored azide motifs on the T-cell surface as ligands and then coated the lentiviral surface with the complementary functional moiety dibenzocyclooctyl (DBCO)-conjugated PEI1.8 K (PEI-DBCO) so that the virus coupled with T cells as a nanocomplex via DBCO/azide bioorthogonal chemistry (Fig. 4D). The virus–T-cell interaction helped the lentivirus increase transduction efficiency from 20% to 80%, without affecting T-cell function. Tay and coworkers combined nanomaterials and electroporation to improve the transfection of a relatively large DNA plasmid (12.5 kbp) into human primary T cells [138]. The researchers developed a platform called nano-electro-injection (NEI) that uses highly localized electric fields created by high aspect-ratio nanostructures to transiently form pores on the membrane and directly inject mRNA or DNA. The findings showed a stable net transfection efficiency (efficiency × viability) of 50% in the absence of cell selection, and the researchers thought that NEI was a powerful transfection technique that allows for mass production of engineered T cells at a low cost with a short time frame.
In addition, nanocarriers can deliver DNA efficiently and precisely, and facilitate the endosomal escape of DNA [139]. Zheng et al. combined the polymeric nanomicelle carrier with electroporation and the transposon system to transport the CAR gene into primary human T cells [140]. The researchers developed novel stimuli-sensitive cationic nanomicelles based on the block copolymer of methoxy poly(ethylene glycol)-poly(l-aspartate-aminoethyl-disulphide-g-heptafluorobutyric) (mPEG-P(Asp-AED-g-HFB)), denoted PAEF, to efficiently deliver and release DNA to the expected site. PAEF carried the piggyBac transposon containing the CAR gene in target cells with the help of electroporation, which promoted persistent expression of the transgene (up to 55%). With the widespread use of CAR-T immunotherapy, increasingly high transfection efficiency is needed. The methods mentioned above can work in this regard.
CAR-T cells are massively expanded to meet the requirements for reinfusion into the body. Three proven and good manufacturing practice (GMP)-compliant culture systems: WAVE bioreactor [141-144], CliniMACS Prodigy culture system [145], and G-Rex bioreactor [141, 146, 147], are currently used the most to expand therapeutic T cells. However, the first two damage cells due to hydrodynamic stresses during production [142, 148, 149], while the latter has no hydrodynamic stresses but a lower capacity [146]. Nanotechnology, especially nanomaterials, can alleviate these deficiencies.
Gel-like materials with unique physical properties have been demonstrated to significantly reduce hydrodynamic stresses and increase the cell expansion capacity. Lin and coworkers demonstrated that T cells were expanded substantially in microscale alginate hydrogel tubes [150]. In the study, T cells were suspended in a tube that had no hydrodynamic stresses so that cells with a high growth rate, low DNA damage, high cell viability, and high yield were harvested. Given the simplicity, low cost, excellent compatibility, and automation of the expansion system, a miniature device for automated T-cell production is possible in the future. Apart from alginate hydrogels, other gel-like materials composed of fibrin can promote the mass expansion of T cells. Coon et al. used magnetron sputtering to fabricate a nitinol-thin-film-coated fibrin gel where CAR-T cells fostered rapid expansion. After the film loaded with CAR-T cells was implanted into the tumor tissue, high densities of CAR-T cells exerted powerful antitumor effects [151]. Currently, CAR-T cells expanded by this method are used for the local treatment of solid tumors; further research is needed to achieve systemic reinfusion.
Nanomaterials have also been used to mimic the T-cell in vivo environment to promote the expansion of CAR-T cells. Interaction of antigen-specific lymphocytes with antigen-presenting cells (APCs) can effectively promote T-cell activation and expansion. Fadel et al. employed a carbon nanotube-polymer composite (CNP) as an artificial APC to perform environmental simulations [152]. CNP was assembled from a neutravidin-absorbed carbon nanotube, biotinylated T-cell stimuli (MHC-Ⅰ and αCD28), and loaded poly(lactic-co-glycolic) acid (PLGA) nanoparticles that encapsulated IL-2, biotin and magnetite. Due to many surface defects and a pronounced aspect ratio, CNP could enhance the interaction of cells. Utilizing CNPs, Fadel and colleagues expanded T cells in vitro comparable to clinical standards with 1000-fold less soluble IL-2. Moreover, in a mouse model of melanoma, the expanded T cells significantly slowed tumor progression. In addition, Cheung and coworkers designed a system that mimics natural APC [153]. The system consists of a fluid lipid bilayer supported by mesoporous silica microrods formed at a high aspect ratio that absorb cytokines and antibodies to stimulate T-cell expansion. The scaffold enabled a 5-fold expansion of CD19 CAR-T cells compared to commercial expansion beads (Dynabeads), with similar efficacy in the lymphoma model. The biomimetic application of nanomaterials minimizes toxicity while expanding CAR-T cells. In the future, nanobionics is a good direction for CAR-T therapy.
Despite the great success of CAR-T cells in the treatment of hematological tumors, CAR-T immunotherapy for solid tumors remains a challenge. Compared to hematological tumors, solid tumors must overcome more difficulties in CAR-T therapy, such as a few CAR-T cells trafficking to the tumor lesion [22], heterogeneous antigens on tumor cells [154], and immunosuppression of the TME (Fig. 5) [155, 156]. CAR-T cells reaching the tumor foci are associated with chemokines, such as CXCL-9, CXCL-10, and CXCL-11 [157]. It was demonstrated that, in many human cancers, a few T cells infiltrated solid tumor foci due to the mismatch between chemokines from tumor cells and CAR-T cells [158, 159]. Additionally, some studies have shown that an inhibitory pathway involving the A2A adenosine receptor (A2AR) on the surface of activated T cells is one of the underlying mechanisms for T-cell loss of function, in both solid and hematological tumors [160-162]. Importantly, the TME prevents CAR-T cells from contacting tumor cells through dense tissues on the one hand and inhibits the cytotoxic effect of CAR-T cells by changing the local physiological conditions (e.g., hypoxia, low pH, immunosuppression) on the other hand [163]. Moreover, the antigen on solid tumor cells is more complex than that on hematological tumor cells. Therefore, conventional CAR-T cells do not work well in solid tumors.

In hematological tumors, CAR-T immunotherapy also faces failure caused by antigen escape or lineage switching, which contributes to relapse with unclear specific mechanisms [164-168]. Available CAR-T products showed that non-expression of CD19 and low expression of CD22 were detected in 11%−25% and 66% of relapsed patients, respectively [44, 169-173]. In addition to the above factors, studies have suggested that excessive MDSCs may aid the immune escape of acute myeloid leukemia (AML) via PD-L1 binding to PD-1 expressed on CAR-T cells [174, 175]. Due to the disappointing solid tumor outcomes and relapsed hematologic tumors after CAR-T therapy, many innovative approaches, including oncolytic viruses [176-179], genome editing [180-185] and various nanotechnologies [186-188], have been developed in CAR-T-cell manufacturing to enhance antitumor effect. Here, we primarily reviewed nanotechnologies that enhance CAR-T immunotherapy.
Nanoparticles are used widely in CAR-T immunotherapy. In addition to promoting T-cell transfection, nanoparticles improve the antitumor effect of CAR-T cells in various ways. Drug-loaded nanoparticles are most commonly used (Table 3). These drug-loaded nanoparticles are used to reverse immunosuppression by disrupting cytokines and immunosuppressor cells scattered in the TME [189-194]. Zhang et al. developed a nanoparticle loaded with a tumor-targeting peptide, PI3K inhibitor, and T-cell stimulant that could improve the suppression of the TME and continuously activate T cells. In a mouse model of breast cancer infused with drug-loaded nanoparticles, CAR-T cells had an enhanced ability to infiltrate tumors and expand; the immunosuppressed state of the TME was also reversed. Compared with conventional CAR-T immunotherapy, this sequential CAR-T therapy based on nanoparticle-loaded drugs doubled the overall survival [195]. Researchers have also conducted studies to address the current issues of CAR-T therapy for hematologic tumors. Siriwon et al. reported that adjuvant drug-loaded cross-linked multilamellar liposomes (cMLVs) were maleimide-functionalized to the surface of CAR-T cells to enhance the efficacy of CAR-T therapy [196]. The cMLV loaded with the A2AR-specific small molecule antagonist, SCH-58261 (SCH), blocked the inhibition of adenosine on CD4+ and CD8+ T cells. In the K562 cell line experiment, it was demonstrated that this drug delivery system could efficiently transport drugs to the TME and rescue hypofunctional CAR-T cells. In addition to conventional forms, nanoparticles combine with drugs in other forms. Wang et al. synthesized microneedle patch-coupled anti-PD-1 antibodies for the treatment of melanoma. The microneedle is composed of biocompatible hyaluronic acid integrated with pH-sensitive dextran nanoparticles. Under acidic conditions, nanoparticles degrade and continuously release anti-PD-1 antibodies and thus improve the TME and T-cell trafficking [197]. In another study, Wang and coworkers conjugated PD-L1 antibodies with platelet-derived microparticles, and then T-cell trafficking was improved. The researchers also found that PD-L1 siRNA coated with folic-acid-modified PEI could reduce the expression of PD-L1 on the surface of tumor cells and enhance the contact between T cells and tumor cells in vitro [198].
Additionally, some other drug-loaded nanoparticles have been demonstrated to target immunosuppressive cells around the tumor tissue and have some potential for enhancing CAR-T therapy. Han et al. designed an M2-like TAM-targeted nanocomplex that encapsulates baicalin and melanoma antigen Hgp peptide fragments. The complex was loaded with cytosine-phosphate-guanine (CpG) fragments and conjugated M2pep and α-pep peptides on the surface based on PLGA nanoparticles as the backbone. The researchers found that the nanocomplexes were effectively ingested by M2-like TAMs in vitro and in vivo, and reversed the M2-like TAMs into the M1-like phenotype, which could release cytokines and suppress tumor angiogenesis. In tumor-bearing mice, it was shown that the nanocomplex could retard tumor growth and further remodel the TME to kill tumor cells [199]. Apart from TAMs, Treg cells are another very important negative regulatory target in tumor immune regulation. Ou et al. developed tLyp1 peptide-conjugated hybrid nanoparticles for targeting Treg cells in the TME. The hybrid nanoparticle targeted the downregulation of Treg cells via inhibition of STAT3 and STAT5 phosphorylation. In the in vivo experiment, the researchers observed enrichment of the tLyp1 peptide-modified hybrid nanoparticles around the tumor, decreased Treg cells and elevated CD8+ T cells. Specifically, mice bearing melanoma lived longer after receiving the nanoparticle combined with the immune checkpoint blockade. The authors thought that hybrid nanoparticles targeting Treg cells could improve antitumor immunotherapy by modulating the TME [200].
Hydrogels are special materials formed by large amounts of water and a cross-linked polymer network. A high water content results in excellent biocompatibility. Hydrogels can be formed into almost any size and shape with a high density of internal pores and a large surface area [201]. As a new type of drug delivery material, hydrogels are characterized by high efficiency and low toxicity, prolonged tumor site retention, and customizable physicochemical properties [202, 203], all of which can free hydrogels from the limitations of traditional drug delivery systems. Additionally, hydrogels provide a three-dimensional growth space and spatiotemporal examination of infiltration and cytotoxicity for CAR-T cells [204]. Many studies have focused on nanoplatform-based hydrogels (Table 4).
In 2008, hydrogels were first demonstrated to be excellent carriers in immunotherapy [205]. Over the years, hydrogels combined with CAR-T therapy have also been greatly developed with the support of nanotechnology [206-211]. Atik et al. recently synthesized a hyaluronic acid-based low viscosity hydrogel carrier (LVHydrogel) [212]. The researchers found that CAR-T-cells are suspended in the hydrogel without sedimentation, which greatly improves the efficiency of CAR-T-cell delivery in vivo. Hu and coworkers took the research a step further. They encapsulated CAR-T cells, IL-15, and anti-PD-L1 using hyaluronic hydrogel, which efficiently transported CAR-T cells to target organs but also promoted CAR-T-cell proliferation in vivo and reduced the immunosuppression of the TME [213].
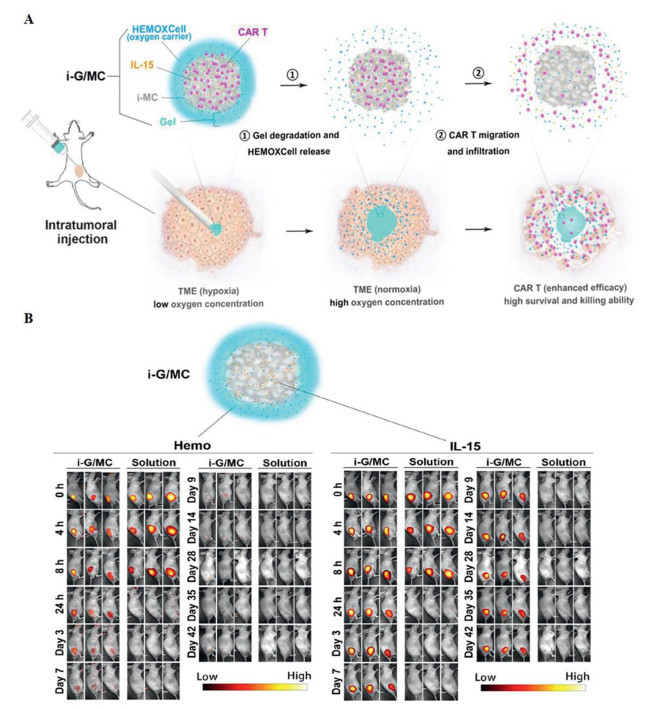
Luo et al. designed a more complicated and effective delivery system [214]. The authors prepared the injectable alginate hydrogel and used it to coat CAR-T cells, HEMOXCell (marine extracellular hemoglobin with a high oxygen storage capacity), and IL-15 (i-G/MC). Once contacting tumors, the outer hydrogel layer quickly degraded. HEMOXCell was released to supply oxygen to reduce immunosuppression in hypoxic environments, and IL-15, a potent cytokine that promotes the proliferation and memory of T cells, offered a condition suitable for immunization to enhance the function of CAR-T cells. The researchers found that iG/MC enabled drugs to stay in vivo longer and confirmed that, in ovarian cancer models, the i-G/MC system can significantly improve the survival, proliferation of CAR-T cells and the effect of tumor-killing (Fig. 6). Hydrogel is an excellent nano-delivery system that fits well with CAR-T-cell therapy, and has almost no effect on CAR-T cells themself and enhances the efficacy of CAR-T cells to a certain extent. Considering that hydrogels are easy to prepare, they may be used on a large scale soon in the future.
Many nanomaterials have the photothermal property and this property can be used to kill tumor cells, which is known as photothermal therapy (PTT). PTT damages tumor cells based on the heat generated by photothermal agents that can absorb near-infrared (NIR) light (650–1350 nm) [215-218]. Hyperthermia (> 42 ℃) enables cell membrane rupture and DNA chain inhibition so that tumor cells undergo apoptosis, while healthy tissue is not significantly affected [219-221]. Tumor cells are more sensitive to heat than normal cells [222], and the tumor-killing mechanism does not result in resistance [223, 224]. PTT can also stimulate the immune system to some extent [225, 226]. However, due to the limited penetration depth for NIR light [227] and reduced photothermal efficiency after laser irradiation [216], PTT alone has low efficacy in treating cancer and is generally used in combination with immunotherapy and nanomaterials [228]. Recent studies on the combination of CAR-T immunotherapy and nanoplatform-based PTT are listed in Table 5.
It was demonstrated that nanomaterial-based PTT does enhances CAR-T immunotherapy. NIR dye is needed as a photothermal agent to trigger the photothermal effect, which is modified based on nanoplatforms [207, 229, 230]. Chen and colleagues created PLGA nanoparticles loaded with indocyanine green (PLGA-ICG) as a photothermal agent [231]. In the in vivo experiment, tumor-bearing mice were injected with PLGA-ICG nanoparticles on the tumor site and then exposed to infrared radiation and finally infused with CAR-T cells. Compared to the mice receiving PTT or CAR-T therapy alone, mice receiving PTT combined with CAR-T therapy presented with significant suppression of the tumor for up to 20 days (Fig. 7). The tumor mass was not observable with the naked eye in two mice.

Other novel nanoparticles are also used as photothermal agents to trigger the PTT effect and thus enhance CAR-T therapy. Zhu and coworkers reported that a combination with nanozymes enhances CAR-T therapy for non-small-cell lung cancer (NSCLC) by remodeling the TME [232]. Nanozymes, as photothermal agents, were synthesized from the pegylation of Cu2+, S2– and hyaluronic acid. After laser irradiation, nanozymes induced reactive oxygen species (ROS) production and damaged the tumor extracellular matrix so that CAR-T cells could effectively infiltrate tumor lesions. In the NSCLC model, tumor eradication and the overall survival of nanozymes combined with CAR-T therapy were twice as much as those of CAR-T therapy alone.
In addition to this sequential administration of PTT and CAR-T cells, another approach is the conjugation of CAR-T cells with a nanophotosensitizer via nanoplatforms. Recently, one study reported that CAR-T cells enhanced the efficacy of lymphoma via bioorthogonal conjugation with a nanophotosensitizer (indocyanine green nanoparticles, INPs) [233]. In this study, researchers immobilized N3 and DBCO on CAR-T cells and INPs, respectively, and then bound these two by a click chemistry reaction (CT-INPs). CT-INPs successfully elicited a photothermal effect upon stimulation with an NIR laser, which disintegrated the protective barrier of the solid tumor and boosted CAR-T therapy. The CAR-T biohybrid creates an immunologically active TME conducive to the behavior of CAR-T cells.
Cytokines play a vital role in CAR-T therapy-associated toxicities [234]. A large number of cytokines are produced when CAR-T cells recognize and kill tumor cells [235]. Additionally, some cytokines are injected to maintain the proliferation and viability of CAR-T cells [236]. Of all the adverse events during CAR-T therapy, CRS and CRES occur the most frequently [38]. Studies demonstrated that CRS was associated with the level of IL-1 or IL-6 that was induced to be released by damage-associated molecular pattern molecules (DAMPs) from tumor cell pyroptosis [237-239], and despite unknown mechanisms, CRES was shown to be associated with the accumulation of cytokines and T cells in the cerebrospinal fluid, which was triggered by proinflammatory cytokines disrupting the blood–brain barrier [39, 40, 240]. Currently, tocilizumab (IL-6 receptor antagonist) [241-243], siltuximab (IL-6 antagonist) [244], infliximab (TNF-α antagonist) [245], and etanercept (TNF antagonist) [246] are employed to reduce CRS, and high-dose corticosteroids are used with a delay against CRES [247]. Additionally, IL-1 antagonist [248] and granulocyte-macrophage colony stimulating factor (GM-CSF) inhibition [249] have been shown to somewhat mitigate CRS and CRES. Nevertheless, these drugs also bring other toxicities. For example, one study reported that tocilizumab may cause cytopenias and infections among patients with rheumatoid arthritis [250].
In addition to the antagonists of cytokines, researchers are considering modifying the structure of CAR to mitigate the toxicity of CAR-T therapy. CARs on the surface of engineered T cells are adjusted to inactivate pathways downstream in some ways [251] and can fundamentally prevent toxicity. Weber et al. degraded CARs by binding CARs with a small molecule [252]; Juillerat et al. disabled CARs under hypoxic conditions [253]. In these systems, the status of CAR-T cells is "off" by default. When the constraints are removed, CAR-T cells are activated upon antigen stimulation. Another way to prevent toxicity is by implementing the suicide gene. Herpes simplex virus thymidine kinase (HSV-TK) was designed to be expressed on CAR-T cells, and is eliminated once it comes in contact with ganciclovir [254]. Nevertheless, HSV-TK itself can result in some immune responses. In addition, it was demonstrated that the inducible caspase 9 (iCasp9) suicide gene could remove > 90% of CAR-T cells within a short time [255]. AP1930, an otherwise bioinert small-molecule dimerizing drug, can also activate fused caspase 9 and induce the apoptosis of cells expressing the iCasp9 protein [256]. However, transgene integration still has the risk for carcinogenicity.
The modification of monoclonal antibodies or the delivery of cytokines by the nanoparticle can also effectively mitigate cytokine-induced toxicity. Since 2013, Irvine's team has done a lot of work on this approach. At first, they anchored anti-CD137 and IL-2, which can promote T-cell proliferation, to the surfaces of PEGylated liposomes [257]. It was demonstrated in the melanoma model that, compared to soluble anti-CD137 and IL-2, anti-CD137 liposomes and IL-2 liposomes did not cause systemic toxicity with significant tumor suppression. This team then continued to apply a similar approach to the TGF-β inhibitor, and a reduction in toxicity was also observed in murine models [258]. Subsequently, Irvine and colleagues took the approach a step further. They bound nanogels containing cytokines as a backpack directly to T cells [259]. A redox-responsive nanogel was prepared through chemical cross-linking of cytokines and then linked to T cells via a click chemistry reaction. When the T-cell surface reduction potential increased upon antigen recognition, cytokines were released from the backpack, which ensures that cytokines do not work in circulation, decreases the level of various cytokines, and subsequently reduces the potential risk of systemic cytokine release. This nanogel-encapsulated approach was successfully used in the delivery of IL-15 [260], IL-21, and IL-2 [261]. Compared to free cytokines, nanogel-encapsulated cytokines achieved a higher safe dose with no overt toxicity in the mouse model.
The systemic toxicity of CAR-T therapy can also be alleviated by optimizing administration methods for solid tumors, such as local treatment. Biodegradable scaffolds are common forms of local administration of CAR-T cells. Biodegradable scaffolds have good biocompatibility and release T cells locally after degradation in vivo, thus limiting systemic CRS. Smith et al. synthesized an alginate scaffold loaded with CAR-T cells and stimulator of IFN genes (STING) agonists and placed the scaffold at the lesion in a mouse model of pancreatic cancer. No significant organ toxicity was observed while triggering host antitumor immunity [262]. In another study, Stephan and coworkers created macroporous scaffolds by polymerized alginate integrated with GFOGER (a synthetic collagen-mimetic peptide (CMP) that binds to lymphocytes via the α2β1 collagen receptor) and lymphocytes. In a mouse model of breast cancer, localized elevated cytokine levels did not cause systemic CRS [263]. Additionally, the thin film was used for local delivery of CAR-T cells. Coon et al. developed thin films with complex designs, high structural resolution and excellent biocompatibility. Thin films loaded with CAR-T cells were implanted in the mouse model of ovarian cancer. Thin films released numerous CAR-T cells locally once they contacted the tumor. The results indicated that there were no significant changes in blood biochemical parameters while improving the efficacy [151]. Although many studies have confirmed that local delivery can significantly reduce the incidence of systemic toxicities, for hematological tumors and metastatic tumors, the local approach is not desirable since tumor cells spread all over the body.
Currently, a clinical trial aimed at mitigating CRS is recruiting patients (NCT03854994). In this trial, a KD-019 CAR with FMC63, which is demonstrated to release a relatively low level of cytokines, was integrated into synthetic biology optimizing nanovector T cells. We are looking forward to the results on how nanotechnology performs in CAR-T clinical trials, which we expect to produce positive results.
After CAR-T cells are imported back into patients, doctors are eager to know the distribution, visualization, and survival of these cells in vivo to improve dosing regimens. To achieve this purpose, researchers usually take samples from serially sampled tissues or peripheral blood to undertake quantitative PCR and flow cytometry, which, despite the novel technique of droplet digital polymerase chain reaction for absolute quantification and higher precision [264, 265], do not provide real-time monitoring of CAR-T cells in vivo [266]. Imaging of genetically engineered T cells by positron emission tomography-computed tomography (PET-CT) or magnetic resonance imaging (MRI) has been used for trials of CAR-T cells in vivo [267-269]. Engineered T cells are modified with a direct or indirect label to be recognized by imaging devices [270]. For the direct labeling of cells, exogenous markers are transfected into the cytoplasm of T cells with either a transfection agent or a positively charged peptide. However, imaging will diminish over time due to dilution during cell division [271]. For the indirect labeling of cells, reporter genes, which encode receptors, fluorescent proteins, or enzymes to activate the imaging probe, modified cells via transfection with a vector [272-275]. Although imaging does not decay over time, complications arising from transfection with viral vectors are not ignored [276].
It has been demonstrated that nanoparticles can be coupled to nuclides as tracers to track CAR-T cells. Bhatnagar and colleagues functionalized gold nanoparticles (GNP) with 64Cu2+ using the macrocyclic chelator (1, 4, 7, 10-tetraazacyclododecane-1, 4, 7, 10-tetraacetic acid, DOTA) and polyethyleneglycol (GNP–64Cu/PEG2000). Then, they electrotransferred the nanoparticle into the prepared CAR-T cells that contained luciferase [277]. Researchers have found that CAR-T cells can be tracked in real-time using µPET/CT. Bhatnagar et al. also highlighted that the procedure can be seamlessly integrated into the current CAR-T-cell manufacturing for phase I/II trials, despite the impact of an electroporation process on T-cell death and the contribution of free GNP–64Cu/PEG2000 released from necrotic cells. Similarly, Li et al. transferred surface-modified gold nanoparticles radiolabeled with 111In or 64Cu into T cells via electroporation [278]. The researchers subsequently demonstrated that imaging of the cells injected into nude mice was observed using micro-SPECT/CT, although they also admitted that the functions of these T cells should be further investigated, such as their ability to release specific nanoparticle payloads.
Furthermore, a study demonstrated that the technetium-99m pertechnetate (99mTcO4−) radiotracer helped SPECT/CT imaging track CAR-T cells [279]. After human sodium iodide symporters (hNIS) were incorporated into a CAR-encoding vector, the transduced T cells acquired radiotracer kinetics, and SPECT/CT imaging with 99mTcO4− radiotracer confirmed the methodology for CAR-T-cell tracing in prostate cancer models. Nonetheless, this approach cannot be used for thyroid and stomach tumors due to organ-specific tracer uptake by hNIS.
In addition to PET/CTs, MRIs can be used to monitor CAR-T-cell movement, despite the need for a specially designed MRI contrast agent. Chapelin and coworkers adopted nanoemulsion and nuclear magnetic resonance (NMR) with the nuclide of 19F to trace CAR-T cells [280]. The researchers employed a kind of perfluorocarbon (PFC) nanoemulsion tracer that labels cells by coculturing with CAR-T cells. After CAR-T cells were reinfused into mice with subcutaneous human U87 glioblastomas, the biodistribution and pharmacokinetics of cells were quantitated. However, the label level of PFC and the sensitivity of NMR were subject to limitations due to a small cytoplasmic volume and the weak phagocytosis of lymphocytes. Therefore, for the sake of boosting cell labeling, in another study, the researchers incorporated the transactivator of transcription (TAT), which helped PFC nanoemulsions display a cell‐penetrating peptide, into the nanoemulsion [281]. Compared to the PFC nanoemulsion without TAT, the cell-penetrating peptide enables more fluorine atoms to enter the CAR-T cells (more than 8-fold) while maintaining the high cell viability (~84%) and intact antitumor effect of the cells.
In addition to nanoemulsions, radiolabeled nanoparticles are expected to be used for CAR-T-cell tracking via MRI. Liu and coworkers first reported IOPC-NH2 series particles, superparamagnetic nanosized iron-oxide particles coated with PEG, with high transverse relaxivity (250 L mmol−1 s−1) [282]. They found that IOPC-NH2 nanoparticles could label human T cells at over 90% efficiency with no measurable effects on T-cell properties. A further study in a rat model undergoing heart-lung transplantation demonstrated that MRI could detect T cells labeled with IOPC-NH2 nanoparticles. This finding showed that nanosized IOPC-NH2 particles serving as contrast agents have the potential for effective tracking of CAR-T cells.
Other nanoparticles, such as "reporter nanoparticles", also have the potential for in vivo tracking of CAR-T cells. Reporter nanoparticles contain a polymeric backbone conjugated to a cytotoxic chemotherapeutic agent or an immunotherapy agent as an effector element and an enzyme-activatable reporter element engineered from a quenched fluorescent dye [283]. The study reported that the effector and reporter elements in a single nanoparticle enhanced the maximum fluorescence signal and captured apoptosis with time, which could present antitumor efficacy in real time via an IVIS. In addition to monitoring efficacy, the reporter nanoparticle efficiently promoted the delivery of the effector to tumors. The findings confirmed that reporter nanoparticles promote T-cell trafficking and a strong fluorescence signal in the immunotherapy. Thus, reporter nanoparticles have the potential to track CAR-T cells. Nonetheless, an IVIS, unlike CT and MRI, which are imaging devices that can be used for patients, is only currently used for research. It also remains unclear whether various fluorescent dyes are harmful to the human body. Thus, reporter nanoparticles have a long way to go as transactivators used to clinically monitor CAR-T cells.
The sophisticated nano-CAR-T characterization technique has advanced the understanding of the interaction of nanotechnology and CAR-T cells and the benefits it brings. However, the weakness of any novel treatment cannot be ignored at the beginning, and nano-CAR-T therapy is no exception. As a result of the stronger migration, proliferation, and activation of nano-CAR-T cells, the severity and frequency of the corresponding toxicities should first be considered. Some studies have reported that nanotechnology enhances the function of cells or drugs while causing toxicity to cells in vitro [284-289]. Although there are no reports of significant toxicity in animal experiments, we do not know whether nanomaterials or nanotechnology-treated cells are harmful to humans or whether there are any long-term cumulative toxic effects because different species have different organ accumulations of nanomaterials [290, 291] and responses [292, 293]. Currently, due to the lack of relevant studies, whether nanomaterials themselves impair the human body is not yet clear. Second, after entering the body, nanoparticles are inevitably attenuated by various blood components [294, 295] and the reticuloendothelial system during delivery [296, 297]. Under these circumstances, nanoparticles must increase the accumulation in the tumor foci via the improvement of specific targeting [298, 299]. Last, the cost of nano-CAR-T therapy should also be considered. CAR-T therapy on the market today can cost hundreds of thousands of dollars. Nano-CAR-T cells are also at a disadvantage compared to existing techniques in terms of experience and mechanistic understanding. It will be a long time before this medical product is approved for clinical use. Researchers still need to spend a great deal of labor, material resources and financial powers to fill this gap. Moreover, in the future, it is also important that marketed nano-CAR-T therapy be affordable for each patient. Although nanotechnology can help to construct universal CARs, simplify the manufacturing of CAR-T cells, and achieve mass production of CAR-T cells, the price orientation of future marketed CAR-T therapy remains an issue worthy of careful consideration.
Although a combination of nanotechnology with CAR-T immunotherapy is still developing, the enthusiasm of researchers and doctors will not wane. Numerous CAR-T immunotherapies combined with nanotechnology have already entered the clinical trial stage. In addition to the nanotechnologies presented above, other nanotechnologies that perform well in combination with other immunotherapies (vaccines, immunoadjuvants, immune checkpoint blockades, monoclonal antibodies, etc.) have not been introduced into CAR-T therapy for the time being. These nanotechnologies include various cell membrane-coated nanoparticle cores that deliver drugs [300-303], photodynamic therapy that generates ROS to kill cancer cells [304-307], and some biomaterials, such as nanoscale metal-organic frameworks, Fe-TBP (constructed from Fe3O clusters and 5, 10, 15, 20-tetra(p-benzoato)porphyrin) [308], CpG DNA nanococoons [309], PEGylated and imidazoylated poly(β-amino esters) [310], polydopamine-coated spiky gold nanoparticles [220], and others that all induce tumor cell death by enhancing tumor-specific immunogenicity. These nanotechnologies have great potential for CAR-T-cell manufacturing, trafficking, and tracing. Further studies will help to understand the interactions between CAR-T cells, the immune system and nanosystems in vivo, accelerate CAR-T-cell manufacturing in nanoplatforms, and broaden the scope of nano-CAR-T immunotherapy. Once nano-CAR-T-cell therapy completes GMP-level sublimation and the corresponding preparation process is well established, we believe that nano-CAR-T-cell therapy has better prospects for clinical application in the future.
CAR-T immunotherapy is a prime example of basic research translated into clinical practice. Currently, some immunotherapies with nanotechnologies have already entered the clinical study phase. The manufacturing, delivery and efficacy of CAR-T cells have developed rapidly, while nanotechnology has been introduced into CAR-T therapy in recent years. To improve CAR-T immunotherapy, nanotechnology can be seen in almost every step of manufacturing. Initially, nanoparticles improved transfection efficiency as vectors, and degradable biomaterials increased the production of CAR-T cells. As CAR-T cells return to the body, the delivery system based on nanomaterials achieves efficacy enhancement and toxicity mitigation by utilizing the special effects of nanoparticles, such as the EPR effect. After CAR-T immunotherapy is completed, nanomaterials help facilitate endocytosis or themselves act as radiological markers to assess the long-term efficacy of the treatment. There is no doubt that nanotechnology is already boosting CAR-T immunotherapy. A deepening understanding of CAR-T therapy and increasing advances in nanotechnology will be increasingly combined more intricately and will help patients achieve better outcomes. We therefore believe that nano-CAR-T immunotherapy will bring about a radical change in cancer treatment in the future.
The authors declared that they have no conflicts of interest to this work. We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work.
This work was supported by the National Natural Science Foundation of China (Nos. 31930067, 31771096, 31700868, 31700869, and 31525009), China Postdoctoral Science Foundation (Nos. 2018M633367 and 2018T110977), Post-Doc Research Project, West China Hospital, Sichuan University (No. 2020HXBH165), and 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (No. ZYGD18002).
P. Vyas, S.E. Jacobsen, Cell Stem Cell 8 (2011) 242–244. doi: 10.1016/j.stem.2011.02.015
C. Sordo-Bahamonde, M. Vitale, S. Lorenzo-Herrero, A. López-Soto, S. Gonzalez, Cancers 12 (2020) 893. doi: 10.3390/cancers12040893
J.N. Kochenderfer, S.A. Rosenberg, Nat. Rev. Clin. Oncol. 10 (2013) 267–276. doi: 10.1038/nrclinonc.2013.46
C.H. June, M. Sadelain, New Engl. J. Med. 379 (2018) 64–73. doi: 10.1056/nejmra1706169
M.V. Maus, Nature 543 (2017) 48–49. doi: 10.1038/nature21506
M.V. Maus, C.H. June, Clin. Cancer Res. 22 (2016) 1875–1884. doi: 10.1158/1078-0432.CCR-15-1433
S. Stoiber, B.L. Cadilha, M.R. Benmebarek, et al., Cells 8 (2019) 472. doi: 10.3390/cells8050472
Z. Walsh, Y. Yang, M.E. Kohler, Immunol. Rev. 290 (2019) 100–113. doi: 10.1111/imr.12794
M.C. Burger, C. Zhang, P.N. Harter, et al., Front. Immunol. 10 (2019) 2683. doi: 10.3389/fimmu.2019.02683
Y. Hu, Z.G. Tian, C. Zhang, Acta Pharmacol. Sin. 39 (2018) 167–176. doi: 10.1038/aps.2017.125
Z. Eshhar, T. Waks, G. Gross, D.G. Schindler, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 720–724. doi: 10.1073/pnas.90.2.720
D. Moritz, W. Wels, J. Mattern, B. Groner, Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 4318–4322. doi: 10.1073/pnas.91.10.4318
I. Stancovski, D.G. Schindler, T. Waks, et al., J. Immunol. 151 (1993) 6577–6582. doi: 10.4049/jimmunol.151.11.6577
C. Imai, K. Mihara, M. Andreansky, et al., Leukemia 18 (2004) 676–684. doi: 10.1038/sj.leu.2403302
A.A. Hombach, J. Heiders, M. Foppe, M. Chmielewski, H. Abken, Oncoimmunology 1 (2012) 458–466. doi: 10.4161/onci.19855
M. Chmielewski, A.A. Hombach, H. Abken, Immunol. Rev. 257 (2014) 83–90. doi: 10.1111/imr.12125
V. Hoyos, B. Savoldo, C. Quintarelli, et al., Leukemia 24 (2010) 1160–1170. doi: 10.1038/leu.2010.75
B. Hu, J. Ren, Y. Luo, et al., Cell Rep. 20 (2017) 3025–3033. doi: 10.1016/j.celrep.2017.09.002
M.C. O'Leary, X. Lu, Y. Huang, et al., Clin. Cancer Res. 25 (2019) 1142–1146. doi: 10.1158/1078-0432.ccr-18-2035
N. Bouchkouj, Y.L. Kasamon, R.A. de Claro, et al., Clin. Cancer Res. 25 (2019) 1702–1708. doi: 10.1158/1078-0432.ccr-18-2743
C.H. June, R.S. O'Connor, O.U. Kawalekar, S. Ghassemi, M.C. Milone, Science 359 (2018) 1361–1365. doi: 10.1126/science.aar6711
A. Schmidts, M.V. Maus, Front. Immunol. 9 (2018) 2593. doi: 10.3389/fimmu.2018.02593
K. Newick, S. O'Brien, E. Moon, S.M. Albelda, Annu. Rev. Med. 68 (2017) 139–152. doi: 10.1146/annurev-med-062315-120245
S. Rafiq, C.S. Hackett, R.J. Brentjens, Nat. Rev. Clin. Oncol. 17 (2020) 147–167. doi: 10.1038/s41571-019-0297-y
T. Schioppa, B. Uranchimeg, A. Saccani, et al., J. Exp. Med. 198 (2003) 1391–1402. doi: 10.1084/jem.20030267
G.W. Tormoen, M.R. Crittenden, M.J. Gough, Adv. Radiat. Oncol. 3 (2018) 520–526. doi: 10.1016/j.adro.2018.08.018
S. Wilkie, S.E. Burbridge, L. Chiapero-Stanke, et al., J. Biol. Chem. 285 (2010) 25538–25544. doi: 10.1074/jbc.M110.127951
E.I. Buchbinder, A. Desai, Am. J. Clin. Oncol. 39 (2016) 98–106. doi: 10.1097/COC.0000000000000239
L. Labanieh, R.G. Majzner, C.L. Mackall, Nat. Biomed. Eng. 2 (2018) 377–391. doi: 10.1038/s41551-018-0235-9
S.L. Buchan, L. Dou, M. Remer, et al., Immunity 49 (2018) 958–970 e957. doi: 10.1016/j.immuni.2018.09.014
R. Parihar, C. Rivas, M. Huynh, et al., Cancer Immunol. Res. 7 (2019) 363–375. doi: 10.1158/2326-6066.cir-18-0572
M. Ruella, M. Klichinsky, S.S. Kenderian, et al., Cancer Discov. 7 (2017) 1154–1167. doi: 10.1158/2159-8290.CD-16-0850
S.A. Grupp, M. Kalos, D. Barrett, et al., New Engl. J. Med. 368 (2013) 1509–1518. doi: 10.1056/NEJMoa1215134
J.C. Fitzgerald, S.L. Weiss, S.L. Maude, et al., Crit. Care Med. 45 (2017) e124–e131. doi: 10.1097/CCM.0000000000002053
S.J. Schuster, J. Svoboda, E.A. Chong, et al., New Engl. J. Med. 377 (2017) 2545–2554. doi: 10.1056/NEJMoa1708566
D.W. Lee, B.D. Santomasso, F.L. Locke, et al., Biol. Blood Marrow Transplant. 25 (2019) 625–638. doi: 10.1016/j.bbmt.2018.12.758
H. Klingemann, Oncoimmunology 3 (2014) e28147. doi: 10.4161/onci.28147
L.B. Kennedy, A.K.S. Salama, CA Cancer J. Clin. 70 (2020) 86–104. doi: 10.3322/caac.21596
J. Gust, K.A. Hay, L.A. Hanafi, et al., Cancer Discov. 7 (2017) 1404–1419. doi: 10.1158/2159-8290.CD-17-0698
B.D. Santomasso, J.H. Park, D. Salloum, et al., Cancer Discov. 8 (2018) 958–971. doi: 10.1158/2159-8290.cd-17-1319
K.R. Parker, D. Migliorini, E. Perkey, et al., Cell 183 (2020) 126–142 e117. doi: 10.1016/j.cell.2020.08.022
J.N. Brudno, J.N. Kochenderfer, Blood Rev. 34 (2019) 45–55. doi: 10.1016/j.blre.2018.11.002
M. Namuduri, R.J. Brentjens, Expert Rev. Hematol. 9 (2016) 511–513. doi: 10.1080/17474086.2016.1183479
S.L. Maude, N. Frey, P.A. Shaw, et al., New Engl. J. Med. 371 (2014) 1507–1517. doi: 10.1056/NEJMoa1407222
D. Kalyane, N. Raval, R. Maheshwari, et al., Mater. Sci. Eng. C Mater. Biol. Appl. 98 (2019) 1252–1276. doi: 10.1016/j.msec.2019.01.066
J. Wu, J. Pers. Med. 11 (2021) 771. doi: 10.3390/jpm11080771
M. Zhao, D. van Straten, M.L.D. Broekman, V. Préat, R.M. Schiffelers, Theranostics 10 (2020) 1355–1372. doi: 10.7150/thno.38147
C. Hamers-Casterman, T. Atarhouch, S. Muyldermans, et al., Nature 363 (1993) 446–448. doi: 10.1038/363446a0
P. Safarzadeh Kozani, A. Naseri, S.M.J. Mirarefin, et al., Biomark. Res. 10 (2022) 24. doi: 10.1186/s40364-022-00371-7
J.R. Ingram, F.I. Schmidt, H.L. Ploegh, Annu. Rev. Immunol. 36 (2018) 695–715. doi: 10.1146/annurev-immunol-042617-053327
H. Revets, P. De Baetselier, S. Muyldermans, Expert Opin. Biol. Ther. 5 (2005) 111–124. doi: 10.1517/14712598.5.1.111
S. Muyldermans, Annu. Rev. Biochem. 82 (2013) 775–797. doi: 10.1146/annurev-biochem-063011-092449
J. Jayaraman, M.P. Mellody, A.J. Hou, et al., EBioMedicine 8 (2020) 102931. doi: 10.1016/j.ebiom.2020.102931
W. Sun, J. Xie, H. Lin, et al., Protein Expr. Purif. 83 (2012) 21–29. doi: 10.1016/j.pep.2012.02.006
S. Muyldermans, J. Biotechnol. 74 (2001) 277–302.
B.M. Tijink, T. Laeremans, M. Budde, et al., Mol. Cancer Ther. 7 (2008) 2288–2297. doi: 10.1158/1535-7163.MCT-07-2384
C. Bao, Q. Gao, L.L. Li, et al., Biomolecules 11 (2021) 238. doi: 10.3390/biom11020238
M.M. Harmsen, H.J. De Haard, Appl. Microbiol. Biotechnol. 77 (2007) 13–22. doi: 10.1007/s00253-007-1142-2
F.J. Iri-Sofla, F. Rahbarizadeh, D. Ahmadvand, M.J. Rasaee, Exp. Cell Res. 317 (2011) 2630–2641. doi: 10.1016/j.yexcr.2011.08.015
S. Khaleghi, F. Rahbarizadeh, D. Ahmadvand, M.J. Rasaee, P. Pognonec, Int. J. Hematol. 95 (2012) 434–444. doi: 10.1007/s12185-012-1037-6
Z. Sharifzadeh, F. Rahbarizadeh, M.A. Shokrgozar, et al., Cancer Lett. 334 (2013) 237–244. doi: 10.1016/j.canlet.2012.08.010
S.H. Bakhtiari, F. Rahbarizadeh, S. Hasannia, et al., Hybridoma (2005) 28 (2009) 85–92. doi: 10.1089/hyb.2008.0079
N. Li, H. Fu, S.M. Hewitt, D.S. Dimitrov, M. Ho, Proc. Natl. Acad. Sci. U. S. A. 114 (2017) E6623–e6631. doi: 10.1073/pnas.1700536114
M. Hassani, F. Hajari Taheri, Z. Sharifzadeh, et al., J. Cell. Biochem. 120 (2019) 10787–10795. doi: 10.1002/jcb.28370
Y.J. Xie, M. Dougan, N. Jailkhani, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 7624–7631. doi: 10.1073/pnas.1817147116
F. Hajari Taheri, M. Hassani, Z. Sharifzadeh, et al., IUBMB Life 71 (2019) 1259–1267. doi: 10.1002/iub.2019
F.R. Jamnani, F. Rahbarizadeh, M.A. Shokrgozar, et al., Biochim. Biophys. Acta 1840 (2014) 378–386. doi: 10.1016/j.bbagen.2013.09.029
N. An, Y.N. Hou, Q.X. Zhang, et al., Mol. Pharm. 15 (2018) 4577–4588. doi: 10.1021/acs.molpharmaceut.8b00584
J. Xu, L.J. Chen, S.S. Yang, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 9543–9551. doi: 10.1073/pnas.1819745116
S. De Munter, A. Van Parys, L. Bral, et al., Int. J. Mol. Sci. 21 (2020) 833. doi: 10.3390/ijms21030833
T. Zhang, T. Wang, F. You, et al., Transpl. Immunol. 71 (2022) 101538. doi: 10.1016/j.trim.2022.101538
M. Zhang, D. Chen, X. Fu, et al., Clin. Cancer Res. (2022) 2830–2843. doi: 10.1158/1078-0432.ccr-21-4097
D. Chen, F. You, S. Xiang, et al., Am. J. Cancer Res. 11 (2021) 5263–5281.
M.A. Nix, K. Mandal, H. Geng, et al., Cancer Discov. 11 (2021) 2032–2049. doi: 10.1158/2159-8290.cd-20-0242
Z. Feng, X. He, X. Zhang, et al., Nat. Cancer (2022) 581–594. doi: 10.1038/s43018-022-00344-7
F. Mo, S. Duan, X. Jiang, et al., Signal Transduct. Target. Ther. 6 (2021) 80. doi: 10.1038/s41392-021-00462-1
L. Han, J. Zhou, K. Zhou, et al., J. Immunother. Cancer 8 (2020) e000927. doi: 10.1136/jitc-2020-000927
S. De Munter, J. Ingels, G. Goetgeluk, et al., Int. J. Mol. Sci. 19 (2018) 403. doi: 10.3390/ijms19020403
H. Wang, L. Wang, Y. Li, et al., Cancer Cell Int. 21 (2021) 450. doi: 10.1186/s12935-021-02151-z
R.C. Larson, M.V. Maus, Nat. Rev. Cancer 21 (2021) 145–161. doi: 10.1038/s41568-020-00323-z
S. Albert, C. Arndt, A. Feldmann, et al., Oncoimmunology 6 (2017) e1287246. doi: 10.1080/2162402X.2017.1287246
S. Albert, C. Arndt, S. Koristka, et al., Oncotarget 9 (2018) 25597–25616. doi: 10.18632/oncotarget.25390
K. L A.R.K. Kumar, Y. Shou, B. Chan, A. Tay, Adv. Mater. 33 (2021) e2007421. doi: 10.1002/adma.202007421
B.L. Levine, J. Miskin, K. Wonnacott, C. Keir, Mol. Ther. Methods Clin. Dev. 4 (2017) 92–101. doi: 10.1016/j.omtm.2016.12.006
V. Poletti, F. Mavilio, Mol. Ther. Methods Clin. Dev. 8 (2018) 31–41. doi: 10.1016/j.omtm.2017.10.001
C.H. June, B.R. Blazar, J.L. Riley, Nat. Rev. Immunol. 9 (2009) 704–716. doi: 10.1038/nri2635
P. Vormittag, R. Gunn, S. Ghorashian, F.S. Veraitch, Curr. Opin. Biotechnol. 53 (2018) 164–181. doi: 10.1016/j.copbio.2018.01.025
H. Nakai, E. Montini, S. Fuess, et al., Nat. Genet. 34 (2003) 297–302. doi: 10.1038/ng1179
A. Donsante, D.G. Miller, Y. Li, et al., Science 317 (2007) 477. doi: 10.1126/science.1142658
A. Tay, N. Melosh, Adv. Ther. 2 (2019) 1900133. doi: 10.1002/adtp.201900133
Y.C. Wu, T.H. Wu, D.L. Clemens, et al., Nat. Methods 12 (2015) 439–444. doi: 10.1038/nmeth.3357
M.P. Stewart, A. Sharei, X. Ding, et al., Nature 538 (2016) 183–192. doi: 10.1038/nature19764
S. Sun, V.B. Rao, M.G. Rossmann, Curr. Opin. Struct. Biol. 20 (2010) 114–120. doi: 10.1016/j.sbi.2009.12.006
E. Neumann, M. Schaefer-Ridder, Y. Wang, P.H. Hofschneider, EMBO J. 1 (1982) 841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x
T.Y. Tsong, Biophys. J. 60 (1991) 297–306. doi: 10.1016/S0006-3495(91)82054-9
X. Xu, D. Gao, P. Wang, et al., Sci. Rep. 8 (2018) 11649. doi: 10.1038/s41598-018-30227-w
J.T. Sengel, M.I. Wallace, Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 5281–5286. doi: 10.1073/pnas.1517437113
J.C. Weaver, Y.A. Chizmadzhev, Bioelectrochem. Bioenerg. 41 (1996) 135–160. doi: 10.1016/S0302-4598(96)05062-3
D. Morita, N. Nishio, S. Saito, et al., Mol. Ther. Methods Clin. Dev. 8 (2018) 131–140. doi: 10.1016/j.omtm.2017.12.003
S. Saito, Y. Nakazawa, A. Sueki, et al., Cytotherapy 16 (2014) 1257–1269. doi: 10.1016/j.jcyt.2014.05.022
I. Querques, A. Mades, C. Zuliani, et al., Nat. Biotechnol. 37 (2019) 1502–1512. doi: 10.1038/s41587-019-0291-z
Q. Gao, X. Dong, Q. Xu, et al., Cancer Med. 8 (2019) 4254–4264. doi: 10.1002/cam4.2257
L. Mátés, M.K. Chuah, E. Belay, et al., Nat. Genet. 41 (2009) 753–761. doi: 10.1038/ng.343
X. Xue, X. Huang, S.E. Nodland, et al., Blood 114 (2009) 1319–1330. doi: 10.1182/blood-2009-03-210005
T.T. Smith, S.B. Stephan, H.F. Moffett, et al., Nat. Nanotechnol. 12 (2017) 813–820. doi: 10.1038/nnano.2017.57
C.J. McKinlay, N.L. Benner, O.A. Haabeth, R.M. Waymouth, P.A. Wender, Proc. Natl. Acad. Sci. U. S. A. 115 (2018) E5859–E5866.
B. Mostaghaci, J. Susewind, G. Kickelbick, C.M. Lehr, B. Loretz, ACS Appl. Mater. Interfaces 7 (2015) 5124–5133. doi: 10.1021/am507193a
L. Raes, S.C. De Smedt, K. Raemdonck, K. Braeckmans, Biotechnol. Adv. 49 (2021) 107760. doi: 10.1016/j.biotechadv.2021.107760
B.R. Olden, Y. Cheng, J.L. Yu, S.H. Pun, J. Control. Release 282 (2018) 140–147. doi: 10.1016/j.jconrel.2018.02.043
W.P. Verdurmen, R. Wallbrecher, S. Schmidt, et al., J. Control. Release 170 (2013) 83–91. doi: 10.1016/j.jconrel.2013.05.001
S.A.B. Riedl, P. Kaiser, A. Raup, et al., Processes 6 (2018) 188. doi: 10.3390/pr6100188
W. Nawaz, S. Xu, Y. Li, et al., Acta Biomater. 109 (2020) 21–36. doi: 10.1016/j.actbio.2020.04.015
H. Yin, R.L. Kanasty, A.A. Eltoukhy, et al., Nat. Rev. Genet. 15 (2014) 541–555. doi: 10.1038/nrg3763
Y. Song, J. Hormes, C.S. Kumar, Small 4 (2008) 698–711. doi: 10.1002/smll.200701029
H. Dong, M. Zhu, J.A. Yoon, et al., J. Am. Chem. Soc. 130 (2008) 12852–12853. doi: 10.1021/ja8038097
J. Xie, S. Peng, N. Brower, et al., Pure Appl. Chem. 78 (2006) 1003–1014. doi: 10.1351/pac200678051003
O. Boussif, F. Lezoualc'h, M.A. Zanta, et al., Proc. Natl. Acad. Sci. U. S. A. 92 (1995) 7297–7301. doi: 10.1073/pnas.92.16.7297
J.J. Virgen-Ortíz, J.C.S. Dos Santos, Á. Berenguer-Murcia, et al., J. Mater. Chem. B 5 (2017) 7461–7490. doi: 10.1039/C7TB01639E
J. Fan, Q. He, Z. Jin, W. Chen, W. Huang, RSC Adv. 8 (2018) 14975–14982. doi: 10.1039/c8ra02133c
Q. Yu, M. Zhang, Y. Chen, et al., Int. J. Nanomed. 15 (2020) 483–495. doi: 10.2147/ijn.s229858
I.A. Khalil, K. Kogure, H. Akita, H. Harashima, Pharmacol. Rev. 58 (2006) 32–45. doi: 10.1124/pr.58.1.8
K.A. Mislick, J.D. Baldeschwieler, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 12349–12354. doi: 10.1073/pnas.93.22.12349
A. Harizaj, M. Wels, L. Raes, et al., Adv. Funct. Mater. 31 (2021) 2102472. doi: 10.1002/adfm.202102472
Y. Zhao, L. Huang, Adv. Genet. 88 (2014) 13–36.
D. Luo, W.M. Saltzman, Nat. Biotechnol. 18 (2000) 33–37. doi: 10.1038/71889
A. Yen, Y. Cheng, M. Sylvestre, et al., Mol. Pharm. 15 (2018) 2268–2276. doi: 10.1021/acs.molpharmaceut.8b00134
S. Warashina, T. Nakamura, Y. Sato, et al., J. Control. Release 225 (2016) 183–191. doi: 10.1016/j.jconrel.2016.01.042
T. Nakamura, M. Kuroi, Y. Fujiwara, et al., Sci. Rep. 6 (2016) 37849. doi: 10.1038/srep37849
Y. Sawalha, A.S. Advani, Int. J. Hematol. Oncol. 7 (2018) Ijh02. doi: 10.2217/ijh-2017-0023
E. Samaridou, J. Heyes, P. Lutwyche, Adv. Drug. Deliv. Rev. 154-155 (2020) 37–63. doi: 10.1016/j.addr.2020.06.002
X. Zhao, J. Chen, M. Qiu, et al., Angew Chem. Int. Ed. 59 (2020) 20083–20089. doi: 10.1002/anie.202008082
M.M. Billingsley, N. Singh, P. Ravikumar, et al., Nano Lett. 20 (2020) 1578–1589. doi: 10.1021/acs.nanolett.9b04246
M.M. Billingsley, A.G. Hamilton, D. Mai, et al., Nano Lett. (2021) 533–542.
H.F. Moffett, M.E. Coon, S. Radtke, et al., Nat. Commun. 8 (2017) 389. doi: 10.1038/s41467-017-00505-8
K. Garber, Nat. Biotechnol. 36 (2018) 777–778. doi: 10.1038/nbt0918-777
H. Pan, P. Zhang, D. Gao, et al., ACS Nano 8 (2014) 5468–5477. doi: 10.1021/nn501028b
H. Pan, P. Li, G. Li, et al., Adv. Funct. Mater. 29 (2019) 1807528. doi: 10.1002/adfm.201807528
A. Tay, N. Melosh, Small 17 (2021) e2103198. doi: 10.1002/smll.202103198
L.H. Wang, D.C. Wu, H.X. Xu, Y.Z. You, Angew. Chem. Int. Ed. 55 (2016) 755–759. doi: 10.1002/anie.201508695
Y. Zheng, Z.R. Li, R. Yue, et al., Am. J. Transl. Res. 11 (2019) 7126–7136.
R.P. Somerville, M.E. Dudley, Oncoimmunology 1 (2012) 1435–1437. doi: 10.4161/onci.21206
R.P. Somerville, L. Devillier, M.R. Parkhurst, S.A. Rosenberg, M.E. Dudley, J. Transl. Med. 10 (2012) 69. doi: 10.1186/1479-5876-10-69
B.L. Levine, Cancer Gene Ther. 22 (2015) 79–84. doi: 10.1038/cgt.2015.5
D. Hollyman, J. Stefanski, M. Przybylowski, et al., J. Immunother. 32 (2009) 169–180. doi: 10.1097/CJI.0b013e318194a6e8
A.D. Kaiser, M. Assenmacher, B. Schröder, et al., Cancer Gene Ther. 22 (2015) 72–78. doi: 10.1038/cgt.2014.78
P. Bajgain, R. Mucharla, J. Wilson, et al., Mol. Ther. Methods Clin. Dev. 1 (2014) 14015. doi: 10.1038/mtm.2014.15
J. Jin, M. Sabatino, R. Somerville, et al., J. Immunother. 35 (2012) 283–292. doi: 10.1097/CJI.0b013e31824e801f
U. Mock, L. Nickolay, B. Philip, et al., Cytotherapy 18 (2016) 1002–1011. doi: 10.1016/j.jcyt.2016.05.009
M. Apel, M. Bruening, M. Granzin, et al., Chem. Ing. Tech. 85 (2013) 103–110. doi: 10.1002/cite.201200175
H. Lin, Q. Li, O. Wang, et al., Adv. Healthc. Mater. 7 (2018) e1701297. doi: 10.1002/adhm.201701297
M.E. Coon, S.B. Stephan, V. Gupta, C.P. Kealey, M.T. Stephan, Nat. Biomed. Eng. 4 (2020) 195–206.
T.R. Fadel, F.A. Sharp, N. Vudattu, et al., Nat. Nanotechnol. 9 (2014) 639–647. doi: 10.1038/nnano.2014.154
A.S. Cheung, D.K.Y. Zhang, S.T. Koshy, D.J. Mooney, Nat. Biotechnol. 36 (2018) 160–169. doi: 10.1038/nbt.4047
Y. Wang, F. Luo, J. Yang, C. Zhao, Y. Chu, Front. Immunol. 8 (2017) 1934. doi: 10.3389/fimmu.2017.01934
M. Binnewies, E.W. Roberts, K. Kersten, et al., Nat. Med. 24 (2018) 541–550. doi: 10.1038/s41591-018-0014-x
W. Zou, Nat. Rev. Cancer 5 (2005) 263–274. doi: 10.1038/nrc1586
J.R. Groom, A.D. Luster, Immunol. Cell Biol. 89 (2011) 207–215. doi: 10.1038/icb.2010.158
E.M. Hanson, V.K. Clements, P. Sinha, D. Ilkovitch, S. Ostrand-Rosenberg, J. Immunol. 183 (2009) 937–944. doi: 10.4049/jimmunol.0804253
C.Y. Slaney, M.H. Kershaw, P.K. Darcy, Cancer Res. 74 (2014) 7168–7174. doi: 10.1158/0008-5472.CAN-14-2458
A. Ohta, E. Gorelik, S.J. Prasad, et al., Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 13132–13137. doi: 10.1073/pnas.0605251103
C. Cekic, J. Linden, Cancer Res. 74 (2014) 7239–7249. doi: 10.1158/0008-5472.CAN-13-3581
P.A. Beavis, M.A. Henderson, L. Giuffrida, et al., J. Clin. Invest. 127 (2017) 929–941. doi: 10.1172/JCI89455
J. Li, W. Li, K. Huang, et al., J. Hematol. Oncol. 11 (2018) 22. doi: 10.1504/ijhpcn.2018.10025202
R.G. Majzner, S. Heitzeneder, C.L. Mackall, Cancer Cell 31 (2017) 476–485. doi: 10.1016/j.ccell.2017.03.002
C.J. Turtle, L.A. Hanafi, C. Berger, et al., J. Clin. Invest. 126 (2016) 2123–2138. doi: 10.1172/JCI85309
R. Gardner, D. Wu, S. Cherian, et al., Blood 127 (2016) 2406–2410. doi: 10.1182/blood-2015-08-665547
E. Jacoby, S.M. Nguyen, T.J. Fountaine, et al., Nat. Commun. 7 (2016) 12320. doi: 10.1038/ncomms12320
R.G. Majzner, C.L. Mackall, Cancer Discov. 8 (2018) 1219–1226. doi: 10.1158/2159-8290.cd-18-0442
S.L. Maude, D.T. Teachey, S.R. Rheingold, et al., J. Clin. Oncol. 34 (2016) 3011. doi: 10.1200/JCO.2016.34.15_suppl.3011
S.L. Maude, T.W. Laetsch, J. Buechner, et al., New Engl. J. Med. 378 (2018) 439–448. doi: 10.1056/nejmoa1709866
R. Gardner, O. Finney, H. Smithers, et al., Blood 128 (2016) 219. doi: 10.1182/blood.v128.22.219.219
D.W. Lee, M. Stetler-Stevenson, C.M. Yuan, et al., Blood 128 (2016) 218. doi: 10.1182/blood.V128.22.218.218
T.J. Fry, N.N. Shah, R.J. Orentas, et al., Nat. Med. 24 (2018) 20–28. doi: 10.1038/nm.4441
A.R. Pyzer, D. Stroopinsky, H. Rajabi, et al., Blood 129 (2017) 1791–1801. doi: 10.1182/blood-2016-07-730614
R. Jitschin, D. Saul, M. Braun, et al., J. Immunother. Cancer 6 (2018) 116. doi: 10.1186/s40425-018-0432-9
A.K. Park, Y. Fong, S.I. Kim, et al., Sci. Transl. Med. 12 (2020) eaaz1863. doi: 10.1126/scitranslmed.aaz1863
N. Nishio, G. Dotti, Oncoimmunology 4 (2015) e988098. doi: 10.4161/21505594.2014.988098
E.K. Moon, L.S. Wang, K. Bekdache, et al., Oncoimmunology 7 (2018) e1395997. doi: 10.1080/2162402X.2017.1395997
A. Aalipour, F. Le Boeuf, M. Tang, et al., Mol. Ther. Oncolytics 17 (2020) 232–240. doi: 10.1016/j.omto.2020.03.018
J. Ren, X. Liu, C. Fang, et al., Clin. Cancer Res. 23 (2017) 2255–2266. doi: 10.1158/1078-0432.CCR-16-1300
J.R. Hamilton, C.A. Tsuchida, D.N. Nguyen, et al., Cell Rep. 35 (2021) 109207. doi: 10.1016/j.celrep.2021.109207
M. Hale, B. Lee, Y. Honaker, et al., Mol. Ther. Methods Clin. Dev. 4 (2017) 192–203. doi: 10.1016/j.omtm.2016.12.008
J. Eyquem, J. Mansilla-Soto, T. Giavridis, et al., Nature 543 (2017) 113–117. doi: 10.1038/nature21405
L.J. Rupp, K. Schumann, K.T. Roybal, et al., Sci. Rep. 7 (2017) 737. doi: 10.1038/s41598-017-00462-8
T. Nakazawa, A. Natsume, F. Nishimura, et al., Cells 9 (2020) 998. doi: 10.3390/cells9040998
K. Reinhard, B. Rengstl, P. Oehm, et al., Science 367 (2020) 446–453. doi: 10.1126/science.aay5967
M.N. Garas, S.V. Tillib, O.V. Zubkova, et al., Acta Nat. 6 (2014) 95–105. doi: 10.32607/20758251-2014-6-2-95-105
Y. Zhai, W. Ran, J. Su, et al., Adv. Mater. (2018) e1802378.
G.B. Kim, V. Aragon-Sanabria, L. Randolph, et al., Bioact. Mater. 5 (2020) 624–635. doi: 10.1016/j.bioactmat.2020.04.011
V. de Turris, M. Cardoso Trabuco, G. Peruzzi, et al., Nanoscale 9 (2017) 647–655. doi: 10.1039/C6NR07129E
C.T. Jiang, K.G. Chen, A. Liu, et al., Nat. Commun. 12 (2021) 1359. doi: 10.1038/s41467-021-21497-6
X. Huang, J.Z. Williams, R. Chang, et al., Nat. Nanotechnol. 16 (2021) 214–223. doi: 10.1038/s41565-020-00813-z
L. Ma, T. Dichwalkar, J.Y.H. Chang, et al., Science 365 (2019) 162–168. doi: 10.1126/science.aav8692
D. Schmid, C.G. Park, C.A. Hartl, et al., Nat. Commun. 8 (2017) 1747. doi: 10.1038/s41467-017-01830-8
F. Zhang, S.B. Stephan, C.I. Ene, et al., Cancer Res. 78 (2018) 3718–3730. doi: 10.1158/0008-5472.can-18-0306
N. Siriwon, Y.J. Kim, E. Siegler, et al., Cancer Immunol. Res. 6 (2018) 812–824. doi: 10.1158/2326-6066.cir-17-0502
C. Wang, Y. Ye, G.M. Hochu, H. Sadeghifar, Z. Gu, Nano Lett. 16 (2016) 2334–2340. doi: 10.1021/acs.nanolett.5b05030
C. Wang, W. Sun, Y. Ye, et al., Nat. Biomed. Eng. 1 (2017) 0011. doi: 10.1038/s41551-016-0011
S. Han, W. Wang, S. Wang, et al., Theranostics 11 (2021) 2892–2916. doi: 10.7150/thno.50928
W. Ou, R.K. Thapa, L. Jiang, et al., J. Control. Release 281 (2018) 84–96. doi: 10.1016/j.jconrel.2018.05.018
Q. Liu, D. Zhang, H. Qian, et al., Int. J. Nanomed. 15 (2020) 3669–3680. doi: 10.2147/ijn.s249174
J. Li, D.J. Mooney, Nat. Rev. Mater. 1 (2016) 16071. doi: 10.1038/natrevmats.2016.71
S.W. Jung, S.H. Oh, I.S. Lee, J.H. Byun, J.H. Lee, Tissue Eng. Regen. Med. 16 (2019) 479–490. doi: 10.1007/s13770-019-00206-x
Y. Ando, E.L. Siegler, H.P. Ta, et al., Adv. Healthc. Mater. 8 (2019) e1900001. doi: 10.1002/adhm.201900001
Y. Hori, A.M. Winans, C.C. Huang, E.M. Horrigan, D.J. Irvine, Biomaterials 29 (2008) 3671–3682. doi: 10.1016/j.biomaterials.2008.05.033
K. Wang, Y. Chen, S. Ahn, et al., Nat. Cancer 1 (2020) 990–997. doi: 10.1038/s43018-020-00119-y
Y. Dong, S. Li, X. Li, X. Wang, Int. J. Biol. Macromol. 190 (2021) 693–699. doi: 10.1016/j.ijbiomac.2021.09.037
Y. Cao, Y. Zhou, Z. Chen, et al., Adv. Healthc. Mater. 10 (2021) e2100814. doi: 10.1002/adhm.202100814
C. Liu, P. Leon-Plata, M. Zaroudi, et al., ACS Appl. Bio. Mater. 3 (2020) 7357–7362. doi: 10.1021/acsabm.0c00992
E.A. Ogunnaike, A. Valdivia, M. Yazdimamaghani, et al., Sci. Adv. 7 (2021) eabg5841. doi: 10.1126/sciadv.abg5841
X. Dong, A. Yang, Y. Bai, D. Kong, F. Lv, Biomaterials 230 (2020) 119659. doi: 10.1016/j.biomaterials.2019.119659
A.F. Atik, C.M. Suryadevara, R.M. Schweller, et al., J. Clin. Neurosci. 56 (2018) 163–168. doi: 10.1016/j.jocn.2018.06.005
Q. Hu, H. Li, E. Archibong, et al., Nat. Biomed. Eng. 5 (2021) 1038–1047. doi: 10.1038/s41551-021-00712-1
Z. Luo, Z. Liu, Z. Liang, et al., ACS Appl. Mater. Interfaces 12 (2020) 56712–56722. doi: 10.1021/acsami.0c15239
T. Shang, X. Yu, S. Han, B. Yang, Biomater. Sci. 8 (2020) 5241–5259. doi: 10.1039/d0bm01158d
X. Xu, T. Li, S. Shen, et al., Theranostics 9 (2019) 7889–7905. doi: 10.7150/thno.38583
R. Ahmad, J. Fu, N. He, S. Li, J. Nanosci. Nanotechnol. 16 (2016) 67–80. doi: 10.1166/jnn.2016.10770
Y. Huang, G. Zeng, Q. Xin, et al., MedComm 1 (2020) 202–210. doi: 10.1002/mco2.28
T. Yata, Y. Takahashi, M. Tan, et al., Biomaterials 146 (2017) 136–145. doi: 10.1016/j.biomaterials.2017.09.014
J. Nam, S. Son, L.J. Ochyl, et al., Nat. Commun. 9 (2018) 1074. doi: 10.1038/s41467-018-03473-9
A.L. Oei, L.E. Vriend, J. Crezee, N.A. Franken, P.M. Krawczyk, Radiat. Oncol. 10 (2015) 165. doi: 10.1186/s13014-015-0462-0
S. Wang, Q. Zhang, X.F. Luo, et al., Biomaterials 35 (2014) 9473–9483. doi: 10.1016/j.biomaterials.2014.07.064
H. Yang, H. He, Z. Tong, et al., J. Colloid Interface Sci. 565 (2020) 186–196. doi: 10.1016/j.jcis.2020.01.026
Z. Liao, W. Zhang, Z. Qiao, et al., J. Colloid Interface Sci. 562 (2020) 81–90. doi: 10.1016/j.jcis.2019.11.055
X. Zhang, J. Tang, C. Li, et al., Bioact. Mater. 6 (2021) 472–489. doi: 10.1016/j.bioactmat.2020.08.024
F. Zhang, G. Lu, X. Wen, et al., J. Control. Release 326 (2020) 131–139. doi: 10.1016/j.jconrel.2020.06.015
Q. Yang, J. Peng, K. Shi, et al., J. Control. Release 308 (2019) 29–43. doi: 10.1016/j.jconrel.2019.06.031
X. Yi, Q.Y. Duan, F.G. Wu, Research 2021 (2021) 9816594.
I.C. Miller, A. Zamat, L.K. Sun, et al., Nat. Biomed. Eng. (2021) 1348–1359. doi: 10.1038/s41551-021-00781-2
W. Ma, D. Zhu, J. Li, et al., Theranostics 10 (2020) 1281–1295. doi: 10.7150/thno.40291
Q. Chen, Q. Hu, E. Dukhovlinova, et al., Adv. Mater. 31 (2019) e1900192. doi: 10.1002/adma.201900192
L. Zhu, J. Liu, G. Zhou, et al., Small (2021) e2102624.
Z. Chen, H. Pan, Y. Luo, et al., Small 17 (2021) e2007494. doi: 10.1002/smll.202007494
H.J. Jackson, S. Rafiq, R.J. Brentjens, Nat. Rev. Clin. Oncol. 13 (2016) 370–383. doi: 10.1038/nrclinonc.2016.36
S.S. Neelapu, S. Tummala, P. Kebriaei, et al., Nat. Rev. Clin. Oncol. 15 (2018) 47–62. doi: 10.1038/nrclinonc.2017.148
B. Wolf, S. Zimmermann, C. Arber, et al., Drug Saf. 42 (2019) 315–334. doi: 10.1007/s40264-018-0779-3
M. Norelli, B. Camisa, G. Barbiera, et al., Nat. Med. 24 (2018) 739–748. doi: 10.1038/s41591-018-0036-4
T. Giavridis, S.J.C. van der Stegen, J. Eyquem, et al., Nat. Med. 24 (2018) 731–738. doi: 10.1038/s41591-018-0041-7
Y. Liu, Y. Fang, X. Chen, et al., Sci. Immunol. 5 (2020) eaax7969. doi: 10.1126/sciimmunol.aax7969
A. Taraseviciute, V. Tkachev, R. Ponce, et al., Cancer Discov. 8 (2018) 750–763. doi: 10.1158/2159-8290.cd-17-1368
S.J. Schuster, J. Svoboda, E.A. Chong, et al., New Engl. J. Med. 377 (2017) 2545–2554. doi: 10.1056/NEJMoa1708566
S.L. Maude, N. Frey, P.A. Shaw, et al., New Engl. J. Med. 371 (2014) 1507–1517. doi: 10.1056/NEJMoa1407222
M.L. Davila, I. Riviere, X. Wang, et al., Sci. Transl. Med. 6 (2014) 224 ra25.
C.J. Turtle, K.A. Hay, J. Gust, et al., J. Clin. Oncol. 35 (2017) 3020. doi: 10.1200/JCO.2017.35.15_suppl.3020
D.W. Lee, R. Gardner, D.L. Porter, et al., Blood 124 (2014) 188–195. doi: 10.1182/blood-2014-05-552729
C.J. Turtle, L.A. Hanafi, C. Berger, et al., J. Clin. Invest. 126 (2016) 2123–2138. doi: 10.1172/JCI85309
J.N. Brudno, J.N. Kochenderfer, Blood Rev. 34 (2019) 45–55. doi: 10.1016/j.blre.2018.11.002
T. Giavridis, S.J.C. van der Stegen, J. Eyquem, et al., Nat. Med. 24 (2018) 731–738. doi: 10.1038/s41591-018-0041-7
R.M. Sterner, R. Sakemura, M.J. Cox, et al., Blood 133 (2019) 697–709. doi: 10.1182/blood-2018-10-881722
A. Shetty, R. Hanson, P. Korsten, et al., Drug Des. Devel. Ther. 8 (2014) 349–364.
M. Hong, J.D. Clubb, Y.Y. Chen, Cancer Cell 38 (2020) 473–488. doi: 10.1016/j.ccell.2020.07.005
E.W. Weber, R.C. Lynn, K.R. Parker, et al., bioRxiv (2020), https://doi.org/10.1101/2020.01.26.920496. doi: 10.1101/2020.01.26.920496
A. Juillerat, A. Marechal, J.M. Filhol, et al., Sci. Rep. 7 (2017) 39833. doi: 10.1038/srep39833
M. Casucci, L. Falcone, B. Camisa, et al., Front. Immunol. 9 (2018) 507. doi: 10.3389/fimmu.2018.00507
A. Di Stasi, S.K. Tey, G. Dotti, et al., New Engl. J. Med. 365 (2011) 1673–1683. doi: 10.1056/NEJMoa1106152
K.C. Straathof, M.A. Pulè, P. Yotnda, et al., Blood 105 (2005) 4247–4254. doi: 10.1182/blood-2004-11-4564
B. Kwong, S.A. Gai, J. Elkhader, K.D. Wittrup, D.J. Irvine, Cancer Res. 73 (2013) 1547–1558.
Y. Zheng, L. Tang, L. Mabardi, S. Kumari, D.J. Irvine, ACS Nano 11 (2017) 3089–3100. doi: 10.1021/acsnano.7b00078
Y.Q. Xie, H. Arik, L. Wei, et al., Biomater. Sci. 7 (2019) 1345–1357. doi: 10.1039/c8bm01556b
L. Tang, Y. Zheng, M.B. Melo, et al., Nat. Biotechnol. 36 (2018) 707–716. doi: 10.1038/nbt.4181
Y. Zhang, N. Li, H. Suh, D.J. Irvine, Nat. Commun. 9 (2018) 6. doi: 10.1038/s41467-017-02251-3
T.T. Smith, H.F. Moffett, S.B. Stephan, et al., J. Clin. Invest. 127 (2017) 2176–2191. doi: 10.1172/JCI87624
S.B. Stephan, A.M. Taber, I. Jileaeva, et al., Nat. Biotechnol. 33 (2015) 97–101. doi: 10.1038/nbt.3104
S.K. Härmälä, R. Butcher, C.H. Roberts, Methods Mol. Biol. 1654 (2017) 135–149. doi: 10.1007/978-1-4939-7231-9_9
L.B. Pinheiro, V.A. Coleman, C.M. Hindson, et al., Anal. Chem. 84 (2012) 1003–1011. doi: 10.1021/ac202578x
E.D. Nair-Gill, C.J. Shu, C.G. Radu, O.N. Witte, Immunol. Rev. 221 (2008) 214–228. doi: 10.1111/j.1600-065X.2008.00585.x
E.T. Ahrens, J.W. Bulte, Nat. Rev. Immunol. 13 (2013) 755–763. doi: 10.1038/nri3531
H. Singh, A.M. Najjar, S. Olivares, et al., Leukemia 23 (2009) 620–622. doi: 10.1038/leu.2008.256
G. Dotti, M. Tian, B. Savoldo, et al., Mol. Imaging 8 (2009) 230–237.
J. Grimm, M.F. Kircher, R. Weissleder, Radiologe 47 (2007) 25–33. doi: 10.1007/s00117-006-1449-5
R. Zhou, D.H. Thomas, H. Qiao, et al., J. Nucl. Med. 46 (2005) 816–822.
R.G. Blasberg, J. Gelovani, Mol. Imaging 1 (2002) 280–300. doi: 10.1162/153535002760235472
V. Ponomarev, M. Doubrovin, I. Serganova, et al., Eur. J. Nucl. Med. Mol. Imaging 31 (2004) 740–751. doi: 10.1007/s00259-003-1441-5
B.A. Tannous, J. Grimm, K.F. Perry, et al., Nat. Methods 3 (2006) 391–396. doi: 10.1038/nmeth875
R. Weissleder, A. Moore, U. Mahmood, et al., Nat. Med. 6 (2000) 351–355. doi: 10.1038/73219
D.B. Kohn, M. Sadelain, J.C. Glorioso, Nat. Rev. Cancer 3 (2003) 477–488. doi: 10.1038/nrc1122
P. Bhatnagar, Z. Li, Y. Choi, et al., Integr. Biol. (Camb) 5 (2013) 231–238. doi: 10.1039/c2ib20093g
H. Li, L. Diaz, D. Lee, et al., Radiol. Med. 119 (2014) 269–276. doi: 10.1007/s11547-013-0335-2
N. Emami-Shahri, J. Foster, R. Kashani, et al., Nat. Commun. 9 (2018) 1081. doi: 10.1038/s41467-018-03524-1
F. Chapelin, S. Gao, H. Okada, et al., Sci. Rep. 7 (2017) 17748. doi: 10.1038/s41598-017-17669-4
D.V. Hingorani, F. Chapelin, E. Stares, et al., Magn. Reson. Med. 83 (2020) 974–987. doi: 10.1002/mrm.27988
L. Liu, Q. Ye, Y. Wu, et al., Nanomedicine 8 (2012) 1345–1354. doi: 10.1016/j.nano.2012.02.017
A. Kulkarni, P. Rao, S. Natarajan, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) E2104–2113. doi: 10.1073/pnas.1510959113
M. Horie, K. Nishio, K. Fujita, et al., Chem. Res. Toxicol. 22 (2009) 543–553. doi: 10.1021/tx800289z
P. Chandran, J.E. Riviere, N.A. Monteiro-Riviere, Nanotoxicology 11 (2017) 507–519. doi: 10.1080/17435390.2017.1314036
E. Preedia Babu, A. Subastri, A. Suyavaran, et al., Sci. Rep. 7 (2017) 4203. doi: 10.1038/s41598-017-04440-y
H. Huang, W. Lai, M. Cui, et al., Sci. Rep. 6 (2016) 25518. doi: 10.1038/srep25518
T. Wang, X. Jiang, ACS Appl. Mater. Interfaces 7 (2015) 129–136. doi: 10.1021/am503865g
H.L. Karlsson, P. Cronholm, Y. Hedberg, et al., Toxicology 313 (2013) 59–69. doi: 10.1016/j.tox.2013.07.012
X. He, W.G. Aker, H.M. Hwang, Nanotoxicology 8 (Suppl. 1) (2014) 185–195. doi: 10.3109/17435390.2013.874050
A. Alalaiwe, G. Roberts, P. Carpinone, J. Munson, S. Roberts, Drug Deliv. 24 (2017) 591–598. doi: 10.1080/10717544.2017.1282554
Y. Chen, C. Ren, S. Ouyang, X. Hu, Q. Zhou, Environ. Sci. Technol. 49 (2015) 10147–10154. doi: 10.1021/acs.est.5b02220
J.R. Velicogna, E.E. Ritchie, R.P. Scroggins, J.I. Princz, Nanotoxicology 10 (2016) 1144–1151. doi: 10.1080/17435390.2016.1181807
S.M. Moghimi, A.C. Hunter, T.L. Andresen, Annu. Rev. Pharmacol. Toxicol. 52 (2012) 481–503. doi: 10.1146/annurev-pharmtox-010611-134623
D.E. Owens 3rd, N.A. Peppas, Int. J. Pharmacol. 307 (2006) 93–102. doi: 10.1016/j.ijpharm.2005.10.010
S. Nie, Nanomedicine 5 (2010) 523–528. doi: 10.2217/nnm.10.23
S.D. Li, L. Huang, Biochim. Biophys. Acta 1788 (2009) 2259–2266. doi: 10.1016/j.bbamem.2009.06.022
L. Brannon-Peppas, J.O. Blanchette, Adv. Drug. Deliv. Rev. 56 (2004) 1649–1659. doi: 10.1016/j.addr.2004.02.014
O. Veiseh, J.W. Gunn, M. Zhang, Adv. Drug. Deliv. Rev. 62 (2010) 284–304. doi: 10.1016/j.addr.2009.11.002
H. Cao, Z. Dan, X. He, et al., ACS Nano 10 (2016) 7738–7748. doi: 10.1021/acsnano.6b03148
H. Cao, B. Jiang, Y. Yang, et al., J. Colloid Interface Sci. 604 (2021) 596–603. doi: 10.1016/j.jcis.2021.07.004
T. Kang, Q. Zhu, D. Wei, et al., ACS Nano 11 (2017) 1397–1411. doi: 10.1021/acsnano.6b06477
Y. Guo, D. Wang, Q. Song, et al., ACS Nano 9 (2015) 6918–6933. doi: 10.1021/acsnano.5b01042
D. Gao, X. Guo, X. Zhang, et al., Mater. Today Bio 5 (2020) 100035. doi: 10.1016/j.mtbio.2019.100035
X. Yu, H. Zheng, M.T.V. Chan, W.K.K. Wu, Environ. Sci. Pollut. Res. Int. 25 (2018) 20569–20574. doi: 10.1007/s11356-018-2426-z
H.S. Hwang, H. Shin, J. Han, K. Na, J. Pharm. Investig. 48 (2018) 143–151. doi: 10.1007/s40005-017-0377-x
X. Xu, H. Lu, R. Lee, Front. Bioeng. Biotechnol. 8 (2020) 488. doi: 10.3389/fbioe.2020.00488
G. Lan, K. Ni, Z. Xu, et al., J. Am. Chem. Soc. 140 (2018) 5670–5673. doi: 10.1021/jacs.8b01072
D.W. Zheng, F. Gao, Q. Cheng, et al., Nat. Commun. 11 (2020) 1985. doi: 10.1038/s41467-020-15927-0
Y. Wang, Y.X. Lin, S.L. Qiao, et al., Biomaterials 112 (2017) 153–163. doi: 10.3901/JME.2017.05.153


Figure 4 Different nanotechnologies for T-cell transfection. (A) Cationic polymers for gene delivery to human T cells. Reproduced with permission [109]. Copyright 2018, Elsevier. (B) Schematic of polydopamine-sensitized photoporation for the delivery of mRNA into cells. Reproduced with permission [123]. Copyright 2021, Wiley. (C) Schematic of the components used to generate lipid nanoparticles (LNPs) via microfluidic mixing. Reproduced with permission [132]. Copyright 2020, American Chemical Society. (D) Bioorthogonal PEI-DBCO/azide–glucose system in CAR-T-cell manufacture and immunotherapy. Reproduced with permission [137]. Copyright 2019, Wiley.

Figure 6 (A) Schematic of the injectable i-G/MC system for the intratumoral delivery of CAR-T cells. (B) Fluorescence IVIS images depicting the release profiles of molecule payloads modeling Hemo and IL-15 in vivo. Reproduced with permission [214]. Copyright 2020, American Chemical Society.

Figure 7 (A) Schematic illustration showing the effects of the mild heating of the tumor. (A) Representative bioluminescence of the WM115 tumors. Reproduced with permission [231]. Copyright 2019, Wiley.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:






 下载:
下载:


 下载:
下载: