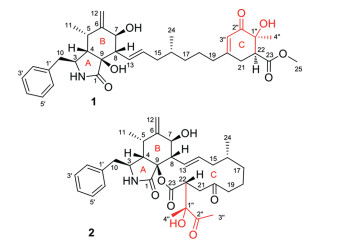
Figure 1.
Chemical structures of compounds 1 and 2.
Vercytochalasins A and B: Two unprecedented biosynthetically related cytochalasins from the deep-sea-sourced endozoic fungus Curvularia verruculosa
Xueyi Hu , Xiaoming Li , Suiqun Yang , Xin Li , Bingui Wang , Linghong Meng

Cytochalasins are a class of natural products derived from fungi and are usually characterized by a fused macrocyclic ester ring coupled with a phenylalaine unit. These compounds showed a variety of structures and broad range of activities [1]. The biosynthesis of cytochalasins are based on the combination of polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) pathway [2]. Cytochalasins attracted great attentions from researchers not only due to the interesting chemical structures but also due to their potent biological activities such as cytotoxic [3, 4], antimicrobial [5], antiparasitic [6], and antiviral [7] properties.
Cytochalasins are produced by fungi from diverse habitats including marine environments such as algae [3], squat lobster [4], soft-coral [8], deep-sea sediment [9], and jellyfish as well [10]. As part of our works to search structurally unique and biologically active natural products from marine sourced fungi [3, 4], Curvularia verruculosa CS-129, a fungus derived from deep-sea squat lobster Shinkaia crosnieri collected from the cold seep area of South China sea was investigated. Initial works on this fungus resulted in the isolation and identification of several cytotoxic cytochalasins [4]. Further works on the remaining portions of the fungus resulted in the characterization of two unprecedentedl cytochalasins, namely, vercytochalasins A (1) and B (2) (Fig. 1). The high structural diversity of previously described cytochalasins is mainly due to the changes of cyclization and substitution [11]. Structurally, compound 1 is an unprecedented secocytochalasin derivative featuring the ester group cleaved between C-9 and C-23, and incorporating an additional oxygenated C4 unit which coupled with the C-20 and C-22 to form a new substituted cyclohexenone moiety, while compound 2 featuring an unusual C4 unit (2‑hydroxy-3-oxobutan-2-yl) at C-22. Compounds 1 and 2 are biosynthetically related congeners and both of them are the first reported cytochalasins coupling with an oxygenated C4 unit. The enzyme inhibitory activity was firstly evaluated for cytochalasins and compound 1 exhibited potent angiotensin-Ⅰ-converting enzyme (ACE) inhibitory activity. The isolation, structural elucidation, bioactivities and plausible biosynthesis pathway of compounds 1 and 2 are reported herein.
Vercytochalasin A (1) was obtained as a colourless amorphous solid and its molecular formula was assigned as C34H45NO7 by HRESIMS. The 1H NMR data (Table 1) showed resonances corresponding to substituted phenyl at δH 7.21 (3H, overlapped, H-2′/H-4′/H-6′) and 7.30 (2H, t, J = 7.4 Hz, H-3′/H-5′), an olefinic methylene at δH 5.02 (1H, s, H-12a) and 4.83 (1H, s, H-12b), one oxygenated methine at δH 3.71 (1H, t, J = 6.6 Hz, H-7), three methyls at δH 0.79 (3H, d, J = 6.7 Hz, H3–11), 0.83 (3H, d, J = 6.6 Hz, H3–24), and 1.19 (3H, s, H3–4′′), one methoxy at δH 3.60 (3H, s, H3–25), and two hydroxy protons at δH 4.80 (1H, d, J = 6.6 Hz, 7-OH) and 5.32 (1H, s, 1′′-OH). The 13C NMR data (Table 1) revealed the presence of four methyls (one methoxy), seven methylenes (one olefinic and six aliphatic), 15 methines (eight olefinic), and eight non-protonated carbons (one keto, two ester or amide carbonyls and two oxygenated sp3-hybridized).
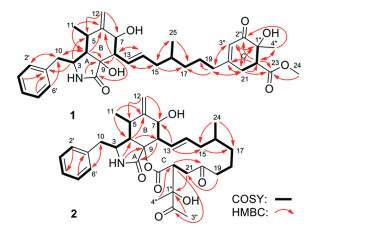
The 1H–1H COSY spectrum of 1 disclosed two proton spin-spin systems in the core structure, namely, H-10/H-3/H-4/H-5/H3–11 and H-7/H-8/H-13 through H-19 (Fig. 2). HMBC correlations from H2–10 to C-2′/6′ indicated that the phenyl group substitutes at C-10 (Fig. 2). Other HMBC correlations from H-4 to C-6 and C-9, from H-8 to C-1 and C-9, from H3–11 to C-4 and C-6, from H2–12 to C-5 and C-7, and from 2-NH to C-9, as well as from H-14 to C-8 determined the cis-fused pattern of rings A and B as well as the substituted position of the long alkene chain (C13 through C19). Further HMBCs from H-19 to C-21 and C-3′′, and vice versa, from H2–21 to C-1′′ and C-3′′, from H-22 to C-20, C-23 and C-2′′, from H-3′′ to C-21 and C-1′′, and from H-4′′ to C-22 and C-2′′, as well as from H-24 to C-23 indicated the presence of substructure ring C and the position of methoxycarbonyl at C-22. Thus, the planar structure of 1 was assigned.
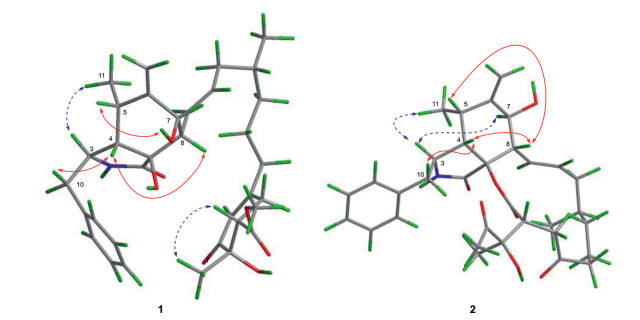
The relative configuration of 1 was determined by NOESY experiment and DP4+ calculation. NOE correlation from H-3 to H3–11 indicated they were cofacial orientation. In contrast, correlations from H-5 to 7-OH and from H-4 to H-8 and H-10b suggested them on the other side (Fig. 3). Thus, the relative configurations of rings A and B (except C-9) were determined. NOE from H3–4′′ to H-22 presented the relative configuration of ring C.
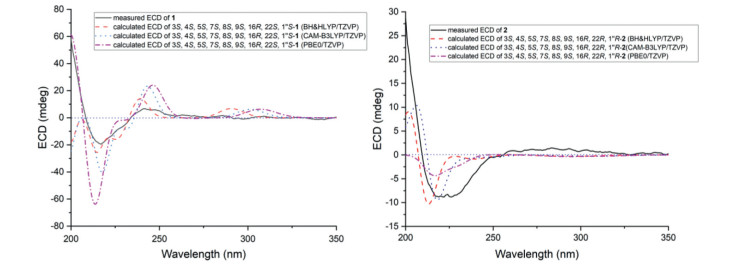
As the protons between rings B and C have no correlations due to the large interatomic spacing and the relative configuration at C-16 could not be determined, DP4+ calculation was performed to confirm the relative configurations at remaining undetermined chiral carbons [12, 13]. This method was used to assign the relative configurations of organic molecules employing GIAO (gauge-independent atomic orbital) NMR shift calculations. Based on a DP4+ protocol [14], both proton and carbon data of eight possible isomers of compound 1 (1a-1h, Fig. S1 in Supporting information) were calculated, and the results were analysed with the experimental values. The statistical comparison showed that the isomer 1f was the equivalent structure with 100% probability (Fig. S1). Based on their unique biosynthesis pathway, this result suggested that the essential elements of 1 have same stereochemistry with most known cytochalasins [15]. The absolute configuration of 1 was determined by ECD calculation (TDDFT) performed on the mPW1PW91/6–311G(d) level of theory. The calculated ECD spectrum for (3S, 4S, 5S, 7S, 8S, 9S, 16R, 22S, 1′′S)−1 is in good accordance with the experimental curve at BH & HLYP/TZVP, CAM-B3LYP/TZVP, and PBE0/TZVP levels (Fig. 4).
Vercytochalasin B (2), originally isolated as a colourless amorphous solid, has a molecular formula of C33H43NO7 which was established by HRESIMS. The 1H and 13C NMR data (Table 1) displayed four methyls, one olefinic and six aliphatic methylenes, 14 methines (seven olefinic), and eight non-protonated carbons (two keto, two ester or amide carbonyls and two oxygenated sp3-hybridized), suggesting that 2 having typical signals of a cytochalasin derivative but with novel modification that different from the known cytochalasin skeletons [1].
The 1H–1H COSY cross-peaks of H-10/H-3/H-4/H-5/H3–11, together with HMBC correlations from H-4 to C-6 and C-8, from H-8 to C-1, from H2–10 to C-2′/6′, from H-11 to C-4 and C-6, and from H2–12 to C-5 and C-7 as well as from 2-NH to C-4 and C-9 determined the fusing pattern of rings A and B as well as the substitute position of phenyl group (Fig. 2). Additional COSY correlations of from H-8 to H-7 and H-13 and from H-13 through H-19 and HMBC correlations from H-13 to C-7 and C-15, from H2–21 to C-1′′, from H-22 to C-20 and C-2′′, from H3–3′′ to C-1′′ as well as from H3–4′′ to C-22 (Fig. 2) revealed the substructure of ring C and the substituent group of C-1′′ through C-4′′.
NOE correlations from H-3 to H-7 and H3–11 indicated the same orientation of these protons, whereas correlations from H-5 to H-8 and from H-4 to H-8 and H-10 placed they were on the opposite face (Fig. 3). Based on the previous reports, most cytochalasins share the same steric configurations due to their unique biosynthesis pathway, i.e., the relative configurations at C-9 and C-16 are both determined as R* [16]. DP4+ calculation was used to confirm the relative configuration at C-22 and C-1′′ and four possible isomers of compound 2 (2a-2d) were involved (Fig. S2 in Supporting information). Comparison of statistics gave the suited structure as 2b with 100% probability. Thus, the relative configuration of 2 was assigned. The absolute configuration of 2 was determined by ECD calculation. The measured ECD spectrum of 2 matched well with calculated spectra of (3S, 4S, 5S, 7S, 8S, 9R, 16R, 22R, 1′′R)−2 at BH&HLYP/TZVP, CAM-B3LYP/TZVP, and PBE0/TZVP levels (Fig. 4).
As compounds 1 and 2 were first examples of fusion of cytochalasin and C4 units, the plausible biosynthetic pathway of compounds 1 and 2 was presented (Scheme 1). It appears that both 1 and 2 share the same precursor cytochalasin B6. Briefly, cytochalasin B6 might be carbonized to form the intermediate Ⅰ, which react with 2, 3-butanedione to yield the key intermediate Ⅱ via aldol condensation reaction. Compound 2 could be obtained by solvation of Ⅱ [17]. In addition, hydrolyzation of compound 2 cleavage of the ester group between C-9 and C-23 would form the intermediate Ⅲ. The key intermediate V was speculated to be produced from Ⅳ, which might be formed from Ⅲ by aldol condensation between C-20 and C-3′′. Reduction of Ⅴ could yield the intermediate Ⅵ, which could be converted to vercytochalasin A (1) by acetylation at C-23.
Compounds 1 and 2 were assayed for their angiotensin-Ⅰ-converting enzyme (ACE) inhibitory and antibacterial activities. In the ACE assay, compound 1 showed potent inhibitory activity with IC50 value 505 nmol/L compared to the positive control captopril (IC50 = 15.0 nmol/L) (Fig. 5). ACE is regarded as a key enzyme that regulates blood pressure and translates angiotensin Ⅰ into angiotensin Ⅱ in the circulating or endocrine system of the human [18]. In the reports describing ACE inhibitory activity, the natural occurring products usually having activity with IC50 values bigger that 4.5 µmol/L [19, 20]. Compound 1, with IC50 value 505 nmol/L, is likely the most active natural products reported so far. In the antibacterial screening, compound 2 showed potent inhibitory activity against aquatic pathogenic bacteria Vibrio anguillarum, V. harveyi, and V. parahaemolyticus with MIC values 32, 8 and 4 µg/mL, respectively (the MIC values of positive control chloromycetin were 0.5, 1 and 2 µg/mL, respectively). The results indicated that compound 2 has potential to be a broad-spectrum antibiotic against aquatic pathogenic bacteria in the aquapathogens of the genus Vibrio.
To explore the intermolecular interaction of compounds 1 and 2 with ACE, the potential binding sites were excavated by molecular docking simulations. The results exhibited significant difference between the potential binding sites of 1 and 2. Compound 1 could form nine hydrogen bonds with residues ASN-66, ASP-358 (two hydrogen bonds), HIS-353, HIS-513, TYR-523, ALA-356, GLU-384 and LYS-522 as well as two π-π stacking interaction of benzene ring with TRP-59 and TYR-62, to interact with ACE (Fig. 6). In contrast, compound 2 could interact with ACE by only two hydrogen bonds with relevant residues. Different combining capacities of these two compounds with ACE might attribute to different ACE inhibitory activities and the potent activity of compound 1 may arising from the hydrogen bonds and π-π stacking interactions.
In summary, vercytochalasins A (1) and B (2), isolated from the deep sea squat lobster Curvularia verruculosa collected from the cold seep environment, represent the first examples of cytochalasins fusing with oxygenated C4 unit, which implies 1 and 2 are representatives of new subclassed of cytochalasin family. In particular, compound 1 possesses an unusual long chain at C-8, which distinguishes it from other known cytochalasin alkaloids. Further, antibiotic and angiotensin-Ⅰ-converting enzyme (ACE) inhibitory activities were tested. Compound 1 exhibited potent angiotensin-Ⅰ-converting enzyme (ACE) inhibitory activity and it was first time that enzyme inhibitory activities of cytochalasins was evaluated. Besides, compound 2 showed antibacterial activity against aquatic pathogenic bacteria in the genus Vibrio. Compound 1 was confirmed to have the capacity to incorporate with ACE by hydrogen bonds and π-π stacking interactions by Molecular docking simulations.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA22050401), the National Natural Science Foundation of China (Nos. U2006203 and 42076090), the Senior User Project of RV KEXUE (No. KEXUE2020GZ02), and the Shandong Provincial Natural Science Foundation (No. ZR2021ZD28). X. Li. appreciates the Basic Applied Research program of Qingdao (No. 19–6–2–40-cg) and B.Wang acknowledges the support of the RV KEXUE of the National Major Science and Technology Infrastructure from the Chinese Academy of Sciences (for sampling) and the Oceanographic Data center at IOCAS (for CPU time).
Supplementary material associated with this article can be found, in the online version, at doi:
K. Scherlach, D. Boettger, N. Remme, et al., Nat. Prod. Rep. 27 (2010) 869–886. doi: 10.1039/b903913a
E. Skellam, Nat. Prod. Rep. 34 (2017) 1252–1263. doi: 10.1039/C7NP00036G
C.M. Cui, X.M. Li, C.S. Li, et al., J. Nat. Prod. 73 (2010) 729–733. doi: 10.1021/np900569t
X.Y. Hu, C.Y. Wang, X.M. Li, et al., J. Nat. Prod. 84 (2021) 3122–3130. doi: 10.1021/acs.jnatprod.1c00907
W. Pongcharoen, V. Rukachaisirikul, S. Phongpaichit, et al., J. Nat. Prod. 69 (2006) 856–858. doi: 10.1021/np0600649
A. Makioka, M. Kumagai, S. Kobayashi, et al., Parasitol. Res. 93 (2004) 68–71. doi: 10.1007/s00436-004-1106-8
T.S. Chen, G.A. Doss, A. Hsu, et al., J. Nat. Prod. 56 (1993) 755–761. doi: 10.1021/np50095a013
C.J. Zheng, C.L. Shao, L.Y. Wu, et al., Mar. Drugs 11 (2013) 2054–2068. doi: 10.3390/md11062054
Z.H. Zhang, X.T. Min, J.J. Huang, et al., Mar. Drugs 14 (2016) 233. doi: 10.3390/md14120233
E.L. Kim, H. Wang, J.H. Park, et al., Bioorg. Med. Chem. Lett. 25 (2015) 2096–2099. doi: 10.1016/j.bmcl.2015.03.080
W.X. Wang, Z.H. Li, T. Feng, et al., Org. Lett. 20 (2018) 7758–7761. doi: 10.1021/acs.orglett.8b03110
C.L. Xie, D. Zhang, K.Q. Guo, et al., Chin. Chem. Lett. 33 (2022) 2057–2059. doi: 10.1016/j.cclet.2021.09.073
J. Cao, X. Zhang, K. Shang, et al., Chin. Chem. Lett. 31 (2020) 427–430. doi: 10.1016/j.cclet.2019.09.020
N. Grimblat, M.M. Zanardi, A.M. Sarotti, J. Org. Chem. 80 (2015) 12526–12534. doi: 10.1021/acs.joc.5b02396
R. Liu, Z. Lin, T. Zhu, et al., J. Nat. Prod. 71 (2008) 1127–1132. doi: 10.1021/np070539b
E.L. Kim, J.L. Li, H.D. Dang, et al., Bioorg. Med. Chem. Lett. 22 (2012) 3126–3129. doi: 10.1016/j.bmcl.2012.03.058
C. Chen, H. Zhu, X.N. Li, et al., Org. Lett. 17 (2015) 644–647. doi: 10.1021/ol503666b
P.R. Liu, X.D. Lan, M. Yaseen, et al., Mar. Drugs 17 (2019) 463. doi: 10.3390/md17080463
K. Chakraborty, S. Dhara, Phytochemistry 195 (2022) 113024. doi: 10.1016/j.phytochem.2021.113024
A.K. Stewart, R. Ravindra, M.V. Wagoner, et al., J. Nat. Prod. 81 (2018) 349–355. doi: 10.1021/acs.jnatprod.7b00829

Figure 3 Key NOE correlations of compounds 1 and 2 (red lines: β-orientation; blue dashed lines: α-orientation).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载: