Scheme 1.
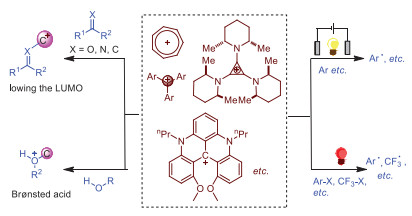
Activation pattern of carbocations.

Discovery of novel catalysts that are efficient and environmentally-friendly is highly desirable. Carbocations are normally considered as unstable and nonisolable intermediates in fundamental transformations such as E1, SN1 and rearrangements [1]. However, this is only partially true as delocalized carbocations can be stable enough to be isolated and utilized without inert conditions or even in aqueous medium [2]. The first stable carbocation discovered was the tropylium cation in 1891 [3]. Since then, several types of carbocations have been extensively studied in terms of reactivity, properties and stability [2]. Despite the fact that carbocations have been known for more than 130 years, it's rather surprising that their applications in organic transformations have been investigated only in recent decades. To date, carbocations, such as tropylium and trityl cation, are commonly used as Lewis acids to activate electrophiles by lowering their LUMO [4]. The utilization of carbocations as Lewis acids can provide a metal-free way to facilitate reactions without precious metals since Lewis acids are normally metals or metalloids [5]. Additionally, the combination of carbocation with other components such as water and alcohol can furnish a stronger Brønsted acid to promote various transformations [6]. Furthermore, electrophoto activation of TAC+ (trisaminocyclopropenium ion) delivers the excited radical dication TAC•2+*, which is a strong oxidant and capable of oxidizing a range of challenging substrates [7]. Recently, a newly developed carbocation, nPr-DMQA+, can be employed as a versatile photocatalyst to catalyze reactions under red light irradiation [8].
In spite of the emergence of carbocation promoted organic reactions in the past decade, few reviews have been published in this area [9, 10]. Herein, this review is aimed to summarize the recent advance in carbocation-catalyzed organic reactions published since 2017, and focuses on the role of carbocation in the reaction process.
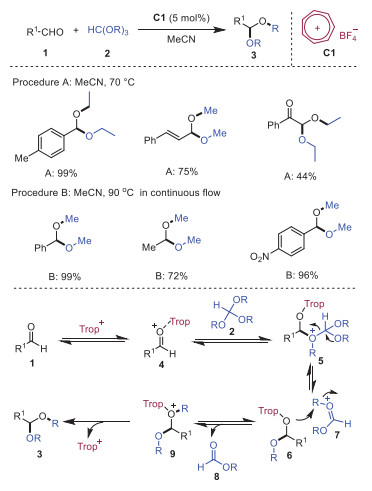
Carbonyl groups are one of the most synthetically important classes of organic substrates due to their versatile and ubiquitous chemical reactivity in organic transformations [11], and acetalization reactions are the popular masking protocol for aldehydes and ketones. In 2017, Nguyen and coworkers reported the tropylium cation catalyzed acetalization reaction with a wide range of aldehydes 1 and trialkyl orthoformate 2 [12]. This metal-free process worked efficiently in both batch and flow conditions. A plausible mechanism is depicted in Scheme 2. It was reasoned that initially, tropylium cation served as a Lewis acid to activate the carbonyl group to form intermediate 4, which went through a nucleophilic addition with trialkyl orthoformate 2 to generate intermediate 5. Subsequently, alkyl group transfer intramolecularly of intermediate 5 gave rise to intermediate 6. Followed by alkyl transfer intermolecularly, intermediate 9 would be provided along with the elimination of alkyl formate 8. Finally, release of tropylium cation in intermediate 9 afforded product 3. This method described an alternative metal-free pathway for the acetalization of carbonyl compounds.
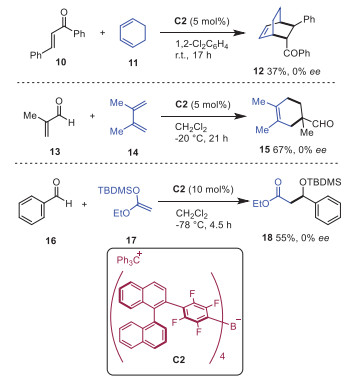
In 2017, Oestreich and coworkers prepared a chiral derivative of [B(C6F5)4]− in which the p-F of each C6F5 was replaced by a 1, 1′-binaphthalen-2-yl group (Scheme 3) [13]. The counteranions were provided as its sodium, lithium and trityl salts. Subsequently, this chiral trityl salt C2 was utilized as a catalyst in counteranion-directed Diels-Alder reactions and Mukaiyama aldol addition. However, no asymmetric induction was achieved.
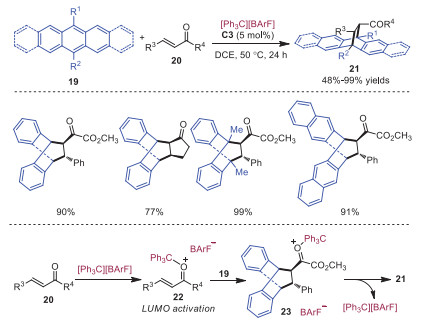
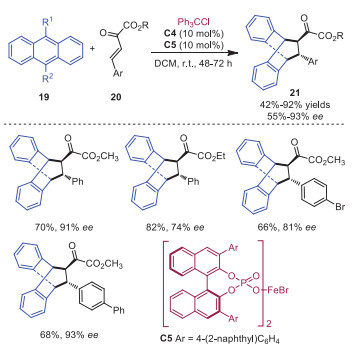
Stoichiometric or excess Lewis acid catalysts were required to promote Diels-Alder reactions of anthracenes via LUMO activation of dienophiles. Luo and coworkers developed a Diels-Alder reaction between α, β-unsaturated carbonyl compounds 20 and anthracene derivatives 19 in 2017 in the presence of a catalytic amount of trityl cation [14]. α, β-Unsaturated ketones 20 bearing various aromatic substituents and cyclic α, β-unsaturated ketones were readily tolerated in this transformation, affording the corresponding cyclic products 21 in good to excellent yields. Pentacene derivatives could also participate smoothly in this reaction. A plausible mechanism is outlined in Scheme 4. The coordination of trityl cation to unsaturated carbonyl compounds 20 formed intermediate 22, which could lower the LUMO of dienophile to enable the Diels-Alder reaction with anthracene derivatives 19. The final product 21 was delivered with the release of catalyst from intermediate 23. In 2019, the same group disclosed that the combination of trityl cation and a chiral weakly coordinating FeⅢ based bisphosphate anion C5 could act as a highly active carbocation Lewis acid catalyst to promote the asymmetric Diels-Alder reaction of anthracenes 19 with α, β-unsaturated carbonyl compounds 20 (Scheme 5) [15]. Control experiments showed that only chiral iron salt C5 or carbocation C4 would be ineffective to promote the reaction, indicating the reaction was catalyzed by the combination of trityl cation and FeⅢ complexed bisphosphate.
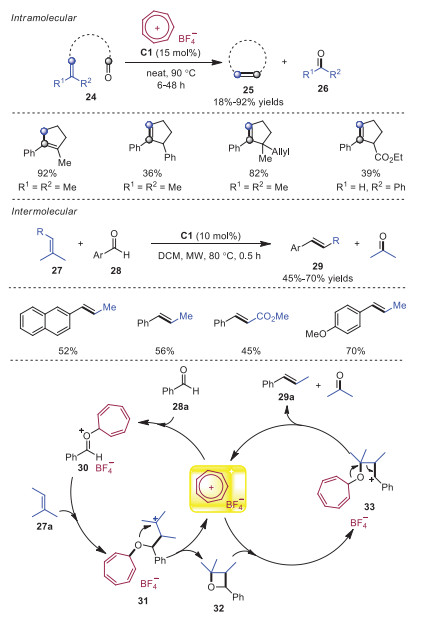
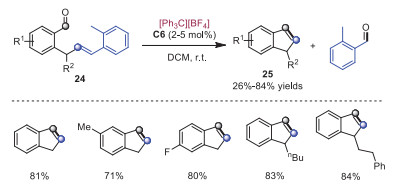
Despite the fact that olefin-olefin metathesis reactions have been extensively studied, the similar carbonyl-olefin metathesis has been rarely investigated. In 2018, Nguyen and coworkers disclosed tropylium cation catalyzed carbonyl-olefin metathesis reactions [16]. A broad range of substrates were applicable to intramolecular and intermolecular reactions. A plausible mechanism was presented in Scheme 6 on the basis of DFT calculations. At the beginning, activation of aldehyde 28a through tropylium cation would form intermediate 30, which lowered the LUMO of aldehyde to enable the electrophilic addition with alkene 27a to furnish intermediate 31. Next, intermediate 31 underwent intramolecular cyclization to generate intermediate 32 along with the release of tropylium cation. Subsequently, tropylium cation would coordinate to the oxygen of intermediate 32 leading to cation intermediation 33, which went through retro [2 + 2] cycloaddition to produce the final product 29a. In the same year, Franzén and coworkers developed analogous trityl cation catalyzed aldehyde-olefin metathesis (Scheme 7) [17]. These methods featured high yields, low catalyst loading, good functional group tolerance and mild reaction conditions.
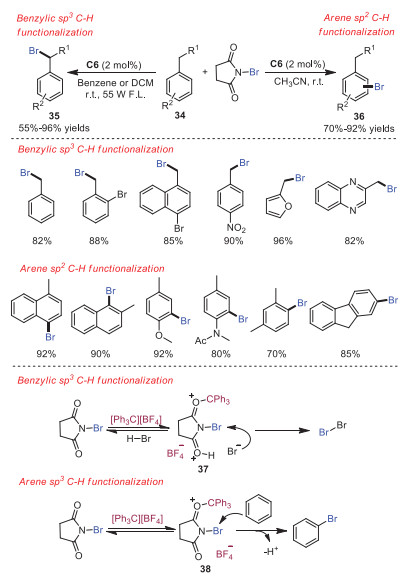
Brominated aromatic compounds are highly valuable in organic synthesis and material science. However, their synthesis usually requires high temperature and toxic radical initiators. In 2018, Franzén and coworkers developed trityl cation catalyzed chemoselective bromination reaction of alkyl arenes under mild conditions [18]. In this contribution, benzylic C(sp3)-H bromination products could be efficiently accessed under common hood light in the presence of trityl cation as the catalyst and N-bromosuccinimide (NBS) as the brominating agent. On the other hand, for electron-rich alkyl arenes, the arene C(sp2)-H bromination products could be obtained by simply alternating the solvent. A possible mechanism is showed in Scheme 8. For the benzylic C(sp3)-H bromination, trityl cation was proposed to assist the production of bromine from NBS and HBr by activation of NBS toward the nucleophilic attack of bromide. For the arene C(sp2)-H bromination, trityl cation was believed to involve in the Lewis acid activation of NBS, leading to a more potent electrophile.
Retro-Claisen rearrangement provides an alternative way to access ester products from carbonyl compounds. This type of reaction usually is promoted by transition metal Lewis acids. In 2018, Nguyen and co-workers developed tropylium cation catalyzed retro-Claisen-type reaction for C−C bond cleavage of diketones 39 under metal-free conditions [19]. A wide range of synthetically valuable esters and amines 41 could be easily afforded through solvolysis of 1, 3-dicarbonyl compounds 39. Furthermore, thio-enol ether product 43 was afforded by using thiols as nucleophiles. A plausible mechanism was proposed as shown in Scheme 9. Firstly, tropylium cation would activate 1, 3-diketones to form intermediate 44. Subsequently, intermediate 44 reacted with nucleophile 40a to generate intermediate 45, which underwent proton transfer and C−C bond cleavage to deliver cation intermediate 46. Next, C−C bond cleavage of intermediate 46 gave rise to intermediate 47, which was followed by isomerization with the release of tropylium cation to furnish the final product 41a.
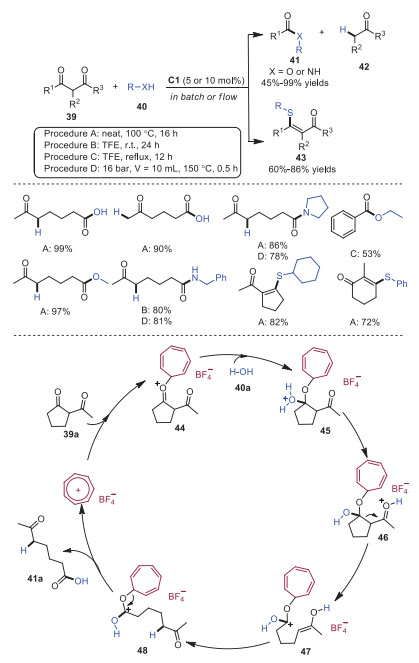
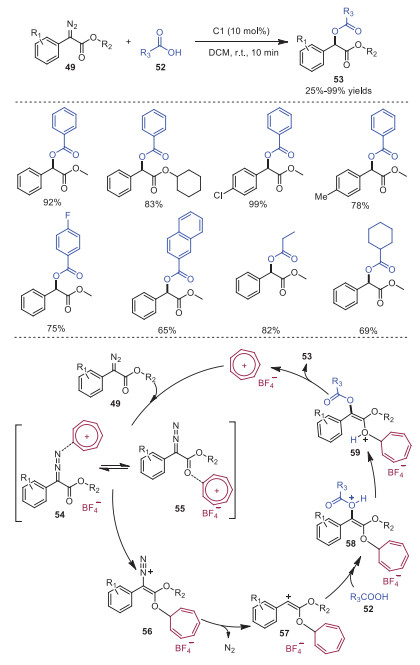
In 2019, Lv and coworkers reported trityl cation catalyzed Roskamp reaction of aldehydes 50 and α-alkyldiazoacetates 49 leading to diverse α-branched β-ketocarbonyls 51 in moderate yields (Scheme 10) [20]. In 2021, Koenigs and coworkers developed an analogous transformation involving tropylium cation catalyzed O−H insertion of diazoalkanes 49 with carboxylic acids 52 [21]. This reaction featured a wide range of carboxylic acid derivatives and diazoalkanes to form α-functionalized esters 53 with high efficiency. A proposed mechanism is depicted in Scheme 11. It was postulated that tropylium cation initially activated diazoalkane 49 via coordination to the carbonyl group or the diazo group to form intermediate 54 or 55, which underwent tautomerization to deliver intermediate 56. Subsequently, an activated carbene intermediate 57 was furnished from intermediate 56 along with the expulsion of N2. Then, carboxylic acid 52 interacted with carbene intermediate 57 to provide intermediate 58, which underwent proton transfer giving rise to the final product 53.

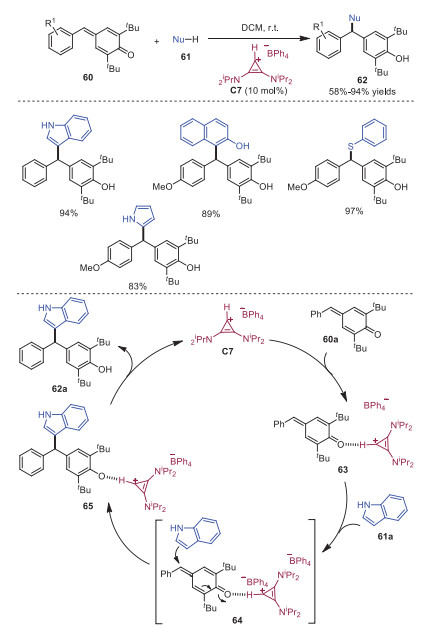
Although cyclopropenium ions are known in the past several decades, their potential as organocatalysts has been investigated only recently. The hydrogen atom attached to the cyclopropane ring is acidic and can serve as a hydrogen-atom donor in organic transformations. In 2021, Anand's group successfully realized bis(amino)cyclopropenium (BAC) ion catalyzed 1, 6-conjugate addition reaction of p-QM with nucleophiles via hydrogen-bonding catalysis [22]. A variety of nucleophiles including indoles, thiols, 2-naphthols and phenols participated in this reaction smoothly, leading to the corresponding products in high yields. The deuterium isotope labeling studies as well as spectropic studies indicated that the hydrogen-atom in the catalyst was indeed responsible for facilitating this transformation. A plausible mechanism is outlined in Scheme 12. Initially, activation of p-QM by catalyst C7 via hydrogen-bonding would provide complex 63. Nucleophilic addition in a 1, 6-fashion with indole 61a would occur via complex 64 to afford intermediate 65, which would then release product 62a and catalyst C7.
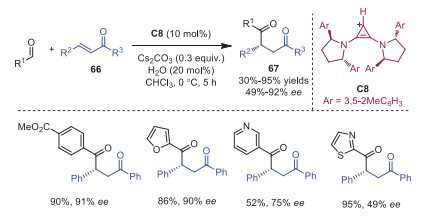
The first asymmetric intermolecular Stetter reaction catalyzed by a chiral bis(amino)cyclopropenium ion C8 was developed by Gravel and coworkers in 2021 (Scheme 13) [23]. A novel C2-symmetric precatalyst possessing restricted rotation around the C−N bond enabled the outcome of high enantioselectivity. Notably, a catalytic amount of water was essential to obtain high enantioselectivities.
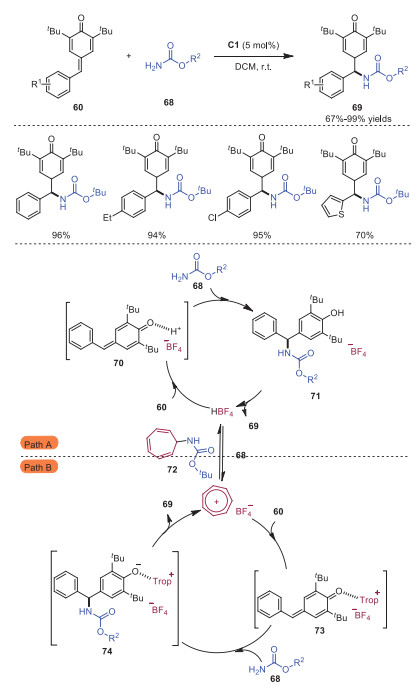
In 2022, Anand and coworkers disclosed tropylium cation catalyzed vinylogous aza-Michael addition of carbamates 68 to p-quinone methides 60 [24]. A wide range of α, α'-diarylmethyl carbamates 68 were afforded in moderate to excellent yields. Two plausible mechanisms involving a hidden Brønsted acid and a Lewis acid catalysis were proposed as outlined in Scheme 14. Path A was based on the hidden Brønsted acid catalysis. At the beginning, tropylium salt reacted with carbamate 68 to provide tropylium-carbamate complex 72 along with the formation of Brønsted acid HBF4, which activated p-QM through hydrogen-bonding to deliver intermediate 70. Subsequently, 1, 6-conjugate addition of carbamate 68 would occur to produce intermediate 71, which decomposed to produce the final product 69 with the regeneration of HBF4. Another alternative possibility was that tropylium cation would coordinate with p-QM through a weak interaction, which resulted in the formation of complex 73. Followed by 1, 6-conjugate addition of carbamate 68, intermediate 74 would be generated, which further underwent proton exchange giving rise to the final product 69 with the release of tropylium cation to complete the catalytic cycle.
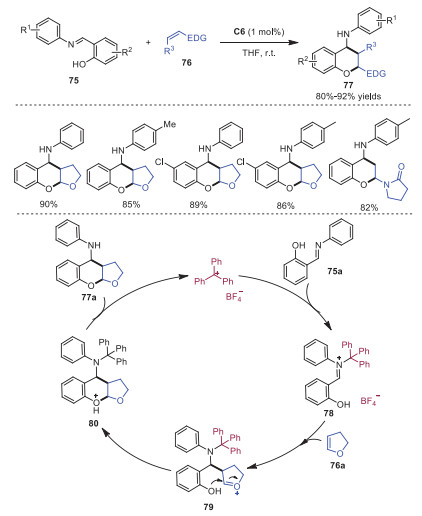
In 2017, Guo and coworkers developed trityl cation promoted interrupted Povarov reactions, affording a series of cis-4-aminobenzodihydropyrans 77 in good yields within 10 min with low catalyst loadings (Scheme 15) [25]. In this reaction, trityl cation performed as a Lewis acid catalyst to activate salicylaldimine 75a, delivering intermediate 78 with increased electrophilicity. Next, intermediate 78 reacted with electron rich 2, 3-dihydrofuran 76a leading to intermediate 79, which went through an intramolecular nucleophilic attack from the hydroxyl group to oxocarbenium carbon giving rise to intermediate 80. Finally, proton transfer of intermediate 80 provided product 77a along with trityl cation to complete the catalytic cycle. This reaction provided an efficient way to access benzodihydropyran skeletons under mild conditions, which were widely existed in bioactive molecules.
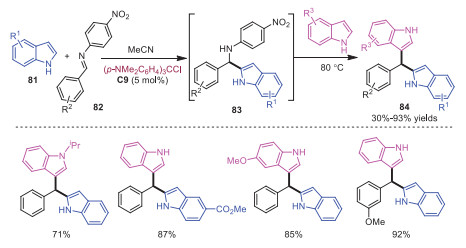
In 2021, Brindle and coworkers developed triarylmethyl cation catalyzed one-pot, three component synthesis of unsymmetrical bisindolylmethanes from two different indoles with N-arylimines 82 (Scheme 16) [26]. This reaction featured commercially available catalyst, high efficiency and mild reaction conditions. The single addition product 83 could be achieved by tuning the stability of carbocation. Followed by one-pot interaction with another molecular indole, unsymmetrical bisindolylmethanes 84 would be formed in moderate to excellent yields.
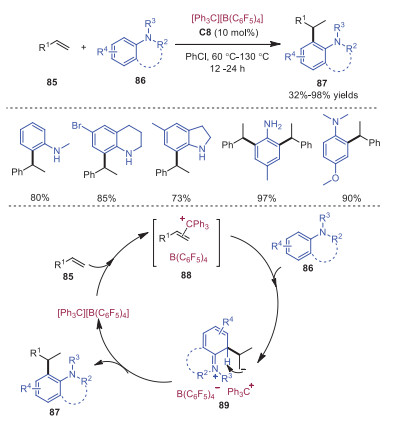
In 2018, Yao and coworkers developed a trityl cation catalyzed hydroarylation reaction of a wide range of alkenes with aromatic primary, secondly and tertiary amines [27]. A broad scope of aniline derivatives 87 were provided in moderate to good yields. A plausible mechanism is shown in Scheme 17. Trityl cation acted as a Lewis acid to activate alkene 85, rendering alkene 85 electron-deficient to allow for the electrophilic attack with aniline 86 leading to intermediate 89. Further proton transfer of intermediate 89 would result in the formation of product 82.
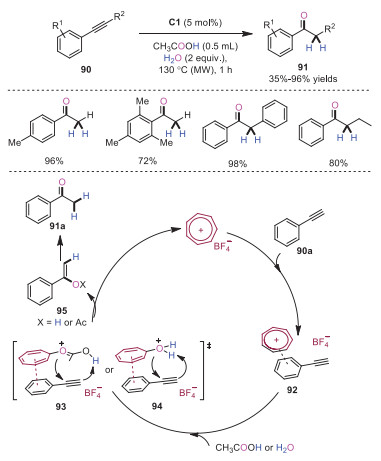
In 2018, Nguyen and coworkers developed tropylium cation promoted hydration reactions of alkynes 90 [28]. A broad scope of alkynes was readily demonstrated in this transformation, providing the corresponding ketone products 84 in moderate to excellent yields. A plausible mechanism based on mechanistic studies was depicted in Scheme 18. It was reasoned that initially, complex 92 was formed through π-π interaction between tropylium cation and phenylacetylene 90a. Subsequently, the covalent interaction between tropylium cation and acetic acid or water would produce intermediate 93 or 94 with enhanced Brønsted acidity, respectively. An intermolecular proton transfer from water or acetic acid with spontaneous acetate addition and hydroxyl addition gave rise to intermediate 95 along with the regeneration of tropylium cation. Finally, enolate species 95 would isomerize to afford ketone products 91a. This protocol provided an alternative way for the hydration of alkynes under metal-free and mild reaction conditions.
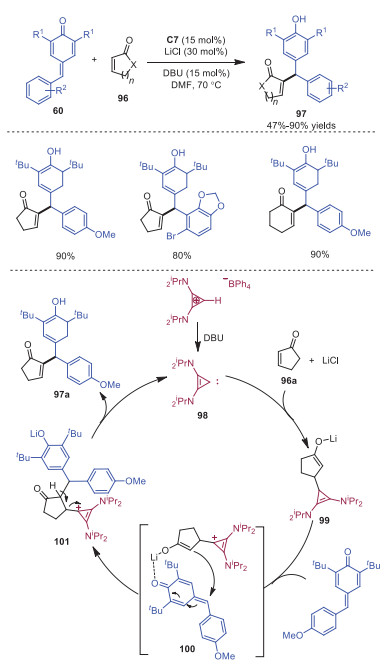
Anand and coworkers explored the intermolecular Rauhut-Currier reaction between p-QM and α, β-unsaturated carbonyl compounds catalyzed by bis(amino)cyclopropenium ion C7 (Scheme 19) [29]. Diverse vinyl diarylmethane derivatives 97 were afforded in moderate to good yields. The reaction started by the abstraction of acidic proton with DBU from C7 to generate carbene intermediate 98, which would react with α, β-unsaturated carbonyl compounds 96a giving rise to enolate 99. The transition state 100 would be formed with p-QM in which the presence of lithium stabilized the enolate and coordinated with the carbonyl group of p-QM to bring it closer to the reaction center. After that, the reaction between enolate and p-QM would take place to form intermediate 101, which further went through proton elimination to furnish the final product 97.
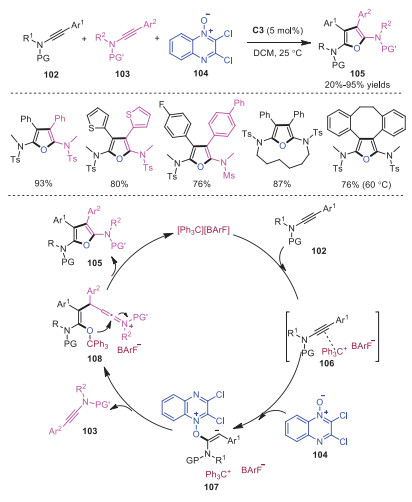
In 2019, Hashmi and coworkers reported trityl cation catalyzed oxidative [2 + 2 + 1] cycloaddition of two molecules of ynamides with 2, 3-dichloroquinoxaline-N-oxide 104 [30]. Various fully substituted symmetric and unsymmetric furan derivatives 105 were rapidly constructed. This protocol could further go through intramolecular macrocyclization of diynamides to convergent assembly of macrocyclic furan derivatives. A plausible mechanism is presented in Scheme 20. At the beginning, the triple bond of ynamide 102 was activated by trityl cation to generate intermediate 106, which was verified by NMR experiments. Subsequently, electrophilic addition of intermediate 106 with N-oxide occurred to produce "umpoled" enolate 107, which underwent an intermolecular C−C coupling with another molecular of ynamide 103 to provide enolonium species 108. Finally, enolonium species 108 went through intramolecular cycloaddition giving rise to fully substituted furans 105 accompanied with the regeneration of trityl cation.
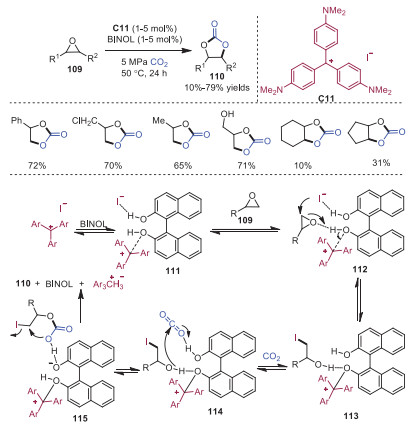
Cyclic carbonates are an important class of industrial intermediates and solvents, and they can be utilized as electrolytes in lithium-ion batteries. In 2017, Belokon and coworkers developed the synthesis of cyclic carbonates from epoxides 109 and carbon dioxide catalyzed by carbocation/polyol. In this transformation, the catalysts were commercially available and less toxic than metal complexes (Scheme 21) [31]. The catalyst system of crystal violet C11 and BINOL could be recycled five times without loss of activity. Mechanistically, carbocation initially acted as a Lewis acid to activate BINOL, forming a stronger Brønsted acid 111. The increased acidity of BINOL allowed the formation of a stronger hydrogen bond between hydroxyl group and epoxide, triggering the epoxide ring opening through the addition of iodide ion leading to intermediate 113. Subsequently, the remaining hydroxyl group in intermediate 113 would coordinate with CO2 to produce intermediate 114, which went through addition and cyclization to provide product 110.
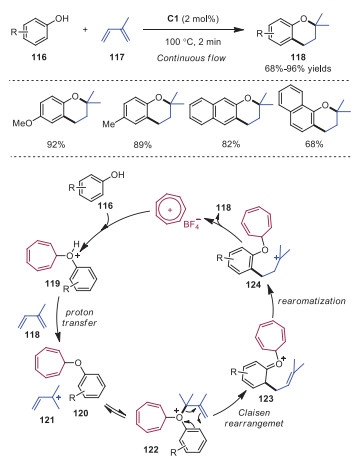
In 2020, Nguyen and coworkers described tropylium cation promoted prenylation reactions of phenols (Scheme 22) [32]. This method was amenable to continuous flow chemistry, enabling an inexpensive pathway to obtain 2, 2-dimethylchromans 118 on multiple-gram scale with high efficiency, short reaction times and simple product purification. Mechanistically, a nucleophilic attack of phenol 116 to tropylium cation afforded the active Brønsted acid 119, which underwent a proton transfer to diene 118 affording intermediates 120 and 121. Subsequently, interaction between intermediates 120 to 121 gave rise to intermediate 122, which went through a Claisen rearrangement and re-aromatization to produce intermediate 124. Further cyclization of intermediate 124 would occur leading to product 118, accompanied with the formation of tropylium cation. This method provided a metal-free and highly efficient approach to access compounds possessing 2-dimethylchroman framework, which was widely existed in bioactive compounds with antitumor, antihypertensive and antithrombotic activities.
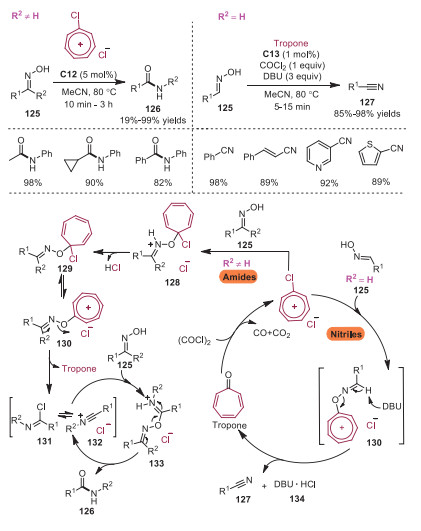
In 2020, Guo and coworkers reported the synthesis of amides and nitriles through chlorotropylium cation promoted transformations of ketoximes and aldoximes, respectively [33]. Amides and nitriles were rapidly assembled in short reaction time with excellent yields. A plausible mechanism is presented in Scheme 23. For the reactions with ketone oximes, complex 128 would be formed with chlorotropylium cation, which eliminated a molecule of HCl to generate intermediate 129. Subsequently, chloride anion was released from intermediate 129 to furnish the key tropylium oxime ether 130. Next, R2 group would migrate to the N atom with the cleavage of O−N bond and formation of C=O bond at the same time. The intermediate nitrilium cation 132, in equilibrium with isomer imidoyl chloride 131, would be attacked by ketoxime leading to intermediate 133. Intermediate 132 was regenerated along with the release of amide product 126. On the other hand, DBU abstracted a proton from the key intermediate 130 to form nitrile product 127 with the explusion of precatalyst tropone. Oxalyl chloride would react with tropone giving rise to chlorotropylium chloride, which would be employed in the catalytic cycle for the synthesis of nitriles. This method provided a mild and efficient way to access amides and nitriles.
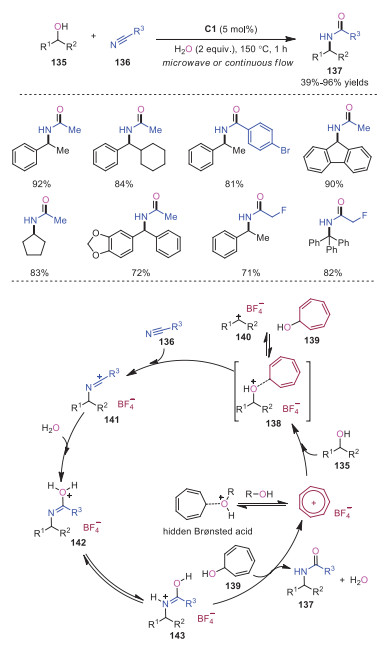
Ritter reactions haven't been frequently used in modern organic synthesis due to the employment of harsh and strong acidic reaction conditions, which may lead to complicated side reactions. In 2021, Nguyen and coworkers developed a new protocol using tropylium cation as a Lewis acid to promote the Ritter reaction, which was amenable to microwave and continuous flow reactors. A variety of alcohol 135 and nitrile substrates 136 were well tolerated, affording the corresponding amides 137 in good to excellent yields. A possible mechanism was proposed as shown in Scheme 24. Tropylium cation coordinated with the oxygen of alcohol, rendering the adjacent carbon electron-deficient to enable the nucleophilic attack of nitrile 136 on alcohol 135. Subsequently, a hydrolysis process would occur, which resulted in the conversion of iminium intermediate 141 into amide intermediate 142. Amide intermediate 142 would further undergo proton transfer and isomerization affording amide product 143. The hidden Brønsted acid, generated from tropylium cation with either alcohol or water, might also promote the reaction.
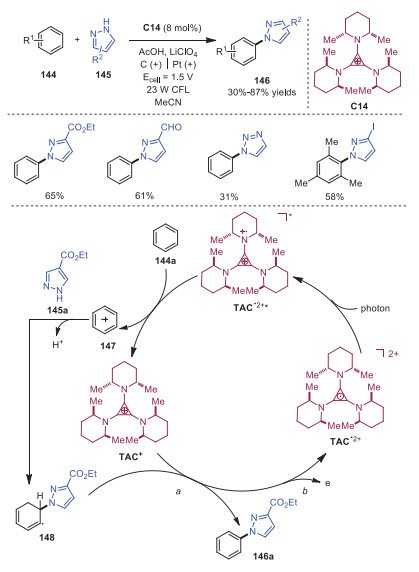
Recently, vigorous activity in the areas of electrocatalysis has enabled a variety of powerful oxidative protocols. TAC+ could be electrochemically oxidized to the air-stable dication TAC•2+, which could further be excited by visible light irradiation to form the excited radical dication TAC•2+*. Notably, the reduction potential of TAC•2+* is 3.33 V (vs. SCE), which is higher than the excited-state reduction potentials of 9-mesityl-10-methylacridinium (E1/2* = 2.06 V), 3-cyano-1-methylquinolinium (E*1/2 = 2.72 V) and even DDQ (E*1/2 = 3.18 V). As a result, TAC•2+* is capable of oxidizing a wide range of challenging substrates. In 2019, Lambert's group realized the electrophotocatalytic coupling of arenes and nitrogen heteroaromatics catalyzed by TAC+ (Scheme 25) [34]. The reaction started with electrochemical oxidation of TAC+ to produce TAC•2+, which was then excited by light irradiation to generate reactive TAC•2+*. Subsequently, TAC•2+* would oxidize benzene 144a via single electron transfer (SET) to afford radical cation 147 with the regeneration of catalyst. The interaction of 147 and pyrazole 145a followed by deprotonation would provide intermediate 148, which would be oxidized by TAC+ or directly at the anode leading to product 146.
Later in 2020, TAC+ was also demonstrated to serve as a hydrogen-atom-transfer catalyst to promote the CH functionalization of ethers via electrophotocatalysis [35]. A variety of ethers 149 went through oxidant-free coupling with isoquinolines and alkenes, leading to the corresponding products 151 with high regioselectivity at the less-hindered α-position. A possible mechanism is described in Scheme 26. Initially, TAC+ went through electrochemical oxidation to form TAC•2+, which was then excited by light irradiation to provide TAC•2+* possessing an aminyl radical character. This TAC•2+* would abstract a hydrogen from ether substrate 149a leading to radical 152 accompanied with the protonated TAC2+−H. After that, radical 152 would react with isoquinoline 150a to generate radical intermediate 153. Followed by a second oxidation and loss of proton, product 151a would be produced. In the meantime, deprotonation of TAC2+−H would regenerate TAC+ to close the catalytic cycle. This method revealed a new reactivity mode for this electrophotocatalyst.
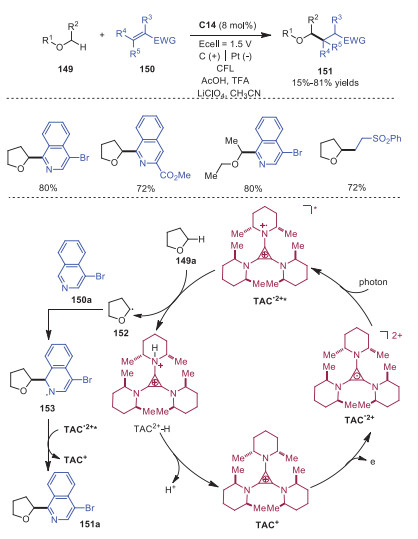
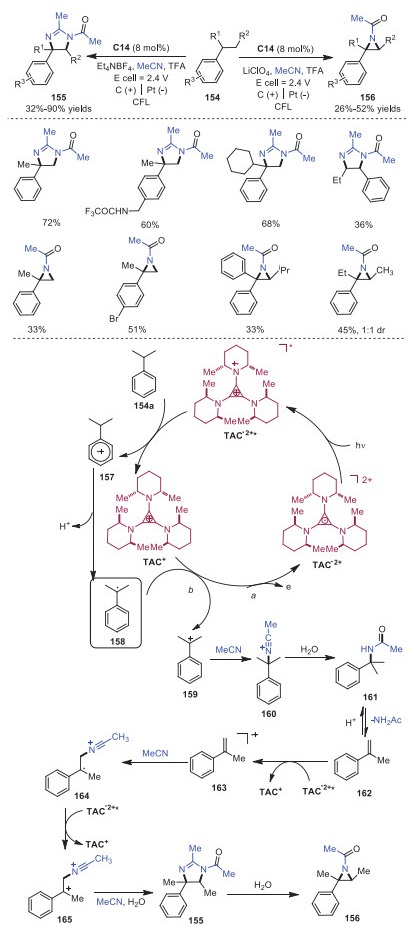
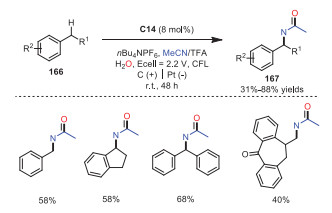
As the first amination diminishes the reactivity of neighbor CH bonds, it remains challenging to access diaminated products via twice CH activation. Electrophotocatalytic diamination of vicinal CH bonds via CH activation catalyzed by TAC+ was realized by Lambert and coworkers in 2021 (Scheme 27) [36]. The reaction started with the generation of photoexcited intermediate TAC•2+* with high oxidizing potential, which could oxidize compound 154a to radical cation 157. Deprotonation of radical cation 157 would produce radical intermediate 158, which would further undergo oxidation to afford carbocation intermediate 159. Cation 159 would react with MeCN leading to intermediate 160, which went through hydrolysis to provide acetamide compound 161. Compound 161 would proceed through a reversible, acid-catalyzed elimination to generate α-methylstyrene 162, which would then undergo a single-electron oxidation, solvent trapping and oxidation giving rise to dihydroimidazole product 155 or aziridine 156. Later, the same group also described Ritter-type CH bond amination via electrophotocatalysis by using TAC+ as the catalyst (Scheme 28) [37].
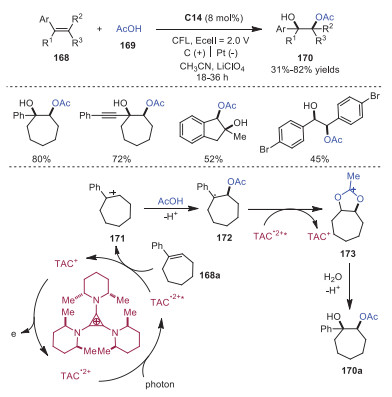
Acetoxyhydroxylation of olefins with high syn selectivity via TAC+ catalysis under electrophotocatalytic conditions was developed by Lambert's group in 2021 (Scheme 29) [38]. The success of this reaction was that the electrochemical potential by itself was unable to oxidize olefin, and the reaction took place only by combination with light irradiation. It was reasoned that TAC+ could be oxidized to radical dication TAC•2+. Upon photoexcitation, TAC•2+* with strongly oxidizing potential was formed, which could oxidize olefin substrate 168a to radical cation 171. Then acid nucleophile 169 trapped 171 leading to radical 172, which was further oxidized to oxocarbenium intermediate 173 and hydrolyzed to provide dioxygenated 170a. This transformation offered an attractive and alternative way for olefin dioxygenation without utilizing transition metal reagents or catalysts.
Photoredox catalysis and has emerged as an effective and environmentally friendly synthetic tool to promote SET process in organic transformations [39, 40]. Compared with the commonly used high-energy white, blue or green light, low-energy red light has advantages including fewer side reactions and less health risks. In 2020, Gianetti and coworkers disclosed that helical carbenium ion C15 could act as a versatile organic photoredox catalyst for red-light-mediated photoredox transformations [41]. Red-light-mediated dual transition-metal/photoredox-catalyzed arylation reaction could be catalyzed by nPr-DMQA through oxidative quenching. Additionally, the potential of nPr-DMQA in photo-oxidation catalysis could be demonstrated by application in red-light-mediated aerobic oxidative hydroxylation of arylboronic acids. Two catalytic cycles involving oxidative or reductive quenching are showed in Scheme 30. The oxidative quenching catalytic cycle was proposed for Pd/nPr-DMQA+ catalyzed C(sp2)-H arylation. At the outset, photoexcitation of nPr-DMQA+ produced nPr-DMQA+*, which reduced aryldiazonium salt 176 to generate an aryl radical 179 with the formation of nPr-DMQA•++. Subsequently, addition of aryl radical 176 to PdⅡ intermediate 180, which was generated by CH activation of aryl substrate 174, afforded PdⅢ species 181. This PdⅢ species 181 was oxidized by nPr-DMQA•++ to regenerate nPr-DMQA+, accompanied with PdⅣ species 182. The final product 176 was obtained by reductive elimination of PdⅣ species 182. On the other hand, a reductive quenching cycle was proposed for nPr-DMQA+-catalyzed aerobic oxidative hydroxylation. Initially, iPr2NEt was oxidized to ammonium radical cation by the excited state nPr-DMQA+* along with nPr-DMQA•. Then, nPr-DMQA• reacted with oxygen to form an O2•− and regenerated nPr-DMQA+. Followed by oxidative attack on aryl boronic acid 177 and hydrolysis, phenol 178 would be produced.
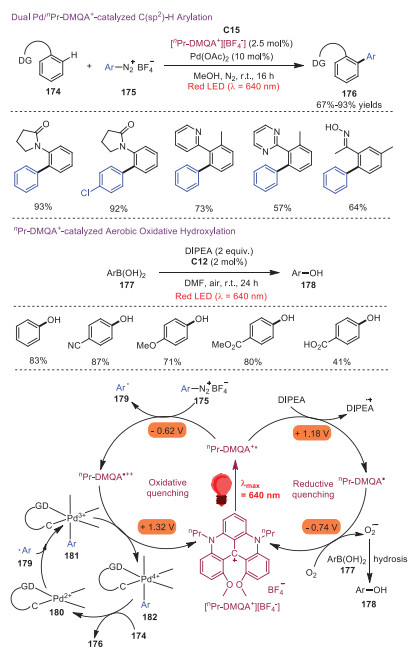
Gianetti and coworkers further extended the above strategy to the construction of CF3-containing spirocyclic indolines in 2021 [42]. A variety of functionalized CF3-containing 3, 3-spirocyclic indoles 185 were afforded in moderate to good yields through a red-light-mediated nPr-DMQA+ catalyzed trifluoromethylation/dearomatization cascade process. A plausible mechanism is presented in Scheme 31. It was proposed that excitation of ground state nPr-DMQA+ would provide nPr-DMQA+* under red light irradiation, which would reduce Umemoto's reagent 184 to generate CF3 radical 186. Subsequently, CF3 radical 186 reacted with indole 183a leading to dearomatized benzyl radical species 187, which went through a SET with nPr-DMQA•++ to afford benzyl carbocation intermediate 188. Further deprotonation of intermediate 188 gave rise to product 185a.

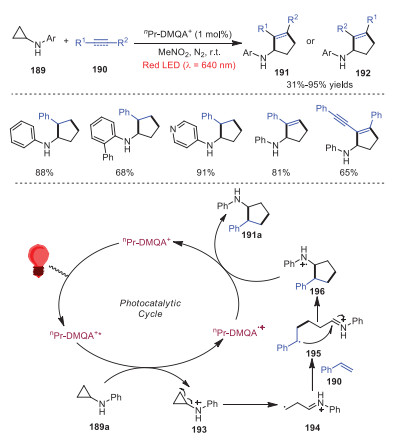
[3 + 2] Cycloaddition of alkenes or alkynes with cyclopropylamines could be achieved as well under red light irradiation by using nPr-DMQA+ as the organic photocatalyst [43]. Diverse cyclopentane and cyclopentene derivatives possessing various functional groups were readily afforded in moderate to good yields under mild conditions. A plausible mechanism is proposed, as shown in Scheme 32. Firstly, red light irradiation of ground state nPr-DMQA+ would produce excited state nPr-DMQA+*, which proceeded through a SET process with cyclopropylamine 189a to provide nitrogen radical cation intermediate 193. Subsequently, intermediate 193 underwent β-scission of cyclopropane ring giving rise to β-carbon radical iminium ion 194, which attacked styrene 190 leading to stabilized radical cation 195. Intramolecular addition of radical to iminium ion of intermediate 195 produced nitrogen radical cation 196, which went through reduction to provide product 191a.
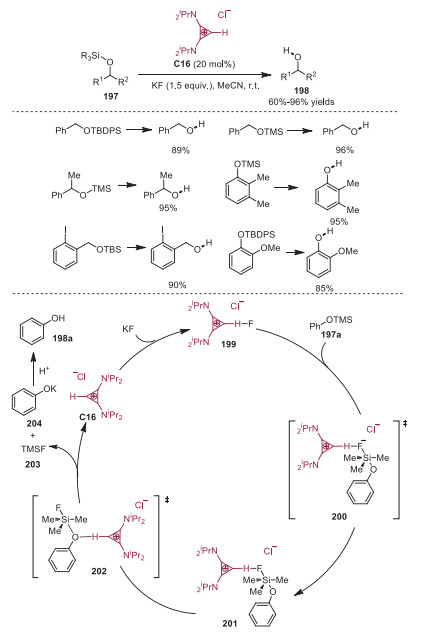
O-silyl ethers are one of the most utilized protecting groups due to the stability under various conditions, functional group tolerance and commercial availability. However, there are several shortcomings employing "naked F−" for silyl deprotection, such as side reactions and requirement of greater amount of "naked F−". In 2017, Dudding and coworkers developed the O-silyl ether deprotection with "naked F−" via bis(amino)cyclopropenium (BAC) ion catalysis [44]. The corresponding deprotection products 198 were afforded in good yields. A plausible mechanism supported by DFT calculation is outlined in Scheme 33. The catalytic cycle was initiated by the counterion ion exchange between bis(amino)cyclopropenium (BAC) ion catalyst C16 with KF leading to active catalyst 199 possessing the H⋯F− bond, in which the cyclopropenium catalyst served as the phase-transfer catalyst. After that, the fluoride added to the Si to form intermediate 201 via transition state 200. Then the Si-O bond cleavage would occur via transition state 202 to furnish the 204, TMSF and regeneration of the catalyst C16. The potential of this H-bonding motif to attenuate fluoride basicity and nucleophilicity might be applicable to other catalytic modes. Later in 2018, the same group further extended the strategy in which the cyclopropenium catalyst served as the phase-transfer catalyst to the benzylic fluorination, the corresponding fluorinated products were provided in high yields (Scheme 34) [45].

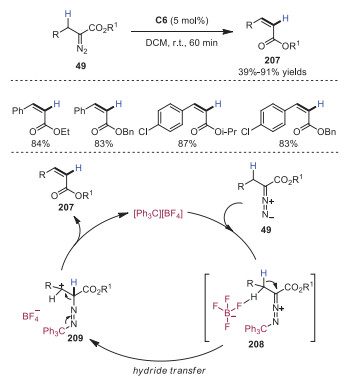
In 2019, Lv and coworkers developed trityl cation catalyzed 1, 2-hydride migration of α-alkyldiazoacetates 49 leading to α, β-unsaturated esters 207 in moderate to excellent yields with high Z selectivity (Scheme 35) [20]. In this reaction process, intermediate 208, generated by coordination of trityl cation with diazene moiety and hydrogen-bonding between fluorine atoms on [BF4]−, converted to intermediate 209 via hydride transfer. Further intramolecular syn elimination of intermediate 209 would afford product 207, with the release of a molecule of N2 and trityl cation.
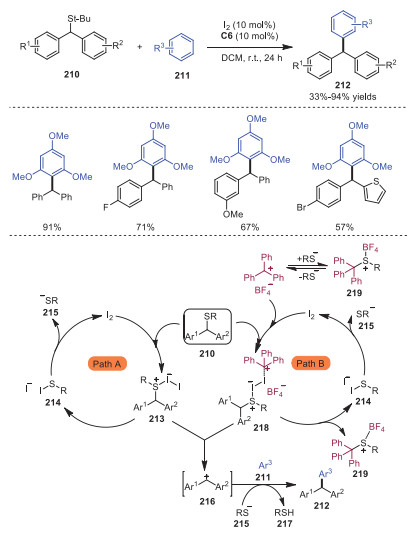
In 2020, Masson and coworkers discovered that the combined Lewis acid catalytic system, formed by molecular iodine and trityl cation, could efficiently catalyze Friedel-Crafts arylation of diarylmethyl triarylmethanes leading to various unsymmetrical triarylmethanes 212 [46]. Two plausible mechanisms are presented in Scheme 36. In the absence of trityl cation, the sulfur atom of compound 210 was attacked by iodine, resulting in the formation of cationic iodosulfonium intermediate 213. Subsequently, cationic iodosulfonium intermediate 213 underwent elimination giving rise to sulfenyl iodide 214 and carbocation 216. Then, sulfenyl iodide went through nucleophilic addition with iodine to produce thiolate 215. Meanwhile, carbocation 215 reacted with electron rich arene 211, with further hydrogen atom abstraction mediated by thiolate 215, affording the desired Friedel-Crafts products 212 and thiol 216. On the other hand, trityl cation might activate iodosulfonium ion via possible Lewis acid assisted Lewis acid model 217 when trityl cation was added in the reaction, thus facilitating the cleavage of C−S bond leading to cation 216 and triphenylmethylium thiolate 219. Triphenylmethylium thiolate 219 could be in equilibrium with thiolate 214 and tritylium tetrafluoroborate to allow the regeneration of co-catalyst.
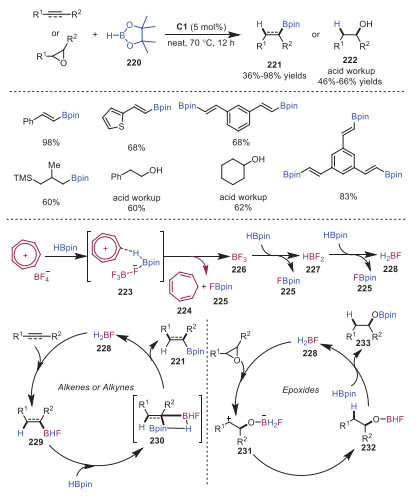
Hydroboration reaction is one of the most frequently used reactions in organic synthesis. Normally, hydroboration was catalyzed by transition-metal or main group element. Therefore, method development of metal-free hydroboration reaction is highly important. In 2021, Nguyen and coworkers reported hydroboration reactions by using tropylium cation as an efficient catalyst [47]. Alkynes, alkenes and epoxides could be readily tolerated, affording the corresponding products in moderate to good yields. DFT calculations and experimental studies suggested that BH2F was the hidden boron catalyst. A proposed mechanism is depicted in Scheme 37. The reaction was triggered by a hydride abstraction of HBpin with tropylium cation to provide cycloheptatriene 224, FBpin 225 and BF3 species 226. Subsequently, BF3 species 226 reacted with HBpin twice leading to BH2F species 228. Next, 1, 2-syn-addition to alkynes or alkenes from BH2F took place giving rise to boron alkene or alkane intermediate 229, which underwent transborylation with HBpin in a stepwise fashion to provide the final product 221. On the other hand, epoxide would undergo ring-opening process with BH2F species 228 to produce zwitterionic species 231, which proceeded through a hydride transfer from boron moiety to the carbocationic center affording intermediate 232. Subsequently, transborylation between intermediate 232 and HBpin gave rise to BH2F species 228 and compound 233, which went through acid workup to generate product 222.
In summary, carbocations such as tropylium and trityl cation, can act as Lewis acids to lower the LUMO of electrophile, thus promoting reactions with nucleophiles. Additionally, interaction between alcohols and carbocations can form Brønsted acids with enhanced acidity. Furthermore, electrophoto activation of TAC+ can furnish TAC•2+*, which is a strong oxidant and capable of oxidizing a variety of challenging substrates. Moreover, nPr-DMQA+ is disclosed as a versatile photoredox catalyst as its excited state can be quenched by both oxidation and reduction. These reported results show that carbocations exhibit great ability toward the activation of various groups, such as carbonyl groups, imines, hydroxyl groups and epoxides. These developed methods provide an environmentally friendly pathway for the synthesis of valuable compounds and will inspire chemists to discover more interesting transformations promoted by carbocations.
Despite the significant achievements of carbocation-catalyzed organic reactions, there are still some challenges to be solved. For example, chiral carbocation catalysts are rarely studied and limited to central, planar chiral cations or chiral counterions. Therefore, developing diverse chiral carbocations such as axial carbocations are highly desirable. We believe that in the near future, more achievements will be obtained for the carbocation-catalyzed organic transformations, and enantioselective reactions involving novel chiral carbocation catalysts will be developed rapidly.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from National Natural Science Foundation of China (No. 21871053), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2020ZD04) is gratefully acknowledged.
J. Clayden, N. Greeves, S. Warren, Organic Chemistry, 2nd Ed., University Press, Oxford, 2012.
G.A. Olah, G.K.S. Prakash, Carbocation Chemistry, Wiley, Hoboken, 2004.
G. Merling, Ber. Dtsch. Chem. Ges. 24 (1891) 3108–3126. doi: 10.1002/cber.189102402151
O. Sereda, S. Tabassum, R. Wilhelm, Top. Curr. Chem. 291 (2010) 349–393.
H. Yamamoto, Lewis Acids in Organic Synthesis, Wiley-VCH, Weinheim, 2000.
J.J. Koenig, M. Breugst, Catalysis by Molecular Iodine, in: S. Huber (Ed.), Halogen Bonding in Solution, Wiley-VCH, Weinheim, 2021, pp. 237–268.
L. Capaldo, L.L. Quadri, D. Ravelli, Angew. Chem. Int. Ed. 58 (2019) 17508–17510. doi: 10.1002/anie.201910348
L. Mei, T. Gianetti, Synlett 32 (2021) 337–343. doi: 10.1055/s-0040-1705942
V.R. Naidu, S.J. Ni, J. Franzen, ChemCatChem 7 (2015) 1896–1905. doi: 10.1002/cctc.201500225
J. Bah, J. Franzen, Chem. Eur. J. 20 (2014) 1066–1072. doi: 10.1002/chem.201304160
W. Xiao, J. Wu, Chin. Chem. Lett. 32 (2021) 2751–2755. doi: 10.1016/j.cclet.2021.03.033
D.J.M. Lyons, R.D. Crocker, D. Enders, T.V. Nguyen, Green Chem. 19 (2017) 3993–3996. doi: 10.1039/C7GC01519D
P. Pommerening, J. Mohr, J. Friebel, M. Oestreich, Eur. J. Org. Chem. 2017 (2017) 2312–2316. doi: 10.1002/ejoc.201700239
Q.C. Zhang, J. Lv, S.J. Li, S.Z. Luo, Org. Lett. 20 (2018) 2269–2272. doi: 10.1021/acs.orglett.8b00619
Q. Zhang, J. Lv, S. Luo, Beilstein J. Org. Chem. 15 (2019) 1304–1312. doi: 10.3762/bjoc.15.129
U.P.N. Tran, G. Oss, D.P. Pace, J. Ho, T.V. Nguyen, Chem. Sci. 9 (2018) 5145–5151. doi: 10.1039/C8SC00907D
S.J. Ni, J. Franzen, Chem. Commun. 54 (2018) 12982–12985. doi: 10.1039/C8CC06734A
S.J. Ni, M. El Remaily, J. Franzen, Adv. Synth. Catal. 360 (2018) 4197–4204. doi: 10.1002/adsc.201800788
M.A. Hussein, V.T. Huynh, R. Hommelsheim, R.M. Koenigs, T.V. Nguyen, Chem. Commun. 54 (2018) 12970–12973. doi: 10.1039/C8CC07329E
W.S. Shang, D.P. Duan, Y.J. Liu, J. Lv, Org. Lett. 21 (2019) 8013–8017. doi: 10.1021/acs.orglett.9b03005
S.H. Doan, M.A. Hussein, T.V. Nguyen, Chem. Commun. 57 (2021) 8901–8904. doi: 10.1039/D1CC02947A
P.K. Ranga, F. Ahmad, P. Nager, et al., J. Org. Chem. 86 (2021) 4994–5010. doi: 10.1021/acs.joc.0c02940
M. Rezazadeh Khalkhali, M.M.D. Wilde, M. Gravel, Org. Lett. 23 (2021) 155–159. doi: 10.1021/acs.orglett.0c03879
S. Sharma Rekha, G. Singh, R. Vijaya Anand, ACS Org. Inorg. Au 2 (2022) 186–196. doi: 10.1021/acsorginorgau.1c00033
J.J. Liu, J.X. Xu, Z.J. Li, et al., Eur. J. Org. Chem. 2017 (2017) 3996–4003. doi: 10.1002/ejoc.201700634
W.J. Patterson, K. Lucas, V.A. Jones, et al., Eur. J. Org. Chem. 2021 (2021) 6737–6742. doi: 10.1002/ejoc.202100946
W.G. Zhu, Q. Sun, Y.R. Wang, D. Yuan, Y.M. Yao, Org. Lett. 20 (2018) 3101–3104. doi: 10.1021/acs.orglett.8b01158
G. Oss, J. Ho, N. Thanh Vinh, Eur. J. Org. Chem. 2018 (2018) 3974–3981. doi: 10.1002/ejoc.201800579
P. Goswami, S. Sharma, G. Singh, et al., J. Org. Chem. 83 (2018) 4213–4220. doi: 10.1021/acs.joc.8b00225
H.M. Jin, M. Rudolph, F. Rominger, A.S.K. Hashmi, ACS Catal. 9 (2019) 11663–11668. doi: 10.1021/acscatal.9b03911
Y.A. Rulev, Z.T. Gugkaeva, A.V. Lokutova, et al., ChemSusChem 10 (2017) 1152–1159. doi: 10.1002/cssc.201601246
K. Omoregbee, K.N.H. Luc, A.H. Dinh, T.V. Nguyen, J. Flow Chem. 10 (2020) 161–166. doi: 10.1007/s41981-020-00082-w
J. Xu, Y. Gao, Z. Li, et al., Eur. J. Org. Chem. 2020 (2020) 311–315. doi: 10.1002/ejoc.201901537
H. Huang, Z.M. Strater, M. Rauch, et al., Angew. Chem. Int. Ed. 58 (2019) 13318–13322. doi: 10.1002/anie.201906381
H. Huang, Z.M. Strater, T.H. Lambert, J. Am. Chem. Soc. 142 (2020) 1698–1703. doi: 10.1021/jacs.9b11472
T. Shen, T.H. Lambert, Science 371 (2021) 620–626. doi: 10.1126/science.abf2798
T. Shen, T.H. Lambert, J. Am. Chem. Soc. 143 (2021) 8597–8602. doi: 10.1021/jacs.1c03718
H. Huang, T.H. Lambert, J. Am. Chem. Soc. 143 (2021) 7247–7252. doi: 10.1021/jacs.1c01967
W. Xiao, X. Wang, R. Liu, J. Wu, Chin. Chem. Lett. 32 (2021) 1847–1856. doi: 10.1016/j.cclet.2021.02.009
W. Xiao, J. Wu, Chin. Chem. Lett. 31 (2020) 3083–3094. doi: 10.1016/j.cclet.2020.07.035
L. Mei, J.M. Veleta, T.L. Gianetti, J. Am. Chem. Soc. 142 (2020) 12056–12061. doi: 10.1021/jacs.0c05507
L. Mei, J. Moutet, S.M. Stull, T.L. Gianetti, J. Org. Chem. 86 (2021) 10640–10653. doi: 10.1021/acs.joc.1c01313
S.M. Stull, L. Mei, T.L. Gianetti, Synlett 33 (2022) 1194–1198. doi: 10.1055/a-1665-9220
R. Mir, T. Dudding, J. Org. Chem. 82 (2017) 709–714. doi: 10.1021/acs.joc.6b02733
K. Dempsey, R. Mir, I. Smajlagic, et al., Tetrahedron 74 (2018) 3507–3511. doi: 10.1016/j.tet.2018.04.083
T. Courant, M. Lombard, D.V. Boyarskaya, L. Neuville, G. Masson, Org. Bio. Chem. 18 (2020) 6502–6508. doi: 10.1039/D0OB01502D
N.N.H. Ton, B.K. Mai, T.V. Nguyen, J. Org. Chem. 86 (2021) 9117–9133. doi: 10.1021/acs.joc.1c01208

Scheme 12 Bis(amino)cyclopropenium catalyzed 1, 6-conjugate addition reaction of p-QM.

Scheme 17 Trityl cation catalyzed hydroarylation reaction of alkenes with aromatic amines.

Scheme 30 Helical carbenium ion catalyzed C−H arylation and aerobic oxidative hydroxylation reactions.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

































 下载:
下载: