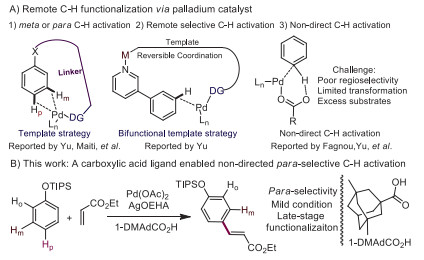
Figure 1.
Remote-selective C—H functionalizations.

Highly regioselective transformation of a C—H bond into carbon-carbon or carbon-heteroatom bond provides a direct route for fine chemical synthesis, because it can reduce the steps of prior functionalizations. Generally, the selective functionalization of a meta- [1-9], or para-position [10,11] C—H bond requires a suitable template that can coordinate with transition-metals to form a cyclometalated intermediate (Fig. 1A1). However, the stoichiometric introduction/removal of the template involves additional steps, thus limiting its application in chemical synthesis. The non-directed C—H activation reaction [12-18] provides a straightforward way for functionalization of arenes, especially for the remote position. However, it usually suffers from poor site selectivity, leading to the ortho, meta, and para regiomers. Recently, noncovalent interaction strategies have been employed to realize the direct C—H borylation [19-22] at meta or para position with iridium as the catalyst. A bifunctional nitrile template that anchors heterocyclic compound to provide a weak coordination center to achieve palladium-catalyzed meta-selective C—H olefination was first reported by the group of Yu (Fig. 1A2) [13]. Until now, there are only a few successful examples of para selective C—H olefinations via a non-directed approach. Fernández-Ibáñez and co-workers firstly developed a para-selective olefination of anilines [23] via a Pd/S, O-ligand-based catalyst. Later, they extend the substrate scopes to indoline and tetrahydroquinolines through the similar catalytic system [24]. In another example, remote site-selective C—H olefination of arene was also achieved by utilizing the steric and electronic effects of 2-pyridone [25]. However, only limited substrates could realize the site selective reaction (Fig. 1A3). Non-directed para-selective C—H functionalization can not only avoid the requirement of additional directing group/template, but can also provide a new model to directly functionalize a specific C—H bond on arene. Here, it is reported for the first time that the carboxylic acid ligand of 3, 5-dimethyladamantane-1-carboxylic acid (1-DMAdCO2H) enables para-selective C—H functionalization of TIPS-protected phenols (Fig. 1B). This new protocol can tolerate a variety of TIPS-protected phenols, including bioactive compounds and drugs. The para-selective olefination was well explored and further successfully extended to para-selective C—H halogenation, and allylation reactions. Preliminary mechanism study revealed that the para-selectivity of this non-directed C—H activation was regulated by steric effect. The protecting group of TIPS enhanced the steric hindrance at ortho and meta positions, while the bulky carboxylic acid ligand assisted C—H activation tended to occur at less hindered position. This combined spatial effect of the carboxylic acid ligand and protecting group resulted in highly para-selective C—H functionalizations.
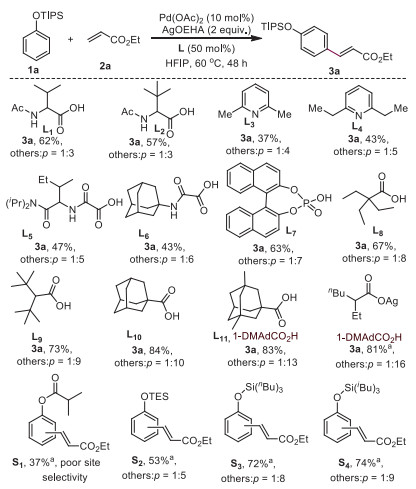
Since the early pioneering work of Fagnou [26-28], the carboxyl group was demonstrated to play an important role during the process of activation of a C—H bond, which is well known as the concerted metalation−deprotonation (CMD) mechanism. Based on our previous discovery [29-31], the steric effect of the ligand attached with transition metal can effectively adjust the site selectivity during the reaction between free radicals and aromatic rings. Thus, it was speculated that the position of C—H bond activation may be controllable by switching different sized carboxylic acid ligands in palladium non-directed C—H activation undergoing a CMD process. Phenol and its derivatives are ubiquitous in various natural products, materials, and pharmaceuticals. Several reports have demonstrated that the site selectivity between ortho and para positions can be modified with different protecting groups in the ligand to promote palladium-catalyzed olefination reactions. For example, the TIPS-protected phenol can provide a 1/4.4 (o/p) selectivity in the 2-pyridone-accelerated non-directed C—H olefination reaction12, while anisole affords a much less selective olefination reaction [32-35] (o: m: p = 1.8/1.0/3.7). In regard to these points, we envision that the combination of spatial factors between a carboxylic acid ligand and a bulk protecting group, a palladium-catalyzed non-direct para-selective C—H activation would be feasible (Scheme 1), which might offer an effective approach to highly para-selective C—H functionalizations. Based on this key point, TIPS-protected phenol (1a) was directly treated with ethyl acrylate (2a, 1.5 equiv.) in the presence of Pd(OAc)2 (10 mol%), N-protected amino acids (30 mol%), and AgOAc (2 equiv.) in HFIP at 60 ℃ for 24 h. Several N-protected amino acids including N-Ac-Val-OH, N-Ac-Ile-OH, and N-Ac-Leu-OH were screened, and good yield of olefinated product 3a was observed. However, none of them provided good selectivity between the para- and others-olefinated products (para/others < 5:1; L1, L2). Next, 2, 6-disubustituted pyridines were further tested, but the site-selectivity was not improved and the yield was also poor (L3, L4). Oxalyl amides (L5, L6), which play an important role in the nickel-enabled para-selective alkylation, were also investigated, but they too displayed poor selectivity. When phosphates (L7) were used, slightly improved selectivity was obtained, and the yields were good too. Encouraged by these results, typical carboxylic acid ligand [36-40] such as L8, L9 and 1-AdCO2H (L10) were subjected to the standard reaction conditions. Reasonably good selectivity (para/others = 10:1) was achieved when 1-AdCO2H was employed as the additive. Although the reason for high para-selectivity is unclear, it is likely that the rigid structure of adamantane enhanced the interaction with the protecting group, leading to the para-selectivity. Gratifyingly, 1-DMAdCO2H (L) was most effective, leading to 81% yield of the product with high para selectivity (para/others = 13:1). Several silver salts were further explored. Among them, silver 2-ethylhexanoate slightly improved the selectivity (para/others = 16:1) and afforded product 3a in 81% yield. Control experiments show that palladium was indispensable for this transformation. It is worth noting that di-olefinated products were observed in less than 5 mol% yield and the products is almost trans and no cis product is observed. Due to the steric hindrance effect between the carboxylic acid ligand and TIPS protecting group, various protected phenols (S1–S4) were subjected to the standard reaction conditions, and it was evident that the selectivity decreased with less bulky protecting groups. These results further support the hypothesis that the high para-selectivity is influenced by the steric repulsion between the bulky carboxylic acid ligand and protecting group (Supporting information).
With the optimized reaction conditions, various ortho-substituted TIPS-protected phenols were subjected to the standard reaction conditions (Scheme 2). Substrates with electron-donating and electron-withdrawing functional groups such as ethyl, isobutyl, tert-butyl, cyclohexyl, chloride, bromide, OCF3, and phenyl (3b–3j) were all well tolerated, leading to the corresponding products in good yields with high para-selectivity. Moreover, ortho-nitro-substituted phenol (3k) was compatible, leading to the corresponding product in acceptable yield. The meta chloride (3l) or fluoride (3m) substituted phenols all provided the olefinated products in good yields with high para-selectivity. When TIPS-protected 3-bromophenol (3n) was used, a slightly poor selectivity was observed, which might be due to the steric hindrance of bromide. A wide variety of di-substituted phenols (3o-3z) were further examined and all of them afforded the corresponding products in moderate to good yields with high site-selectivity, highlighting the synthetic importance of this non-directed para-selectivity olefination reaction. Both tetrahydro-1-naphthol (3aa) and inden-4-ol (3ab) were well tolerated, generating the corresponding para-olefinated products in good yield.

Encouraged by the success of 1-DMAdCO2H enabled para-selective C—H olefination, the scope of olefin coupling partners was evaluated next (Scheme 3). Generally, unsaturated olefins are effective coupling partners for this transformation. Acrylate derivatives (4a–4f) all performed well, yielding the para-olefinated products in good yields. It is worth noting that fluorinated functional group can be indirectly introduced into the aromatic ring. 1, 2-Disubstituted-unsaturated olefins such as methyl crotonoate (4g), ethyl crotonoate (4h), methyl pent-2-enoate (4i), and diethyl fumarate (4j) were all suitable coupling partners in this transformation. The steric effect of these substrates was likely responsible for the low transformation of 1a, leading to low yields of the corresponding olefinated products.

Only 1 equiv. of arene was used in this non-directed para-selective C—H olefination reaction, which can guarantee its late-stage functionalization and scale-up of the bioactive compound (Scheme 4). For example, a gram scale reaction was performed with TIPS-protected thymol (5a), which is a drug molecule, and the olefinated product was isolated in 70% yield with high para-selectivity. The TIPS-protected amylmetacresol (5b), disoprofol (5c), and pyrocatechol (5d) all proceeded well in the reaction, affording the olefinated product in good yields with high para-selectivity. Importantly, the key structure of isoamyl 4-methoxycinnamate and octyl 4-methoxycinnamate (5e, 5f) could be synthesized in one step in good yields.

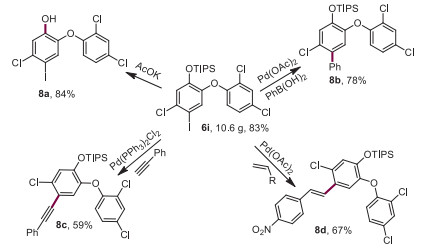
To demonstrate the potential generality of this carboxylic acid ligand enabled non-directed para-selective C—H activation in affording various transformations, non-directed para-selective iodination and allylation reactions were explored (Supporting information). Gratifyingly, a 10-g scale para-selective iodination reaction was also achieved with TIPS-protected triclosan, which is a well-known fungicide. To further demonstrate the synthetic importance of this new strategy, a variety of transformations were carried out through palladium cross-coupling [41-43] with iodinated-triclosan as the starting material (Scheme 5; 8b, 8c, 8d). In addition, the protecting group can be easily removed [44] under basic conditions in excellent yield (8a).
To further understand the role of 1-DMAdCO2H in this non-directed palladium-catalyzed C—H olefination reaction, different TIPS-protected phenols were tested with N-Ac-Gly-OH or acetic acid as the additive (Scheme 6). The results clearly indicate that the site selectivity cannot be controlled without 1-DMAdCO2H as the carboxylic acid ligand.

The para-C-H bond of TIPS-protected 2-ethylphenol substrate was selectively deuterated under the catalysis of palladium acetate in D4-acetic acid, generating the para-deuterated TIPS-protected 2-ethylphenol (Scheme 7a). This result suggests that spatial repulsion factor between the carboxylic acid ligand and the bulky protecting group resulted in the selective C—H activation at the para-position, which rules out the role of olefination coordination in the para-selectivity (Scheme 7b). A kinetic effect of 2.65 was obtained, indicating that C—H activation was the rate determining step and further supporting the above hypothesis. When acetic acid was used as ligand, a kinetic effect of 3.45 was observed, suggesting that the additive 1-DMAdCO2H was more conducive to assist C—H bond activation with a palladium catalyst (Scheme 7c). Pd(1-AdCO2)2 was prepared according to the reported [45] procedure. We found better site selectivity and yield of 3a was observed, when Pd(1-AdCO2)2 was used as a catalyst without adding a carboxylic acid ligand. Interestingly, its site selectivity is nearly identical to the 1-AdCO2H used as a ligand. When the carboxylic acid ligands were added, the yields and selectivity became uniform for both palladium catalysts for 3a. These results may indicate that Pd(1-AdCO2)2 is a key intermediate in the catalytic cycle (Scheme 7d).

In conclusion, this paper reveals for the first time that the bulky carboxylic acid ligand can affect the site selectivity during the C—H activation step when the non-directed C—H functionalizations undergo concerted-metalation deprotonation (CMD) mechanism with a palladium catalyst. Various phenol derivatives including the bioactive molecules of thymol, propofol, and triclosan, were all para-selectively functionalized, leading to the corresponding olefinated, iodinated, or allylated products in moderate to good yields. Moreover, the 10-g scale para-selective iodination reaction proceeded well with the bioactive compound of triclosan, facilitating its late-stage functionalization through cross-coupling reactions. Control experiments show that the use of a bulky carboxylic acid ligand (1-DMAdCO2H) is the key factor to achieve para-selectivity. A preliminary mechanism study revealed that the spatial repulsion factor between carboxylic acid ligand and bulky protecting group resulted in the selective C—H activation happened at the less sterically hindered para-position. This successful example of palladium-catalyzed non-directed para-selective C—H functionalization provides a straightforward route for remote site-selective C—H activation, which would open a new door for other remote site-selective C—H activation reactions.
The authors declare that they have no known competing financial interestsor personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (No. 21772139), the Jiangsu Province Natural Science Found for Distinguished Young Scholars (No. BK20180041), and the PAPD Project. The project was also supported by the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University.
Supplementary material associated with this article can be found, in the online version, at doi:
H.X. Dai, G. Li, X.G. Zhang, A.F. Stepan, J.Q. Yu, J. Am. Chem. Soc. 135 (2013) 7567–7571. doi: 10.1021/ja400659s
L. Wan, N. Dastbaravardeh, J.Q. Yu, J. Am. Chem. Soc. 135 (2013) 18056–18059. doi: 10.1021/ja410760f
S. Lee, K.L. Tan, J. Am. Chem. Soc. 135 (2013) 18778–18781. doi: 10.1021/ja4107034
L. Campeau, K. Fagnou, J. Am. Chem. Soc. 126 (2004) 9186–9187. doi: 10.1021/ja049017y
L. Chu, M. Shang, K. Tanaka, Q. Chen, J.Q. Yu, ACS Cent. Sci. 1 (2015) 394–399. doi: 10.1021/acscentsci.5b00312
M.T. Mihai, G.R. Genov, R.J. Phipps, Chem. Soc. Rev. 47 (2018) 149–171. doi: 10.1039/C7CS00637C
J. Xu, J. Chen, J.Q. Yu, J. Am. Chem. Soc. 141 (2019) 1903–1907. doi: 10.1021/jacs.8b13403
G. Meng, N.Y.S. Lam, E.L. Lucas, et al., J. Am. Chem. Soc. 142 (2020) 10571–10591. doi: 10.1021/jacs.0c04074
S. Porey, X. Zhang, S. Bhowmick, V.K. Singh, D. Maiti, J. Am. Chem. Soc. 142 (2020) 3762–3774. doi: 10.1021/jacs.9b10646
S. Bag, T. Patra, A. Modak, D. Maiti, J. Am. Chem. Soc. 137 (2015) 11888–11891. doi: 10.1021/jacs.5b06793
T. Patra, S. Bag, A. Modak, D. Maiti, Angew. Chem. Int. Ed. 55 (2016) 7751–7755. doi: 10.1002/anie.201601999
P. Wang, P. Verma, J.Q. Yu, Nature551 (2017) 589–593.
Z. Zhang, K. Tanaka, J.Q. Yu, Nature543 (2017) 538–543. doi: 10.1038/nature21418
P. Wedi, M. Gemmeren, Angew. Chem. Int. Ed. 57 (2018) 13016–13027. doi: 10.1002/anie.201804727
H. Chen, P. Wedi, M. Gemmeren, Angew. Chem. Int. Ed. 130 (2018) 2523–2527. doi: 10.1002/ange.201712235
K. Naksomboon, C. Valderas, M. Gomez-Martinez, M.A. Fernandez-Ibanez, ACS Catal. 7 (2017) 6342–6346. doi: 10.1021/acscatal.7b02356
N. Kuhl, J. Wencel-Delord, F. Glorius, Angew. Chem. Int. Ed. 51 (2012) 10236–10254. doi: 10.1002/anie.201203269
S. Kancherla, K.B. Jorgensen, M.A. Fernandez-Ibanez, Synthesis51 (2019) 643–663. doi: 10.1055/s-0037-1610852
Y. Kuninobu, H. Ida, M. Nishi, M. Kanai, Nat. Chem. 7 (2015) 712–717. doi: 10.1038/nchem.2322
Y. Saito, Y. Segawa, K. Itami, J. Am. Chem. Soc. 137 (2015) 5193–5198. doi: 10.1021/jacs.5b02052
H.J. Davis, M.T. Mihai, R.J. Phipps, J. Am. Chem. Soc. 138 (2016) 12759–12762. doi: 10.1021/jacs.6b08164
M.T. Mihai, B.J. Williams, R.J. Phipps, J. Am. Chem. Soc. 141 (2019) 15477–15482. doi: 10.1021/jacs.9b07267
K. Naksomboon, J. Poater, F.M. Bickelhaupt, M.A. Fernandez-Ibanez, J. Am. Chem. Soc. 141 (2019) 6719–6725. doi: 10.1021/jacs.9b01908
W.L. Jia, N. Westerveld, K.M. Wong, T. Morsch, M.A. Fernandez-Ibanez, Org. Lett. 21 (2019) 9339–9342. doi: 10.1021/acs.orglett.9b03505
P. Wang, P. Verma, J.Q. Yu, Nature551 (2017) 489–493. doi: 10.1038/nature24632
L.C. Campeau, S. Rousseaux, K. Fagnou, J. Am. Chem. Soc. 127 (2005) 18020–18021. doi: 10.1021/ja056800x
M. Lafrance, C.N. Rowley, K. Fagnou, J. Am. Chem. Soc. 128 (2006) 8754–8756. doi: 10.1021/ja062509l
D.R. Stuart, K. Fagnou, Science316 (2007) 1172–1175. doi: 10.1126/science.1141956
G. Tu, C. Yuan, Y. Li, J. Zhang, Y. Zhao, Angew. Chem. Int. Ed. 57 (2018) 15597–15601. doi: 10.1002/anie.201809788
W.T. Fan, Y. Li, D. Wang, S.J. Ji, Y. Zhao, J. Am. Chem. Soc. 142 (2020) 20524–20530. doi: 10.1021/jacs.0c09545
Y. Jiang, B. Li, N. Ma, et al., Angew. Chem. Int. Ed. 60 (2021) 19030–19034. doi: 10.1002/anie.202105631
M. Dams, S. Celen, P.A. Jacobs, Angew. Chem. Int. Ed. 42 (2003) 3512–3515. doi: 10.1002/anie.200351524
B. Yin, M. Fu, Q. Zhu, Chem. Commun. 56 (2020) 3293–3296. doi: 10.1039/d0cc00940g
H.T. Kim, E. Kang, J. Joo, Org. Lett. 23 (2021) 3657–3662. doi: 10.1021/acs.orglett.1c01040
V. Sukowski, W.L. Jia, R. Diest, M.A. Fernandez-Ibanez, Eur. J. Org. Chem. 2021 (2021) 4132–4135. doi: 10.1002/ejoc.202100737
T. Fujihara, A. Yoshida, M. Satou, Y. Tsuji, Catal. Commun. (2016) 71–74.
Q. Sun, H. Zhang, Q. Wang, G. Chen, Angew. Chem. Int. Ed. 60 (2021) 19620–19625. doi: 10.1002/anie.202104430
D. Mu, F. Gao, G. Chen, G. He, ACS Catal. 7 (2017) 1880–1885. doi: 10.1021/acscatal.6b03661
Y. Tanji, N. Mitsutake, Y. Tsuji, Angew. Chem. Int. Ed. 57 (2018) 10314–10317. doi: 10.1002/anie.201804566
Y. Tanji, Y. Tsuji, T, Fujihara. Chem. Commun. 56 (2020) 3843–3846. doi: 10.1039/d0cc01129k
C.C. Piras, D.K. Smith, Angew. Chem. Int. Ed. 59 (2020) 853–859. doi: 10.1002/anie.201911404
G. Hamasaka, D. Roy, A. Tazawa, Y. Uozumi, ACS Catal. 9 (2019) 11640–11646. doi: 10.1021/acscatal.9b04593
F. Liu, C. Liu, S. Dai, Green Chem. 18 (2016) 6536–6544. doi: 10.1039/C6GC02237E
B. Wang, H.X. Sun, B. Chena, Z.H. Sun, Green Chem. 11 (2009) 1112–1114. doi: 10.1039/b905443j
D. Willcox, G.N. Chappell, F.K. Hogg, M.J. Gaunt, Science345 (2016) 851–857. doi: 10.1126/science.aaf9621

Scheme 1 Optimization of ligand. Reaction performed on a 0.1 mmol scale with 2a (0.15 mmol), Pd(OAc)2 (10 mol%), AgOAc (2 equiv.), ligand (50 mol%) and HFIP (0.5 mL). a Reaction performed using 1-DMAdCO2H (50 mol%) replaced ligand.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载: