
Figure 1.
Schematic representation of the molecular structures and the landscape of the supramolecular self-assemblies of different types of LTPs. (a) LTP, (b) DTP, (c) type 1, (d) type 2, (e) type 3.
Synergistic regulation of intermolecular interactions to control chiral structures for chiral recognition
Weilin Chen , Lulu Fu , Zhiqiang Zhu , Jingyan Liu , Linxiu Cheng , Zhou Xu , Hao Dong , Jing Ma , Yibao Li , Xiaolin Fan

Chirality is an essential feature of life and plays a vital role in various chemical and biological processes. The double helix of DNA and the triple helix of collagen are all chiral compounds [1-3]. Helical and twisted nanostructures have attracted attention due to their important roles in the fields of catalysis, ion sensing or recognition, fabrication of chiroptical switches, medicine, separation, and biology [4-13]. To obtain chiral nanostructures, supramolecular assemblies are often constructed in a controllable way through noncovalent interactions, such as hydrogen bonding, van der Waals, π-π stacking, and electrostatic interactions. Numerous helical and twisted nanostructures have been successfully synthesized via molecular self-assembly [14-16].
Usually, the construction of chiral structures depends on two factors. First, helical and twisted nanostructures are easily developed from the aggregation of the corresponding chiral building blocks. Hence, the supramolecular chirality of nanostructures depends on the inherent molecular chirality of the building blocks [17-19]. Besides, the building blocks' stacking arrangement plays a crucial role in the supramolecular chirality of the assemblies. Therefore, by manipulating the competitive local chiral interactions and overall geometric constraints in the self-assembled structure, the formation of chiral nanostructures and its structure control can be achieved [20-24].
The building blocks' stacking arrangement is generally dynamic and changes according to external stimuli, such as light, temperature, solvent, pH, rotary stirring, and metal-ions [25-30]. Much work has been done on constructing chiral structures, but very little research has been done on the dynamic processes that produce chiral structures. It remains a fundamental challenge to precisely tailor the intricate balance between the noncovalent forces during the gradual amplification of molecular chirality at different hierarchical processes. Therefore, a detailed understanding of the dynamic changes occurring in the intermolecular noncovalent interactions during the chiral structures' formation is beneficial to constructing chiral structures and their potential applications.
We selected a C3 symmetric 1, 3, 5-benzentricarboxylic amino acid methyl ester (TPE) enantiomer (left-handed LTP and right-handed DTP) as the building block of supramolecular structures to understand the dynamic changes in the intermolecular noncovalent interactions during the formation of chiral systems. We observed that the self-assembly of TPE involves the synergy of various weak intermolecular interactions, including hydrophobic, hydrogen bonding, π-π stacking interactions, among others. Hydrophobic interactions tend to drive the TPE molecules to aggregate in a disordered arrangement to form irregular flower-like structures. The hydrogen bonding and π-π stacking interactions tend to induce TPE assembly to form one-dimensional nano-assemblies with limited architectures, such as nanofibers. Hydrogen bonding and π-π stacking interactions help the TPE molecules assemble to form ordered spiral fiber aggregates (right-handed, P; left-handed, M). Furthermore, the chiral structure constructed from TPE molecules is highly stereoselective for diamine compounds.
In TPE molecules, the three amide bonds are capable of H-bond formation between the neighboring molecules, and the face-to-face π-π stacking interactions provide additional stabilizations. TPEs are highly symmetric compounds and possess an amphiphilic nature because of the concomitant presence of the hydrophobic benzene and the hydrophilic amide bonds, which potentially induce and direct the self-assembly under certain conditions. In addition, the amide bonds of the TPE molecules can rotate freely, indicating that there are many types of interactions between the TPE molecules. Left-handed 1, 3, 5-benzentricarboxylic amino acid methyl esters (LTP) were selected as the subject of research. Three types of detailed self-assembly mechanisms of the LTP molecules are shown in Fig. 1.
As for the type 1 mechanism, in CH3OH, the LTP molecules can be uniformly dispersed. The distance between the molecules is relatively large, making it difficult to generate stable hydrogen bonds and π-π stacking between them. However, even in CH3OH, the molecules form irregular flower-like structures by kinetic aggregation. In the presence of water, the self-assembly occurs dominantly in a thermodynamically controlled fashion. The addition of water (10%) causes a rapid change in the solution's polarity and increased hydrophobic interaction. The LTP molecules aggregate rapidly. For the type 2 mechanism, the intermolecular hydrogen bonding interaction is enhanced when the molecules get aggregated. However, the intermolecular π-π stacking interaction is still weak due to the low content of the poor-solvents. In this case, hydrogen bonding interaction is the main driving force for self-assembly, and the LTP molecules form long-range ordered fibrous structures. For the type 3 mechanism (water 60%), the LTP molecular packing is converted from the displaced packing mode to face-to-face packing mode. In this case, the driving force of molecular self-assembly is the π-π stacking interactions between molecules. The LTP molecules form stable and ordered helical fibers. The three types of self-assembly seen in the LTP molecules indicate that the mode and size of the intermolecular noncovalent interactions play a crucial role in assembling the molecules into various structures.
The morphologies of the LTP molecules were examined by the scanning electron microscopy (SEM) technique. The SEM images corresponding to the patterns of the three different types of noncovalent interactions are shown in Fig. 2. For type 1, hydrogen bonding and π-π stacking interactions between the molecules are weak. The LTP molecule forms irregular flower-like structures (Fig. 2a). Interestingly, in a CH3OH-water binary system with a low amount of water (type 2, 10% water), the addition of hydrogen-bond-forming solvents successfully induced the formation of long-range ordered fibers in LTP molecules (Fig. 2b). Further increase in the water content (type 3, 60% water) enhances the intermolecular hydrogen bond and π-π stacking interactions so that the long-range-ordered left-handed helical fibers were observed (Fig. 2c). As expected, the self-assembly morphology of the DTP molecules was similar to that of the LTP molecules under the same conditions (Fig. S1 in Supporting information). The helical fibers formed different helical orientations due to the different chirality: LTP forms left-handed helical nano-fibers, DTP forms right-handed helical fibers. During solvent evaporation during the drying process, the liquid samples were observed by atomic force microscopy (AFM). Similar morphologies were also observed in AFM images (Fig. S2 in Supporting information). These observations suggested that hydrogen-bonding interactions play a key role in fiber formation. On the other hand, π-π stacking interactions can influence molecules' arrangement, thus inducing the helical fibers' formation.
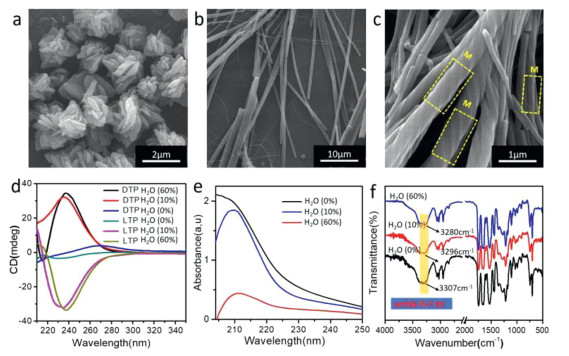
To gain further insight into the secondary structures of the LTP nanostructures, circular dichroism (CD) spectroscopy and Fourier-transform infrared (FT-IR) spectroscopy measurements were performed. CD spectroscopy is a powerful tool for understanding the arrangement of chiral molecules in the assembly. Intermolecular interactions, especially between chromophore molecules, may produce striking chiroptical responses, often much stronger in the assembled state than in their isolated molecular state, and generate CD signals. Concerning the CD analysis of the D-TPE in type 1 self-assembly (Fig. 2d), the irregular flower-like structures were completely CD silent in the spectral range. The red-shift of the wavelength at the maximum intensity of CD spectra suggested that hydrogen bonding and π-π stacking were enhanced. Similar results were observed in the ultraviolet absorption spectra (UV-vis) (Fig. 2e). These spectral changes indicate that the formation of hydrogen bonds and the generation of π-π stacking interactions lead to the change of the stacking arrangement of self-assembling building blocks.
Fourier-transform infrared spectroscopy (FT-IR) analysis (Fig. 2f) showed that the TPE (type 1) exhibited strong adsorption peaks at 3354 cm–1 (amide N-H), which could be ascribed to the isolated amide groups. As the water content was increased to 10% (type 2), the amide N-H peak at 3354 cm–1 showed a red-shift to 3251 cm–1 and the peak became broader. The red-shift and the change in peak shape indicated that the amide groups in the fibers formed weak hydrogen bonds. However, the red-shift of the amide N-H was more pronounced in type 3 interactions. These results suggested that the hydrogen bonds assigned to the amide groups were much stronger in the helical fibers than in the non-helical fibers because of the formation of strong extended hydrogen bonds between the TPE molecules with increasing water content.
Nuclear magnetic resonance (NMR) spectra were also analyzed to elucidate the self-assembly mechanism of the LTP molecules. In type 1, the aromatic protons appeared as sharp peaks, which broadened considerably in the presence of D2O (Fig. S3 in Supporting information). In type 2, a clear up-field shift of the aromatic protons of the benzene-1, 3, 5-tricarboxamide in the TPE molecule was observed, and the protons moved further up-field with further increase in D2O concentration in type 3. This further confirms π-π stacking. Interestingly, the phenylalanine protons showed multiple signals; one part down-field and the other part up-field. These are the split signals arising from three phenylalanine groups. This change in chemical shift demonstrated the de-shielding effect caused by hydrogen bonding between molecules.
To obtain further structural information, we produced LTP single crystals. Detailed crystallographic data can be found in (Table S1 in Supporting information). It should be noted that the crystals could only be obtained in the CH3OH/water mixed solvent system (water 60%). Single-crystal X-ray diffraction (XRD) analyses showed that ordered helical assemblies of LTP are formed through multiple types of non-bonded interactions, including H-bonding, π-π, and C-H···π interactions, between adjacent subunits (Fig. 3). The amide N-H groups on each of the three arms of the subunits formed a hydrogen bond with the carbonyl group on the neighboring subunits, with the bond length being ~2.0 Å between the heavy atoms (Table S2 in Supporting information). Extensive π-π stacking interactions between the central benzene rings of the two adjacent molecules could be observed (in the parallel or the T-shaped configurations). The latter was characterized by C-H···π interactions. Taken together, the versatile noncovalent interactions contribute to a stable chiral helical structure of LTP molecules.

For exploring the formation mechanism of the twisted nanofibers, multi-scale modeling was performed to study the molecular conformation and self-assembly behavior of LTP under different solvent components. Molecular dynamics (MD) simulations were carried out on the LTP helix in solutions using a sander module of AMBER16 CUDA version [31-33]. The Packmol program was used to randomly distribute molecules with a tolerance of 2.0 Å in the periodic box of dimension 70 Å × 70 Å × 70 Å (Fig. S4 and Table S3 in Supporting information). The LTP helix was parameterized using the generalized AMBER force field (GAFF) [34]. The complex was explicitly solvated with TIP3P water and methyl alcohol molecules (at different ratios). After energy minimization, the system was well equilibrated with MD simulations, where the LTP's position was restrained. The temperature of the system was slowly raised to 300 K in 2 ns at NPT conditions. Then, we removed the system's restraints and accumulated the simulations for another 100 ns trajectory.
Time evolution of the centroid distance (Fig. S5a in Supporting information) and the root mean square deviations (RMSD) were calculated to detect the foldamers' intrinsic motion in solution. We found that an increase in the water content of the mixed solvent leads to a decrease in the centroid distance of the adjacent dimers (Figs. S5b-e in Supporting information). The quadruple stranded foldamer LTP showed a small fluctuation over time in aqueous solution than other solvent systems (Fig. S6 in Supporting information), indicating that the helical structure could be well-maintained.
To further investigate the nature of interactions between different solvent molecules and the LTP molecule, the radial distribution function (RDF) of water (oxygen) and methanol (oxygen) molecules around LTP (oxygen and nitrogen) was calculated (Fig. S7 in Supporting information). We found that the solvent does not influence the first peak position for the same system. However, an apparent shift in the peak location of different systems was observed. The first and the second peaks of the atom N in LTP and O in methanol were approximately at 2.1–2.3 Å and 3.0–3.3 Å in the RDFs, respectively. There is only one peak within 4.0 Å of others in the RDF, located approximately at 2.8–3.0 Å, 2.8–3.0 Å, and 3.0–3.3 Å for the atom O in LTP and O in methanol, the atom O in LTP and O in water, and the atom N in LTP and O in water, respectively. The different peak positions suggested that methanol is more likely to form intermolecular interactions with the solute.
We further carried out quantum mechanical (QM) calculations to characterize the 1H NMR spectrum of the LTP structure. From the above-mentioned MD simulations, several clusters containing solvent molecules, within 3.0 Å of the quadruple stranded foldamer LTP, were constructed. Chemical shielding data were calculated using the generalized energy-based fragmentation (GEBF) method [35-37] at the B3LYP (D3BJ) [38]/pcsSeg-1 level, as implemented in low-scaling quantum chemistry (LSQC) [39] employing the gauge-including atomic orbital (GIAO) approach [40,41]. The obtained NMR signals were in line with the experimental results (Tables S4 and S5 in Supporting information), with a deviation of only 0.93 ppm for the benzene-1, 3, 5-tricarboxamide group of LTP.
Meanwhile, we calculated the binding energy of the extracted cluster of LTP and solvent within 3.0 Å with both QM (at the GEBF-B3LYP-D3/6–31G(d, p) level) and molecular mechanics (including the polymer consistent force field (PCFF) [42-44] and COMPASS [45]) methods. Both methods showed that the LTP shows a stronger binding affinity with methanol than with water (Table S6 in Supporting information).
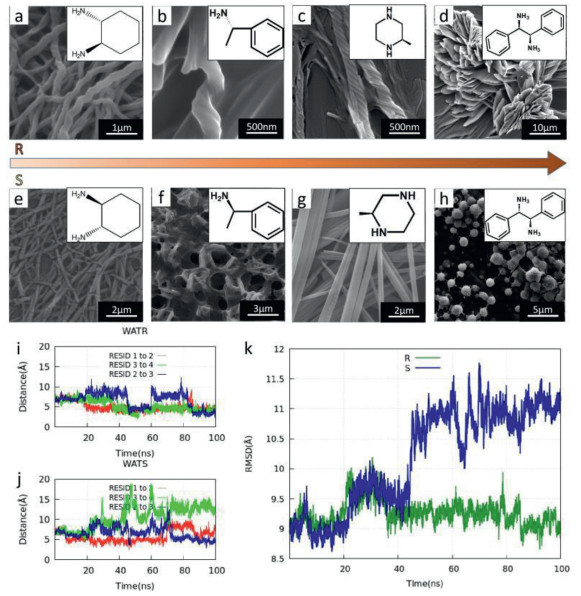
The TPE molecules possess three hydrophilic amide groups and hydrophobic ester groups. Such properties are more likely to show different degrees of van der Waals forces, hydrogen bonds, and other noncovalent interactions, making the TPE molecules suitable for chiral recognition. As shown in Fig. 4a, the LTP self-assembly formed a left-handed helical structure with the R-cyclohexanediamine, but formed an intermittent nanofiber with the S-cyclohexanediamine. Figs. 4b and f show LTP and methyl benzylamine. It was found that the R-type methylamine induced a helical structure in LTP, while the S-type induced the formation of a disordered pore-like structure. Comparing Figs. 4c and g, it was seen that the LTP formed a nano-helical structure with (R)-(-)-2-methyl piperazine, while the (R)-(-)-2-methyl piperazine formed an ordered fibrous structure. These comparisons indicate that the LTP has a more substantial recognition effect on R-type imide.
The UV (Fig. S9 in Supporting information) absorption maxima did not have significant change when the R or S type of 1, 2-cyclohexanediamine was added to the LTP solution. But the intensity of the UV maxima at 210 nm was different at the same concentration, indicating the noncovalent interactions between the LTP molecules and amines of different isomers are different. Compared with the (1S, 2S)-(+)-1, 2-cyclohexanediamine, the LTP and the (1R, 2R)-(−)-1, 2-cyclohexanediamine have stronger interaction, which leads to increased solubility of the molecule, resulting in stronger UV spectral intensity. The difference in the amount of solute in the solution produces a visual representation of the degree of recognition. NMR spectra were analyzed to elucidate the recognition mechanism of the LTP with the 1, 2-cyclohexanediamine. 1H NMR spectra (Fig. S10 in Supporting information) showed a pronounced higher chemical shift value for the aromatic protons in LTP after the addition of (1R, 2R)-(−)-1, 2-cyclohexanediamine (as compared to the addition of (1S, 2S)-(+)-1, 2-cyclohexanediamine).
To study the effect of the conformation of amine on the helical structure of LTP, we also conducted the MD simulations that randomly put (1R, 2R)-(−)-1, 2-cyclohexanediamine and (1S, 2S)-(−)-1, 2-cyclohexanediamine into the aqueous solutions containing LTP. Taking the crystal structure as the reference, the time evolution of the centroid distance and RMSD were analyzed shown in (Figs. 4i–k). The centroid distance and RMSD fluctuated significantly with time, indicating that the LTP's helical structure changes dramatically. The interactions between LTP and (1R, 2R)-(−)-1, 2-cyclohexanediamine as well as (1S, 2S)-(−)-1, 2-cyclohexanediamine have been further studied using the DFT quantum mechanical method at the GEBF-B3LYP-D3/6–31G(d, p) level. The configurations of the LTP and (1R, 2R)-(−)-1, 2-cyclohexanediamine molecules as well as (1S, 2S)-(−)-1, 2-cyclohexanediamine were extracted from the MD simulations (Figs. S11 and S12 in Supporting information). As shown in (Table S7 in Supporting information), the binding energy between LTP and S-cyclohexanediamine is much lower than that between LTP and R-cyclohexanediamine. The results demonstrated that intermolecular interactions are essential for chiral recognition, which may be a major force in destabilizing the helical structure. All these results imply that TPE molecules have potential applications in the field of chiral recognition.
In summary, we constructed a C3-symmetric phenylalanine enantiomer (TPE) to study the dynamic self-assembly processes in the molecule regulated by the solvent. LTP molecules showed different micromorphology in different solvent systems consisting of different proportions of methanol and water. The noncovalent interactions present in LTP molecules (in different types) have been confirmed by CD, UV, FTIR, NMR, and crystallography techniques. The results were consistent with MD simulation results. The formation of the chiral structure is the result of the synergistic effect of multiple noncovalent interaction forces. Furthermore, the chiral structures with three hydrophilic amide groups and hydrophobic ester groups were used for the chiral recognition of chiral diamines, showing good stereoselectivity. This work provides valuable insights for the regulation of chiral supramolecular structures and shines a light on the potential applications of the molecules in the field of chiral recognition.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (No. 21962003), the Natural Science Foundation of Jiangsu Province (No. BK20190056), and the "Fundamental Research Funds for the Central Universities" (No. 021514380014).
Supplementary material associated with this article can be found, in the online version, at doi:
R.A. Hegstrom, D.K. Kondepudi, Sci. Am. 262 (1990) 108-115. doi: 10.1038/scientificamerican0190-108
G M. Whitesides, B.A. Grzybowski, Science 295 (2002) 2418-2421. doi: 10.1126/science.1070821
J. Lehn, Angew. Chem. Int. Ed. 29 (1990) 1304-1319. doi: 10.1002/anie.199013041
M. Liu, L. Zhang, T. Wang, Chem. Rev. 115 (2015) 7304-7397. doi: 10.1021/cr500671p
Y. Wang, W. Qi, R. Huang, et al, J. Am. Chem. Soc. 137 (2015) 7869-7880. doi: 10.1021/jacs.5b03925
C. Zhou, Y. Ren, J. Han, et al, J. Am. Chem. Soc. 140 (2018) 9417-9425. doi: 10.1021/jacs.7b12178
Y. Cai, Z. Zhang, Y. Ding, et al, Chin. Chem. Lett. 32 (2020) 1267-1279 doi: 10.1016/j.cclet.2020.10.036
T. Xiao, J. Wang, Y. Shen, et al, Chin. Chem. Lett. 32 (2020) 1377-1380. doi: 10.1016/j.cclet.2020.10.037
T. Xiao, L. Zhou, X.Q. Sun, et al., Chin. Chem. Lett. 31 (2019) 1-9. doi: 10.1016/j.cclet.2019.05.011
K. Tao, A. Levin, L. Adler-Abramovich, E. Gazit, Chem. Soc. Rev. 45 (2016) 3935-3953. doi: 10.1039/C5CS00889A
L. Li, W. Tuo, Q. Zhu, J. Am. Chem. Soc. 142 (2020) 20583-20587. doi: 10.1021/jacs.0c10901
B. Slater, Z. Wang, S. Jiang, M.R. Hill, B.P. Ladewig, J. Am. Chem. Soc. 139 (2017) 18322-18327. doi: 10.1021/jacs.7b10112
H.L. Qian, C.X. Yang, X.P. Yan, Nat. Commun. 7 (2016) 12104. doi: 10.1038/ncomms12104
Y. Yang, Y. Zhang, Z. Wei, Adv. Mater. 25 (2013) 6039-6049. doi: 10.1002/adma.201302448
G. Basuyaux, A. Desmarchelier, G. Gontard, et al., Chem. Commun. 55 (2019) 8548-8551. doi: 10.1039/c9cc03906f
Y. Zhang, L. Shi, Z. Zhang, Q. Zhao, Y. Yao, Inorg. Chem. 59 (2020) 7924-7927. doi: 10.1021/acs.inorgchem.0c01164
A.R. Palmans, E.W. Meijer, Angew. Chem. Int. Ed. 46 (2007) 8948-8968. doi: 10.1002/anie.200701285
G. Liu, J. Sheng, H. Wu, et al., J. Am. Chem. Soc. 140 (2018) 6467-6473. doi: 10.1021/jacs.8b03309
Y. Fan, L. Cheng, C. Liu, et al., RSC. Adv. 4 (2014) 52245-52249. doi: 10.1039/C4RA06582D
L. Zhang, L. Qin, X. Wang, H. Cao, M. Liu, Adv. Mater. 26 (2014) 6959-6964. doi: 10.1002/adma.201305422
K.U. Jeong, D.K. Yang, M.J. Graham, et al, Adv. Mater. 18 (2006) 3229-3232. doi: 10.1002/adma.200601338
J. Kim, J. Lee, W.Y. Kim, et al., Nat Commun. 6 (2015) 6959. doi: 10.1038/ncomms7959
J. Sun, Y. Li, F. Yan, et al., Nat. Commun. 9 (2018) 2599. doi: 10.1038/s41467-018-05017-7
H. Sun, J. Phy. Chem. B102 (1998) 7338-7364. doi: 10.1021/jp980939v
S.M. Morrow, A.J. Bissette, S.P. Fletcher, Nat. Nanotechnol. 12 (2017) 410-419. doi: 10.1038/nnano.2017.62
S. Das, S. Xu, T. Ben, S. Qiu, Angew. Chem. Int. Ed. 57 (2018) 8629-8633. doi: 10.1002/anie.201804383
J. Wang, K. Liu, L. Yan, et al., ACS Nano 10 (2016) 2138-2143. doi: 10.1021/acsnano.5b06567
C. Liu, Q. Jin, K. Lv, L. Zhang, M. Liu, Chem. Commun. 50 (2014) 3702-3705. doi: 10.1039/c4cc00311j
J. Ren, Y. Hu, C.H. Lu, et al., Chem. Sci. 6 (2015) 4190-4195. doi: 10.1039/C5SC00594A
M.A. Mateos-Timoneda, M. Crego-Calama, D.N. Reinhoudt, Chem. Soc. Rev. 33 (2004) 363-372. doi: 10.1039/b305550g
W. Chen, Z. Zhu, C. Yin, et al., Phys. Chem. Chem. Phys. 20 (2018) 4144-4148. doi: 10.1039/C7CP05845D
R. Salomonferrer, A.W. Gotz, D. Poole, S.L. Grand, R.C. Walker, J. Chem. Theory Comp. 9 (2013) 3878-3888. doi: 10.1021/ct400314y
R. Salomonferrer, D.A. Case, R.C. Walker, WIRES: Comput. Mol. Sci. 3 (2013) 198-210. doi: 10.1002/wcms.1121
W. Gotz, M.J. Williamson, D. Xu, et al., J. Chem. Theory Comp. 8 (2012) 1542-1555. doi: 10.1021/ct200909j
J. Wang, R.M. Wolf, J.W. Caldwell, et al., J. Comp. Chem. 25 (2004) 1157-1174. doi: 10.1002/jcc.20035
S. Li, W. Li, J. Ma, Acc. Chem. Res. 47 (2014) 2712-2720. doi: 10.1021/ar500038z
W. Li, S. Li, Y. Jiang, J. Phy. Chem. A111 (2007) 2193-2199. doi: 10.1021/jp067721q
S. Li, W. Li, T. Fang, J. Am. Chem. Soc. 127 (2005) 7215-7226. doi: 10.1021/ja0427247
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phy. 132 (2010) 154104. doi: 10.1063/1.3382344
W. Li, C. Chen, D. Zhao, S. Li, Inter. J. Quantum Chem. 115 (2015) 641-646. doi: 10.1002/qua.24831
K. Wolinski, J.F. Hinton, P. Pulay, J. Am. Chem. Soc. 112 (1990) 8251-8260. doi: 10.1021/ja00179a005
R. Ditchfield, Mol. Phys. 27 (1974) 789-807. doi: 10.1080/00268977400100711
M. Hwang, T.P. Stockfisch, A.T. Hagler, J. Am. Chem. Soc. 116 (1994) 2515-2525. doi: 10.1021/ja00085a036
H. Sun, Macromolecules 28 (1995) 701-712. doi: 10.1021/ma00107a006
H. Sun, S.J. Mumby, J.R. Maple, A.T. Hagler, J. Phys. Chem. 99 (1995) 5873-5882. doi: 10.1021/j100016a022

Figure 1 Schematic representation of the molecular structures and the landscape of the supramolecular self-assemblies of different types of LTPs. (a) LTP, (b) DTP, (c) type 1, (d) type 2, (e) type 3.

Figure 2 SEM images of LTP nanostructures. (a) type 1, (b) type 2, and (c) type 3 self-assembly. (d) CD spectra of LTP and DTP. (e) UV-vis spectra of LTP. (f) FT-IR spectra of LTP.

Figure 3 The molecular structure of LTP. (a) Crystal structure. (b) The left-handed helical conformation. (c) The subunit. (d) Non-bonded interactions between subunits. Different interactions between neighboring molecules are shown: H-bonding (black dotted line), C-H···π (black dotted line with blue shadow), and π-π interactions (black dotted line with orange shadow). The central benzene ring is light green, yellow, orange, and dark green, respectively. The atoms O, C, and H with non-bonding interactions are shown in red, deep gray, and white, respectively, whereas the others are represented by pink, grey, and omitted, respectively.

Figure 4 SEM images of LTP molecules and chiral diamine co-assembly. (a, e) LTP and 1, 2-cyclohexanediamine. (b, f) LTP and α-methyl benzylamine. (c, g) LTP and 2-methyl piperazine. (d, h) LTP and 1, 2-diphenylethylenediamine. (i, j) Time evolution of centroids distance. (k) RMSD for helical structure. WATR and WATS represent R-cyclohexanediamine and S-cyclohexanediamine put into the aqueous solutions containing LTP.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: