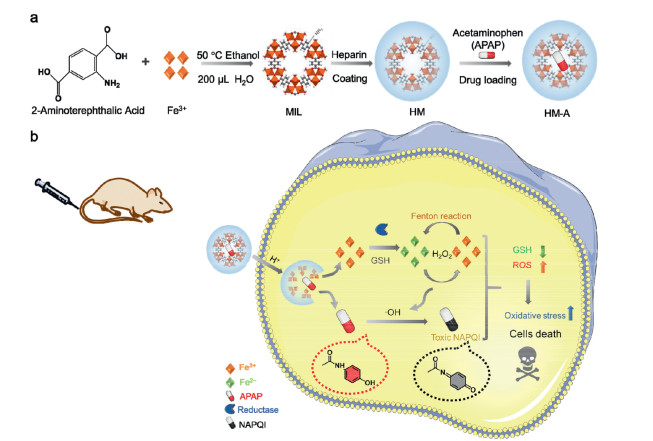
Figure 1.
(a) Schematic illustration of the synthesis of HM-A NPs. (b) Anticancer mechanism of HM-A based on Fenton reaction and APAP metabolism.
Fenton-reaction-triggered metabolism of acetaminophen for enhanced cancer therapy
Fanwen Sun , Yayun Peng , Yanping Li , Menghan Xu , Ting Cai

Acetaminophen (APAP) is a classic nonsteroidal anti-inflammatory drug (NSAID) for treating pain and reducing fever. However, an overdose of APAP can cause severe liver damage [1-3]. In fact, APAP-induced toxicity is the most common cause of acute liver failure in developed countries [4-6]. The metabolic activation of APAP is principally catalyzed by cytochrome P450 enzymes. The highly reactive metabolite of APAP is N-acetyl-p-benzoquinone imine (NAPQI), which reacts rapidly with glutathione (GSH) and binds to proteins in the liver, leading to reactive oxygen species (ROS) generation, mitochondrial dysfunction, and thus liver damage [7-9]. ROS-induced apoptosis is a widely practiced strategy for cancer therapy [10-12]. Thus, we propose that APAP can be used as a prodrug to achieve anti-tumor effects. The APAP metabolism-induced liver injury inspires us to investigate its anti-tumor potentials.
Apart from the cytochromes P450 enzymes abundant in the liver, some enzymes in the tumor cells also exhibit the oxidant activity to APAP. For example, VAD et al. reported that APAP showed selective toxicity towards melanoma cell lines due to the abundant tyrosinase in melanoma cells. APAP can be bioactivated to toxic 4-acetamido-o-benzoquinone by tyrosinase for cancer therapy [13-18]. However, the anticancer therapeutic applications of APAP in the absence of tyrosinase are still underexplored. It is of great importance to develop a more general strategy for APAP to achieve stronger anti-tumor effects.
Fenton reaction can induce oxidative stress of tumor cells by converting intracellular hydrogen peroxide (H2O2) into hydroxyl radicals (•OH), the most harmful ROS [19-25]. Fe-based compounds are common agents for the Fenton reaction [26-30]. Various Fe-based nanomaterials have been reported to trigger the Fenton reaction in tumor microenvironment [31-33]. Among them, Fe-based metal-organic frameworks (MOFs), composed by Fe ions and organic linkers, have become a promising material in the biomedical field owing to their unique properties, including large pore volume, high surface area, tunable pore size, versatile functionality and high drug loading efficiency [34-39]. However, the •OH produced by the Fenton reaction alone is insufficient to kill the tumors [40-43]. Apart from the chemodynamic therapy based on Fenton reactions, Fenton reactions have been widely employed for removing organic pollutants from wastewater [44, 45]. For example, Zhang et al. reported that •OH was able to oxidize APAP to its toxic quinone metabolite, NAPQI [46-48]. This inspires us to explore the potentials of combining Fenton chemodynamic toxicity and APAP metabolic toxicity into a binary delivery system for enhanced cancer therapy.
In this study, we encapsulate APAP in a heparin-coated Fe-based MOF, MIL-88B (abbreviated as MIL) to establish APAP@Hep-MIL-88B nanoparticles (abbreviated as HM-A NPs) for the synergistic cancer therapy (Fig. 1). HM-A NPs remain stable in a normal physiological environment but are highly active in the weakly acidic tumor microenvironment. Fe ions released from HM-A and H2O2 highly expressed in the tumor site trigger the Fenton reaction to produce toxic •OH. The generated •OH convert nontoxic APAP to its toxic metabolite, NAPQI. Meanwhile, NAPQI leads to extensive GSH depletion, which diminishes the antioxidant ability of cancer cells and thus enhances the efficiency of Fenton reaction. Intracellular oxidative stress and toxic NAPQI can provide a synergistic effect through the cascade reaction to kill the cancer cells.
MIL nanoparticles (NPs) were synthesized by a facile and economical method as per the previously reported procedure with some modifications (Fig. 1a) [49]. The powder X-ray diffraction (PXRD) patterns of as-synthesized products agreed well with the calculated patterns for Fe-MIL-88B (Fig. S1 in Supporting information). The MIL NPs were surface-functionalized by heparin to improve the stability and dispersibility [50-52]. The structure of HM was characterized by the Fourier transform infrared (FT-IR) spectroscopy (Fig. 2a). In the FT-IR spectrum of HM, the band at 1585 cm−1 belonged to the stretching vibration of C=O groups of MIL and the band 1053 cm−1 was attributed to –SO3 groups of heparin. Compared to the MIL, a significant change of zeta potential was observed after the heparin coating (Zeta potential: from +9.8 mV to –34.8 mV) (Fig. 2b). In addition, PXRD patterns indicated that the crystalline structure of MIL NPs was not altered after the surface modification (Fig. 2c). Finally, the APAP molecules were loaded into HM by the solvent immersion method. Fig. 2d showed that the characteristic peaks of HM (288 nm) and APAP (240 nm) were displayed in the UV-vis spectrum of HM-A, suggesting the existence of APAP in the HM-A NPs. The drug loading of HM was 59%, as determined by measuring the concentration of APAP in the supernatant after drug loading via high-performance liquid chromatography (HPLC). Scanning electron microscopy (SEM) showed that HM exhibited the spindle-like morphology with 219.80 nm in length and 90.84 nm in width. The morphology of HM did not change after the incorporation of APAP (Figs. 2e and f and Fig. S2 in Supporting information). The dynamic light scattering (DLS) results showed that the hydrodynamic diameters of HM and HM-A were 196 nm and 229 nm respectively. As shown in Fig. S3 (Supporting information), the size of HM-A remained nearly unchanged over one week, suggesting the excellent colloidal stability in RPMI-1640 (containing 10% FBS).
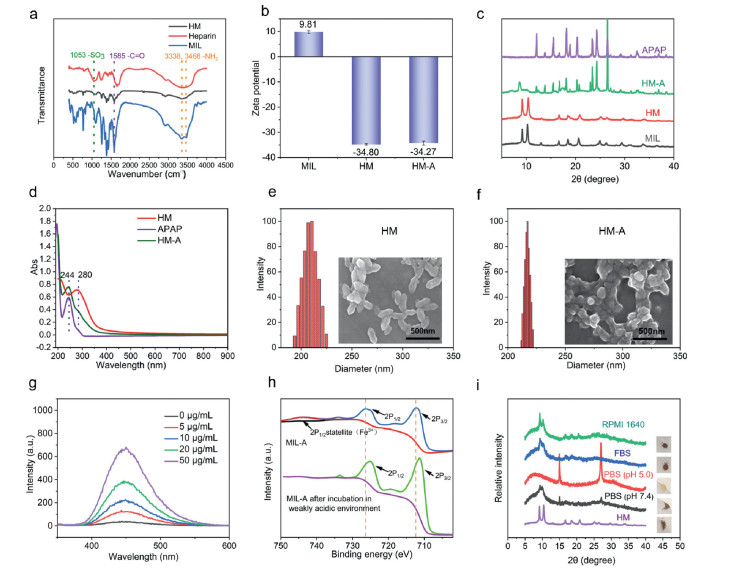
In order to test the peroxidase-mimic activity of HM-A NPs, a classic chromogenic reagent, 3,3′,5,5′-tetramethylbenzidine (TMB) was used. It is often used with peroxidase enzymes for the reduction of H2O2 to H2O [53]. HM-A NPs initially catalyzed H2O2 to produce •OH, followed by oxidizing TMB to form blue ox-TMB with a characteristic peak at 652 nm. From the UV-vis spectrum of TMB, no obvious absorption at 652 nm was observed in the absence of HM-A. However, the absorbance of ox-TMB at 652 nm increased with increasing the concentration of HM-A, suggesting that HM-A was critical for oxidizing TMB (Fig. S4 in Supporting information). The •OH probe (terephthalic acid) was employed to determine the generation of •OH through the Fenton reaction [53-55]. Terephthalic acid can react with •OH to form a highly fluorescent 2-hydroxy terephthalic acid with the maximum emission at 435 nm. As shown in Fig. 2g, the fluorescence intensity increased significantly with increasing the concentration of HM-A. To evaluate the GSH consumption ability of HM-A, we detected the amount of GSH after the addition of HM-A. As shown in Fig. S5 (Supporting information), the increase of the HM-A concentration accelerated the decrease of GSH absorption intensity around 412 nm, suggesting that the GSH was effectively oxidized into GSSG. In order to prove that MIL-88B can trigger the production of NAPQI from APAP via cascade reaction in tumor microenvironment, electrospray mass spectrometry (ESI-MS) of the mixture of MIL-A NPs (200 µg/mL) and H2O2 (100 µmol/L) in acetate buffer (pH 5.0) were conducted using a Waters Q-TOF MicroTM LC-MS. The ESI-MS of the mixture in the negative mode revealed that the most abundant peak at m/z 299 corresponded to the deprotonated APAP dimer, which can be further converted to NAPQI by •OH (Fig. S6 in Supporting information) [16-18]. The results of ESI-MS suggested that HM-A NPs had the ability to generate toxic NAPQI in the presence of •OH. Therefore, we proposed that APAP and Fe3+ ions released from MIL can trigger a redox reaction, where APAP was oxidized to APAP dimer, and Fe3+ ions were partially reduced to Fe2+ ions. Our hypothesis was further tested by X-ray photoelectron spectroscopy (XPS) performed with the HM-A incubated in acetate buffer (pH 5.0) at 37 ℃ for 24 h. The XPS peaks of Fe 2p3/2 and Fe 2p1/2 for the MIL-A and the MIL-A incubated in the acetate buffer were shown in Fig. 2h. MIL-A exhibited the characteristic peaks with binding energies at 712.08 eV and 726.28 eV in the Fe XPS spectrum, as assigned to Fe 2p3/2 and Fe 2p1/2 of FeⅢ [53]. After incubation, the peaks moved to lower values (711.38 eV and 724.9 8eV), suggesting that FeⅢ was partially reduced to FeⅡ in a weakly acidic environment (pH 5.0).
The stability of HM in FBS was conducted to evaluate the feasibility of using this material in animals. More than 90% of Fe was found to stay intact in HM after 144 h of incubation with FBS (Fig. S7 in Supporting information). Meanwhile, DLS measurements showed that there was no significant change on the particle size of HM post culture medium incubation (Fig. S3). PXRD analysis revealed that the structural integrity of HM was maintained post the serum or culture medium treatment. Compared to the neutral environment (pH 7.4), more HM NPs were degraded in the acidic medium (pH 5.0) (Fig. 2i). These results suggest that HM are relatively stable in various biological media.
The cellular uptake of the HM NPs was evaluated by confocal laser scanning microscopy and flow cytometry. As revealed in Figs. 3a and b, a stronger fluorescence was observed in 4T1 cells treated with FITC-labeled HM (abbreviated as HM-FITC) NPs in comparison with the control group, suggesting the excellent cellular uptake efficiency of HM. Similar results were obtained by the flow cytometry analysis (Fig. S8 in Supporting information). The biocompatibility of HM-A NPs was tested in HRGEC, HConEpic, and NIH-3T3 cell lines via MTT assays (Fig. 3c and Fig. S9 in Supporting information). The high survival rates of them proved the good biocompatibility and biosafety of HM-A NPs in the normal cells. For testing the anti-tumor effect of HM-A NPs, the cytotoxicity induced by HM-A NPs to 4T1 tumor cells was quantified by MTT assay. As shown in Fig. S10 (Supporting information) there was no obvious cell death for the APAP groups. The HM carrier showed low cytotoxicity when the concentration of HM was less than 200 µg/mL. It is worth noting that the cytotoxicity increased significantly after the HM combined with APAP (Fig. 3d). In the presence of 400 µg/mL of HM-A, 4T1 cells viability was less than 40% owing to the synergistic effect of intracellular oxidation damage and toxic NAPQI. The enhanced cytotoxicity of HM-A against tumor cells was further confirmed through dead/live staining in which cells were stained with Calcein-AM (green fluorescence) and PI (red fluorescence) to differentiate dead and viable cells. In contrast to the HM or APAP treated groups, the cancer cells treated with HM-A exhibited a strong red fluorescence (Fig. 3e), showing that the HM encapsulated with APAP could cause severe cell apoptosis. In complementary assays, the cytotoxicity of the HM-A was assessed by flow cytometry. After different treatments, cells were stained with Annexin V-FITC, a fluorescent probe of surface-exposed phosphatidyl serine, and propidium iodide (PI), a DNA stain that detects the permeabilization of the plasma membrane. APAP alone caused no significant increase in annexin V and PI staining, indicating that APAP showed negligible toxicity to cells (Fig. 3f). However, cells pretreated with HM-A displayed dual-labeling, as a sign of cell death, due to HM triggering nontoxic APAP to toxic NAPQI. Moreover, the cytotoxicity increased with increasing the concentration of HM-A, which was consistent with the results of MTT assays. (Fig. S11 in Supporting information). The apoptosis rate of HM-A reached 67.33%, due to the production of ROS via Fenton reaction and toxic NAPQI via cascade reaction (Fig. S12 in Supporting information).
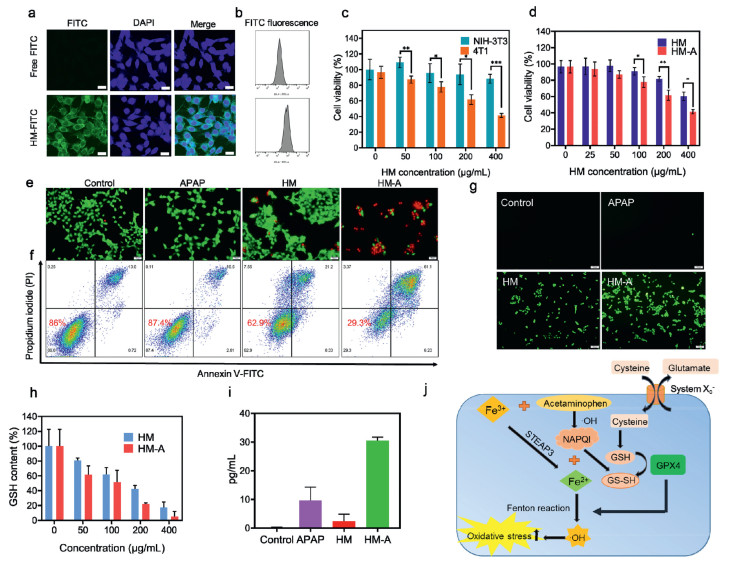
2′,7′-Dichlorofluorescein diacetate (DCFH-DA) probe was used to mark the intracellular ROS generated via Fenton reaction in 4T1 cells. As shown in Fig. 3g and Fig. S13 (Supporting information), no fluorescence signal was observed in the control and APAP groups. The HM group and HM-A group showed the distinct fluorescence signals contributed by •OH via the Fe-mediated Fenton reaction. The results of flow cytometry further confirmed that 4T1 cells treated with HM and HM-A exhibited the higher ROS level compared with other groups (Figs. S14 and S15 in Supporting information). A GSH assay kit was applied for quantitatively analyzing the change of intracellular GSH after different treatments. The distinct absorption attenuation of GSH with increasing the concentration of HM-A indicated that intracellular GSH was oxidized to GSSG effectively. In addition, the results suggested that HM-A consumed more GSH than HM at the same concentration (Fig. 3h). Then NAPQI ELISA Kit was utilized to detect intracellular NAPQI production. As shown in Fig. 3i, 4T1 cells treated with HM-A produced a larger amount of NAPQI than other groups, and the increase of HM-A concentration accelerated the production of NAPQI (Fig. S16 in Supporting information). Fig. 3j showed that APAP was oxidized into NAPQI by •OH generated from HM. NAPQI then caused the extensive GSH depletion, reducing the antioxidant capacity of cancer cells and amplifying the cell-killing efficiency of Fenton reaction. In brief, Fe3+ ions released from HM and prodrug APAP provide a synergistic anti-tumor effect with cascade reactions.
To track the in vivo fate of HM NPs, DiR iodide was used as an imaging probe in 4T1 tumor-bearing mice. All animal experiments were conducted in compliance with the Institutional Animal Care and Use Committee (IACUC) of China Pharmaceutical University. As shown in the in vivo images (Fig. 4a), the fluorescence intensity in tumors of the mice treated with DiR-labeled HM (abbreviated as HM-DiR) increased over time, reaching the maximum level at 24 h post-injection. This implied that the nanoparticles located in the tumor foci via enhanced penetration and retention (EPR) effect [56]. The results also revealed that the nanoparticles were mainly trapped by the reticuloendothelial system (RES) in the liver and spleen. At 24 h post-injection, mice were euthanatized, and the major organs and tumors were dissected for ex vivo biodistribution analysis (Fig. 4b). The prominent distribution of HM nanoparticles in the liver and spleen was 22.28% and 32.65% (Fig. 4c). Meanwhile, the EPR accumulation rate of HM nanoparticles into the tumors was 17.33%, which is high enough to achieve the basic therapeutic doses of HM nanoparticles against malignant tumors. To achieve better anti-tumor effects, the tumor-targeting ability of HM-A can be improved by modifying the NPs with the target ligands such as RGD or F3 peptide in future work.
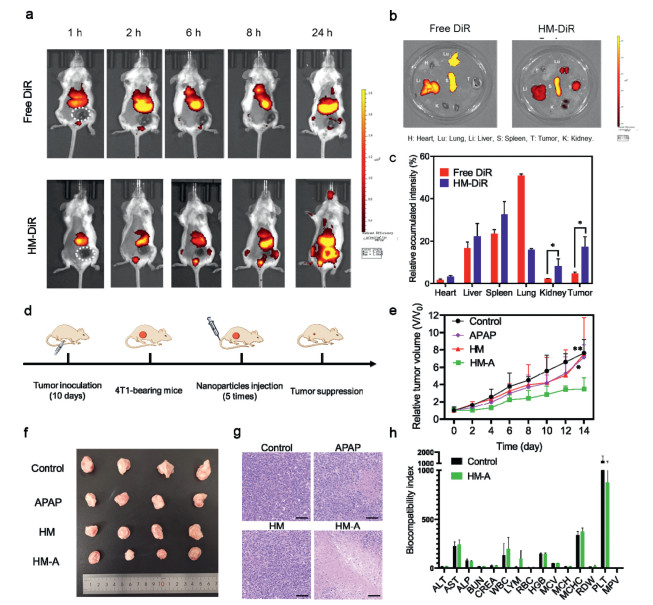
The in vivo antitumor effect of HM-A NPs was evaluated in 4T1 tumor-bearing mice. When the tumor volume reached 150 mm3, the mice were casually allocated into four groups, including saline, APAP, HM, and HM-A. All groups were intravenously injected every two days for 11 days (Fig. 4d). The therapeutic effect, including the relative tumor volume, tumor weight and photographs of those groups was illustrated in Figs. 4e and f, Fig. S17 (Supporting information). APAP or HM alone had a negligible effect on the tumor inhibition. However, when they were administrated simultaneously, the growth of tumor can be significantly suppressed. HM-A showed the strongest inhibitory effect on tumor growth, which can be attributed to the synergetic effect of the Fenton reaction catalyzed by HM and the toxic NAPQI converted from APAP. Furthermore, the therapeutic effect of various treatments was investigated by hematoxylin and eosin (H & E) staining assay (Fig. 4g). Compared to the control group, the other treated groups exhibited more or less cell damage, and the HM-A showed the largest region of necrosis due to the synergetic effect of oxidative stress and toxic NAPQI.
The biocompatibility study of HM-A showed that no obvious hemolytic effects (< 5%) was observed within the tested ranges (Fig. S18 in Supporting information). Little change was observed in body weights for all groups (Fig. S19 in Supporting information), suggesting the high biosafety of HM and APAP treatments. From the H & E staining images of the major organ tissues, there were no obvious pathological changes after the treatments, further supporting the biosafety of this system (Fig. S20 in Supporting information). Moreover, the blood biochemistry and hematology analysis of mice treated with saline, HM, and HM-A showed that there was no significant difference between HM-A group and other groups (Fig. 4h, Figs. S21 and S22 in Supporting information), supporting the feasibility and safety of using HM-A for intravenous injection.
In summary, we construct a nanocomposite that combined the Fe-based MOF with acetaminophen into one system for enhanced antitumor therapy. The Fe-based MOF carrier undergoes pH-responsive decomposition and generates •OH via Fenton reaction in tumor cells selectively. The •OH further trigger APAP into toxic NAPQI, which exhausts GSH rapidly for highly efficient cancer therapy. In vitro and in vivo experiments showed that the anti-tumor efficacy was significantly enhanced by the nanocomposite as the result of the synergistic effect of Fenton activity and NAPQI-induced toxicity. With a growing research effort aimed at turning old drugs into new treatments, the excellent biocompatibility of the acetaminophen-containing delivery system shows great promise as a safe and effective "chemo" drug for clinical translation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors are grateful for financial support of this work by the National Natural Science Foundation of China (No. 81872813), the Outstanding Youth Fund of Jiangsu Province of China (No. BK20190029), the China Postdoctoral Science Foundation (No. 2021M703597) and the Program of State Key Laboratory of Natural Medicines-China Pharmaceutical University (No. SKLNMZZ202031).
Supplementary material associated with this article can be found, in the online version, at doi:
R.T. Stravitz, W.M. Lee, The Lancet 394 (2019) 869–881. doi: 10.1016/S0140-6736(19)31894-X
W. Bernal, J. Wendon, N. Engl. J. Med. 369 (2013) 2525–2534. doi: 10.1056/NEJMra1208937
M.R. Mcgill, C.D. Williams, Y. Xie, et al., Toxicol. Appl. Pharmacol. 264 (2012) 387–394. doi: 10.1016/j.taap.2012.08.015
M.R. Mcgill, H. Yan, A. Ramachandran, et al., Hepatology 53 (2011) 974–982. doi: 10.1002/hep.24132
M.R. Mcgill, H. Jaeschke, Pharm. Res. 30 (2013) 2174–2187. doi: 10.1007/s11095-013-1007-6
W. Bernal, A. Hyyrylainen, A. Gera, et al., J. Hepatol. 59 (2013) 74–80. doi: 10.1016/j.jhep.2013.02.010
X. Wang, Q. Wu, A. Liu, et al., Drug Metab. Rev. 49 (2017) 395–437. doi: 10.1080/03602532.2017.1354014
P. Zhang, S. Chen, H. Tang, et al., Toxicol. Appl. Pharmacol. 410 (2021) 115355. doi: 10.1016/j.taap.2020.115355
Q. Wang, S. Wei, H. Zhou, et al., Cell Death Discov. 5 (2019) 119-119. doi: 10.1038/s41420-019-0198-y
M.P. Murphy, Biochem. J. 417 (2008) 1–13.
S. Zhai, X. Hu, Y. Hu, et al., Biomaterials 121 (2017) 41–54. doi: 10.1016/j.biomaterials.2017.01.002
D. Jia, X. Ma, Y. Lu, et al., Chin. Chem. Lett. 32 (2021) 162–167. doi: 10.1016/j.cclet.2020.11.052
N.M. Vad, G. Yount, D. Moore, et al., J. Pharm. Sci. 98 (2009) 1409–1425. doi: 10.1002/jps.21505
N.M. Vad, S.K. Kudugunti, D. Graber, et al., Int. J. Oncol. 35 (2009) 193–204.
X. Lian, Y. Huang, Y. Zhu, et al., Angew. Chem. Int. Ed. 57 (2018) 5725–5730. doi: 10.1002/anie.201801378
A.K. Sahoo, M.P. Sk, S.S. Ghosh, A. Chattopadhyay, Nanoscale 3 (2011) 4226–4233. doi: 10.1039/c1nr10389j
A.K. Sahoo, S. Sharma, A. Chattopadhyay, S.S. Ghosh, Nanoscale 4 (2012) 1688–1694. doi: 10.1039/c2nr11837h
S. Das, A.K. Sahoo, S.S. Ghosh, A. Chattopadhyay, Langmuir 26 (2010) 15714–15717. doi: 10.1021/la1034867
Y. Zhou, S. Fan, L. Feng, et al., Adv. Mater. 33 (2021) 2104223. doi: 10.1002/adma.202104223
X. Wang, X. Zhong, Z. Liu, L. Cheng, Nano Today 35 (2020) 100946. doi: 10.1016/j.nantod.2020.100946
T. Zhou, Y. Xu, L. Xing, et al., Adv. Mater. 33 (2021) 2100114. doi: 10.1002/adma.202100114
D. Wang, H. Wu, G. Yang, et al., ACS Nano 14 (2020) 13500–13511. doi: 10.1021/acsnano.0c05499
J. Chen, X. Wang, Y. Zhang, et al., Biomaterials 266 (2021) 120457. doi: 10.1016/j.biomaterials.2020.120457
Z. Tang, Y. Liu, M. He, W. Bu, Angew. Chem. Int. Ed. 58 (2019) 946–956. doi: 10.1002/anie.201805664
P. Ji, H. Huang, S. Yuan, et al., Adv. Healthc. Mater. 8 (2019) 1900911. doi: 10.1002/adhm.201900911
M. Liu, B. Liu, Q. Liu, et al., Coord. Chem. Rev. 382 (2019) 160–180. doi: 10.1016/j.ccr.2018.12.015
D. Wang, J. Zhou, R. Chen, et al., Chem. Mater. 29 (2017) 3477–3489. doi: 10.1021/acs.chemmater.6b05215
Y. Liu, W. Zhen, L. Jin, et al., ACS Nano 12 (2018) 4886–4893. doi: 10.1021/acsnano.8b01893
S. Sheng, F. Liu, L. Lin, et al., J. Control. Release 328 (2020) 631–639. doi: 10.1016/j.jconrel.2020.09.029
R. Xu, J. Yang, Y. Qian, et al., Nanoscale Horiz. 6 (2021) 348–356. doi: 10.1039/D0NH00674B
J. Della Rocca, D. Liu, W. Lin, Acc. Chem. Res. 44 (2011) 957–968. doi: 10.1021/ar200028a
Z. Zhou, J. Song, R. Tian, et al., Angew. Chem. Int. Ed. 56 (2017) 6492–6496. doi: 10.1002/anie.201701181
W. Wang, Y. Jin, Z. Xu, et al., WIREs Nanomed. Nanobiotechnol. 12 (2020) e1614.
M. Wu, Y. Yang, Adv. Mater. 29 (2017) 1606134. doi: 10.1002/adma.201606134
Q. Xia, H. Wang, B. Huang, et al., Small 15 (2019) 1803088.
Y. Wang, J. Yan, N. Wen, et al., Biomaterials 230 (2020) 119619. doi: 10.1016/j.biomaterials.2019.119619
Y. Sun, L. Zheng, Y. Yang, et al., Nano-Micro Lett. 12 (2020) 103. doi: 10.1007/s40820-020-00423-3
P. Horcajada, T. Chalati, C. Serre, et al., Nat. Mater. 9 (2010) 172–178. doi: 10.1038/nmat2608
Y. Gu, L. Miao, Y. Yin, et al., Chin. Chem. Lett. 32 (2021) 1491–1496. doi: 10.1016/j.cclet.2020.09.029
B. Yang, J. Shi, J. Am. Chem. Soc. 142 (2020) 21775–21785. doi: 10.1021/jacs.0c09984
X. Wan, L. Song, W. Pan, et al., ACS Nano 14 (2020) 11017–11028. doi: 10.1021/acsnano.9b07789
X. Meng, D. Li, L. Chen, et al., ACS Nano 15 (2021) 5735–5751. doi: 10.1021/acsnano.1c01248
X. Shan, S. Li, B. Sun, et al., J. Control. Release 319 (2020) 322–332. doi: 10.1016/j.jconrel.2020.01.008
S. Zhan, H. Zhang, X. Mi, et al., Environ. Sci. Technol. 54 (2020) 8333–8343. doi: 10.1021/acs.est.9b07245
N. Wang, W. Ma, Y. Du, et al., ACS Appl. Mater. Interfaces 11 (2019) 1174–1184. doi: 10.1021/acsami.8b14987
P. Zhang, J. Guo, C. Wang, J. Mater. Chem. 22 (2012) 21426–21433. doi: 10.1039/c2jm34725c
M.J. Yin, P. Cui, Z. Hu, et al., Adv. Mater. Res. 393-395 (2011) 1173–1176.
A.K. Sahoo, U. Goswami, D. Dutta, et al., ACS Biomater. Sci. Eng. 2 (2016) 1395–1402. doi: 10.1021/acsbiomaterials.6b00334
C. Qiao, R. Zhang, Y. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 16982–16988. doi: 10.1002/anie.202007474
E. Bellido, T. Hidalgo, M.V. Lozano, et al., Adv. Healthc. Mater. 4 (2015) 1246–1257. doi: 10.1002/adhm.201400755
M. Socha, A. Lamprecht, F. El Ghazouani, et al., J. Nanosci. Nanotechno. 8 (2008) 2369–2376. doi: 10.1166/jnn.2008.081
M. Socha, P. Bartecki, C. Passirani, et al., J. Drug Target. 17 (2009) 575–585. doi: 10.1080/10611860903112909
H. Ranji-Burachaloo, F. Karimi, K. Xie, et al., ACS Appl. Mater. Interfaces 9 (2017) 33599–33608. doi: 10.1021/acsami.7b07981
H. Ranji-Burachaloo, P.A. Gurr, D.E. Dunstan, G.G. Qiao, ACS Nano 12 (2018) 11819–11837. doi: 10.1021/acsnano.8b07635
Z. Shen, J. Song, B.C. Yung, et al., Adv. Mater. 30 (2018) 1704007. doi: 10.1002/adma.201704007
Y. Hu, T. Lv, Y. Ma, et al., Nano Lett. 19 (2019) 2731–2738. doi: 10.1021/acs.nanolett.9b01093

Figure 1 (a) Schematic illustration of the synthesis of HM-A NPs. (b) Anticancer mechanism of HM-A based on Fenton reaction and APAP metabolism.

Figure 2 Characterizations of HM-A NPs. (a) FT-IR spectra of Heparin, HM and MIL. (b) Zeta potential of HM-A. (c) PXRD patterns of MIL, HM, APAP, and HM-A. (d) UV-vis absorption spectra of APAP, HM and HM-A. (e) Hydrodynamic size distribution of HM NPs. Inset: SEM image of HM NPs. (f) Hydrodynamic size distribution of HM-A NPs. Inset: SEM image of HM-A NPs. (g) The effect of the HM-A nanoparticles on the formation of hydroxyl radical with terephthalic acid as a fluorescence probe. (h) XPS spectra of HM-A before and after incubation in acetate buffer (pH 5.0). (i) PXRD analysis of HM over different pH values biological media (for 6 days).

Figure 3 HM-A NPs inducing cell death, ROS production, GSH depletion, and NAPQI generation. (a) Confocal fluorescence microscope images of the endocytosis process for HM NPs. Scale bar is 20 µm. (b) Flow cytometry analysis of 4T1 cells after incubation with FITC and HM-FITC. (c) NIH-3T3 and 4T1 cell viability assays of HM-A. (d) 4T1 cell viability assays of HM-A and HM. (e) Live/dead double-staining of 4T1 cells after different treatments indicated by calcein-AM (green, live cells) and PI (red, dead cells). Scale bar: 50 µm. (f) Flow cytometry analysis of 4T1 cells after different treatments stained with annexin V-FITC/PI. (g) Intracellular ROS staining. Microscopic images of 4T1 cells after different treatments with ROS staining. Scale bar: 100 µm. (h) Intracellular GSH depletion with different concentrations of HM-A NPs and HM NPs. (i) Intracellular NAPQI production after different treatments. (j) The proposal signal pathway of synergetic APAP and Fe3+ combo. Values are presented as means ± SD (n = 6), *P < 0.05, **P < 0.01, ***P < 0.001.

Figure 4 In vivo biodistribution and antitumor effect of HM-A. (a) In vivo biodistribution profile of free DiR and HM-DiR NPs in 4T1-tumor-bearing mice at varied time points after one tail-vein injection. (b) Ex vivo fluorescence images of dissected organs and tumor. (c) Ex vivo relative biodistribution percentages of free DiR and DiR conjugated HM NPs in major organs and tumor in 24 h. Values are presented as means ± SD (n = 3 per group). (d) Schematic illustration of the therapeutic process. (e) In vivo 4T1 tumor proliferation curves of mice treated with different groups (Control, APAP, HM NPs and HM-A NPs). (f) Photograph of tumors excised from the mice at the end of the treatments. (g) H & E staining of tumor sections at the end of treatment. Scale bar: 50 µm. Values are presented as means ± SD (n = 6 per group). (h) Hematological, hepatic, and renal biochemical indexes of Balb/c mice after different treatments. Values are presented as means ± SD (n = 3 per group). *P < 0.05, **P < 0.01.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: