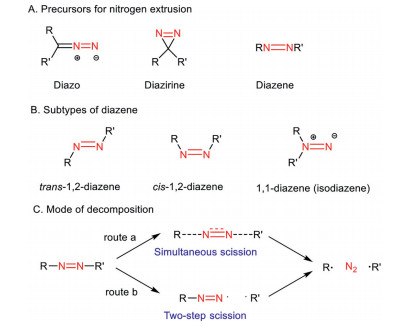
Scheme 1.
Candidates for nitrogen extrusion and various modes of decomposition of diazene.

The construction of chemical bonds lies at the heart of organic synthesis. Much synthetic endeavor has been devoted to efficient new bonds formation, which can be achieved by exploiting new catalytic systems and/or developing concise synthetic strategies [1, 2]. In particular, reactions involving small gaseous molecule extrusion have been used extensively to forge highly-strained and/or sterically-hindered carbon-carbon bonds [3-5]. This process is favorable because the formation of a thermodynamically stable gaseous molecule as a side product and the gained entropy of gas extrusion collectively provides the driving force to the reaction.
Among the reported reactions involving gas extrusion, dinitrogen extrusion has drawn considerable attention owing to the high reactivity and/or chemoselectivity of the transformations [6]. The commonly used reactants and/or synthetic precursors for dinitrogen extrusion involve diazo, diazirine, and diazene (Scheme 1A). Normally, a carbene or a diradical species is generated which will engage in the next synthetic event to form a new chemical bond(s). Insertion of C-H bond or X-H (X = O, N, S) to carbene resulting from diazo or diazirine facilitated by transition-metal catalysis has been well-documented and showed a broad application in complex molecules synthesis [7-11]. On the other hand, the chemistry of diazenes, including 1, 1-diazenes and 1, 2-diazenes, has attracted less attention. Notably, there is a developing research interest in the field of synthetic chemistry to the diazenes chemistry in the past decade, from the innovative approaches of new C-C bond formation via dinitrogen extrusion of diazenes [6, 12, 13] to the methodical renovation of trimethylenemethane (TMM)-diyl cycloaddition applied to natural product synthesis [4, 14]. Motivated by these fascinating and important results, we realize that a concise overview of diazenes and their related chemistry would be of great value to the synthetic community, for instance, synthetic scientists and the medicinal chemists from academia and the pharmaceutical industry. As such, we aspire to highlight the contemporary advancements of the chemistry of diazenes and their synthetic applications. The future directions and opportunities for dinitrogen extrusion reaction from diazenes are discussed in the conclusion.
Diazene, the nitrogen analog of alkene, includes 1, 1-diazene and 1, 2-diazene (cis- and trans-form) depending on the pattern of substitution (Scheme 1B). Diazene compounds lose their nitrogen moiety via the process of dinitrogen extrusion under thermal or photochemical conditions. Although details of the kinetics of radical formation from diazene remain to be unraveled [6], the radical properties of the resultant biradical were well-perceived. In general, there are two possible pathways of dinitrogen extrusion from diazenes to generate carbon-based biradicals, including simultaneous scission of both C-N bonds and step-wise scission via an intermediary diazinyl radical (Scheme 1C) [6]. It is reported that the unsymmetrical nature of the diazene compound is prone to a step-wise scission process [6, 15, 16].
Similar to those reported radical-based reactions, the dinitrogen extrusion of diazenes produces a biradical species, allowing the construction of sterically hindered carbon center(s) with good functional group tolerance [17]. More importantly, the biradical formed via diazene decomposition underwent intramolecular coupling and/or cyclization to give a C-C bond forming product with high stereoselectivity. However, it is noteworthy that undisciplined radical attacks at random may result in the formation of undesired product(s). Therefore, the development of a radical reaction that affords the high yields of the targeted products with minimized side product formation is of paramount importance. This review is mainly categorized into two sections. The first section is dinitrogen extrusion chemistry of 1, 2-dizenes, including ring contraction, homo or hetero-dimerization, and trimethylenemethane (TMM)-diyl cycloaddition. And the second part is nitrogen extrusion chemistry of 1, 1-diazenes. Applications of these classes of reaction in complex natural product synthesis are detailed.
Nitrogen extrusion reaction has been widely applied in organic synthesis. With fewer exceptions such as rhodium-catalyzed nitrenoid insertion [9], most of the nitrogen extrusion reactions facilitate C-C bond formation involving a possible radical-based mechanism. In this section, three types of radical based nitrogen extrusion reaction of 1, 2-diazenes, namely cyclopropanes and/or cyclobutanes formation from 1, 2-diazene, homo- and hetero-dimerization via 1, 2-diazene fragmentation and trimethylenemethane (TMM)-diyl [3 + 2] cycloaddition, are illustrated. Applications of these synthetic methods in natural product synthesis are discussed.
Small carbocycles, such as cyclopropane and cyclobutane, are highly-strained and are present as important scaffolds in many chemical pharmaceutics and bioactive natural products [18-21]. Despite the importance of these structural classes, the limited synthetic methods to forge these small carbocycles make these compounds relatively less accessible compared to the five and six-membered rings [22-26]. Therefore, ring contraction through dinitrogen extrusion from 1, 2-diazene offers a synthetic route to prepare the highly-strained and functionalized cyclopropane or cyclobutane (Scheme 2A).
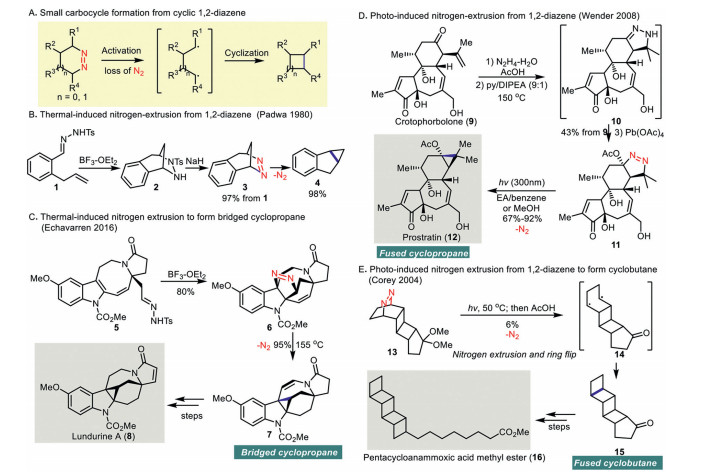
In 1980, a pioneer study from Padwa and co-workers on thermal-induced nitrogen extrusion of 4, 5-dihydro-1, 4-methano-1H-2, 3-benzodiazepine (3) resulted in C-C bond formation to give cyclopropane 4 in 98% yield (Scheme 2B) [27]. Treatment of tosylhydrazone 1 with boron trifluoride etherate led to an intramolecular [3 + 2] cycloaddition to give 3 in 97% yield.
In 2016, Echavarren's synthesis of (−)-lundurine A (8) featured a cyclopropanation via formal [3 + 2] cycloaddition/nitrogen extrusion as a synthetic key step (Scheme 2C) [28]. Treatment of tosyl hydrazone 5 with boron trifluoride etherate produced cycloaddition product 6 in 80% yield. Upon heating of pyrazoline 6 to 155 ℃, cyclopropanation via nitrogen extrusion took place concomitantly with the migration of olefin, presumably via a homodienyl retro-ene/ene rearrangement, to give 7 in 95% yield.
In 2008, a remarkable, 4-step synthesis of prostratin (12) from crotophorbolone (9) reported by Wender and co-workers featured a dinitrogen extrusion of pyrazoline 11 effecting by UV irradiation to give the desired cyclopropane on prostratin (12) (Scheme 2D) [29].
The synthesis of pentacycloanammoxic acid methyl ester (16), which possesses a high angle strain of the ladaderane scaffolds, was accomplished by Corey and co-workers in 2004 (Scheme 2E) [30]. Photoirradiation of pentacyclic azo ketal 13 followed by deketalization afforded the desired ladderane 15 in 6% yield.
The coupling of biradical generated from linear 1, 2-diazenes appears to be more challenging compared to the cyclic congeners. Both self-coupling and cross-coupling are possible once the biradical species is formed from linear 1, 2-diazenes, which may lead to a lower yield of the desired cross-coupling product (Scheme 3A). Therefore, the development of the selective, controlled synthesis of the hetero-coupling method of linear 1, 2-diazenes has become significant.
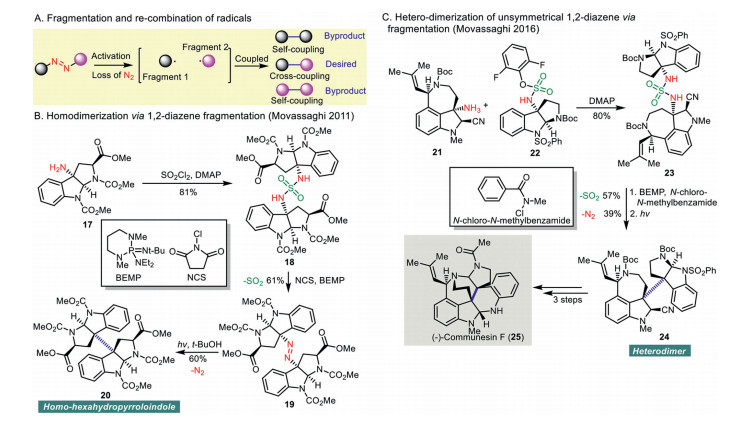
In 2011, Movassaghi and co-workers disclosed a directed and stereocontrolled assembly of carbon-carbon linked homo- and hetero-dimeric hexahydropyrroloindoles (Scheme 3B) [31]. Treatment of amine 17 with sulfuryl chloride in the presence of DMAP gave sulfamide 18 in 81% yield. Oxidation of sulfamide 18 with N-chlorosuccinimide (NCS) in the presence of polystyrene-bound 2-tert-butylimino-2-diethylamino-1, 3-dimethylperhydro-1, 3, 2-diazaphosphorine (BEMP) afforded the desired 1, 2-diazene 19 in 61% yield. Photolysis of the resultant 1, 2-diazene 19 in tert-butanol led to stereocontrolled C-C bond formation and produced homodimer 20 in 60% yield.
Later, a biomimetic enantioselective total synthesis of (-)-communesin F (25) featuring a diazene synthesis from unsymmetric sulfamide/photolysis approach as a key reaction was reported by Movassaghi and co-workers in 2016 (Scheme 3C) [32, 33]. Arylsulfamate 22 was reacted with aminonitrile 21 implemented by DMAP to give unsymmetrical sulfamide 23 in 80% yield. Diazene formation from 23 followed by photolysis gave 24, which was converted to (−)-communesin F (25) in three steps.
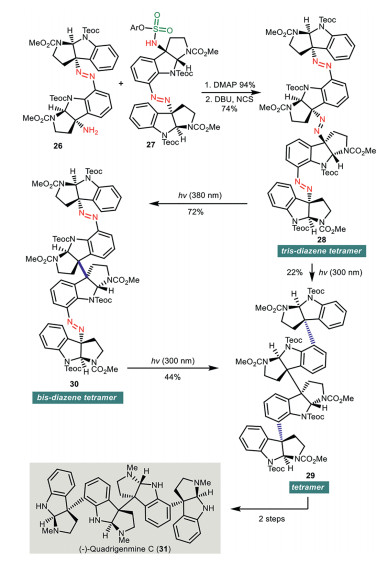
Shortly after, the same research group disclosed a remarkable synthesis of (−)-quadrigemine C (31) employing a multiple photoextrusion of nitrogen from a tris-diazene intermediate as a synthetic key step (Scheme 4) [34]. Sulfonylation of amine 26 with 27 effected by DMAP provided a sulfamide, which was treated with DBU and N-chlorosuccinimide to give tris-diazene 28. Photoirradiation of the resultant tris-diazene 28 at 300 nm led to triple nitrogen extrusion and provided 29 in 22% yield. An additional two-step synthesis from 29 produced quadrigenmine C (31). Alternatively, stepwise photolysis including photoirradiation of tris-diazene 28 at 380 nm provided bis-diazene 30, which was irradiated at 300 nm to give 29 in 44% yield.
Trimethylenemethane (TMM)-diyl cycloaddition is a powerful strategy for annulation to construct polycyclic fused and/angular architectures [4, 35]. TMM-diyl is a transient intermediate, which can be generated by dinitrogen extrusion of a 5-membered 1, 2-diazenes, trapping an olefin via an intramolecular [3 + 2] cycloaddition (Scheme 5A). With well-designed reactants, these methods have been used to prepare complex natural products containing contiguous quaternary carbon centers.
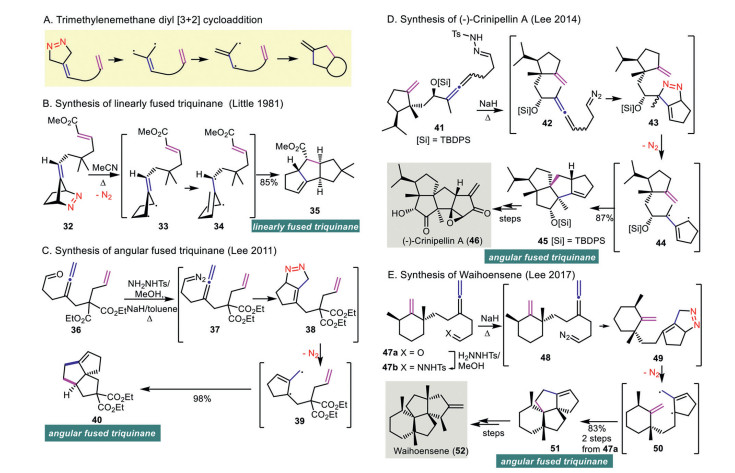
Intramolecular trimethylenemethane (TMM) diyl [3 + 2] cycloaddition through diyl trapping with olefin was reported by Berson's group [36] and Little's group [37, 38] independently. Reflux of diazene compound 32 in acetonitrile generated the proposed biradical intermediate 33 through nitrogen extrusion, which underwent isomerization to 34 followed by intramolecular diyl trapping through a [3 + 2] cycloaddition to give fused tricycle 35 in 85% yield (Scheme 5B). In 2011, Lee and co-workers harnessed allenyl diazene compound 37 to produce diyl 39, which led to the formation of angular fused triquinane 40 in 98% yield (Scheme 5C) [39]. The authors suggested that an intramolecular cycloaddition of the diazene group and allene of 37 affords tetrahydrocyclopentapyrazole 38. Nitrogen extrusion from newly formed 38 produces diyl 39, which undergoes an intramolecular [3 + 2] cycloaddition to give angular fused triquinane 40.
The synthesis of (−)-crinipellin A (46) [40] and waihoensene (52) [41] was achieved by Lee and co-workers making use of their early reported trimethylenemethane (TMM) diyl [3 + 2] cycloaddition [39]. The synthesis of (−)-crinipellin A (46) commenced with the treatment of hydrazone 41 with sodium hydride under reflux to generate the angular fused tetraquinane 45 in 87% yield (Scheme 5D). The authors rationalized that the diazo compound 42 formed undergoes an intramolecular cycloaddition to give 43. Freshly prepared 43 is converted to diyl 44 followed by a [3 + 2] cycloaddition to give the angular tetraquinane 45, which is a key intermediate to (−)-crinipelline A (46). The synthesis of waihoensene (52) by the same group began with the preparation of hydrazone 47b from the corresponding aldehyde 47a followed by treatment with sodium hydride under reflux to give 51 in 83% yield over two steps (Scheme 5E). Here, the authors proposed that the hydrazone 47b is converted to diazo 48, which is subjected to an intramolecular cycloaddition to give adduct 49. Nitrogen extrusion from 49 gives diyl 50, which is subjected to [3 + 2] cycloaddition to give the angular tetracyclic precursor 51 in 83% yield over two steps. The synthesis of waihoensene (52) was completed from 51 in steps.
Novel method development making use of 1, 2-diazenes has gained much attention and many successful applications in organic synthesis have been disclosed. In contrary to 1, 2-diazenes, the reactivity for 1, 1-diazenes chemistry is rarely explored before 2021. One of the pioneer studies of 1, 1-diazenes was reported by Dervan and co-workers (Scheme 6A) [42-45]. 1, 1-Diazene 53, which was derived from the corresponding pyrrolidine, underwent stereospecific ring contraction to give cyclobutane 55 as a result of rapid C-C bond formation. The high stereoretention of the cyclobutane formation can be ascribed to the involvement of thermally generated singlet 1, 4-biradical as a possible intermediate. By-products such as the alkene 57 resulting from β-fragmentation and a small amount of stereo-inverted cyclobutane 56 were also identified.
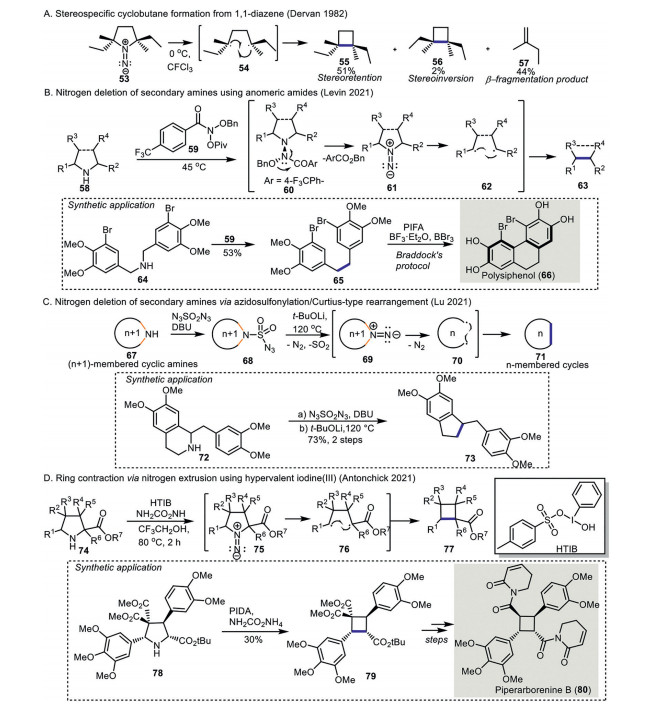
In 2021, Levin's group disclosed the nitrogen deletion of secondary amines making use of an N-anomeric amide (Scheme 6B) [46]. An N-anomeric amide 59 acts as a nitrogen transfer reagent to a secondary amine 58 to produce 1, 1-diazene 61 in-situ, which affords ring contraction product 63, presumably via a biradical intermediate. This new method was applied to the synthesis of natural product polysiphenol (66). To begin with, bibenzylamine 64 was converted to diarylethane 65 using Levin's protocol. Subsequent cyclization of 65 mediated by Boron Lewis acid afforded polysiphenol (66).
Another nitrogen-deletion protocol of secondary amines featuring a Curtius-type rearrangement/nitrogen extrusion was disclosed by Lu's group (Scheme 6C) [47, 48]. A two-step process on a secondary amine 67 including azidosulfonylation to give 68 followed by nitrogen extrusion of 69 produced the nitrogen deletion product 71. An experimental study revealed that 1, 1-diazene 69 is a possible intermediate and gives the nitrogen deletion product via a carbon biradical species 70. Importantly, a wide range of linear and cyclic secondary amines was compatible, and the ring size of the substrates showed almost no effect on reactivity. The synthesis of substituted indene 73 from piperidine derivative 72 was achieved using Lu's elegant procedure.
Later, Antonchick and co-workers disclosed a novel contractive synthesis of multi-substituted cyclobutanes from the pyrrolidines (Scheme 6D) [49]. The authors identified that the in-situ generated iodonitrene from hypervalent iodine(Ⅲ) reagent and ammonia congener works as an electrophilic aminating reagent [50-52], facilitating ring contraction of pyrrolidine to afford cyclobutanes. The mechanistic investigation conducted by the same group revealed that the highly stereoselective nature of the ring contraction could be ascribed to the presence of a 1, 4-biradical intermediate 76. Rapid formation of C-C bond from intermediate 76 affording cyclobutane 77 without loss of stereoselectivity aligns with the proposal of radical mechanism independently suggested by Dervan and Levin (Schemes 6A and B) [42, 46]. Moreover, Antonchick's ring contraction was utilized as a key step to construct the cyclobutane core in the formal synthesis of piperarborenine B (80).
This review highlighted the current states of diazenes' chemistry and its application in organic synthesis. The ability to generate biradical via dinitrogen extrusion from diazenes enables the construction of sterically congested carbon-carbon bond(s) in a stereoselective manner. Many highly-strained and/or sterically congested molecular scaffolds, such as fused and bridged cyclopropanes, fused cyclobutanes, polyhydropyrroloindoles, and linear/angular fused triquinanes, have become accessible from chemically stable 1, 2-diazenes compounds. Besides, nitrogen deletion of secondary amines based on 1, 1-diazenes chemistry prepared ring contraction products of different sizes, complying with the concept of skeletal editing [1, 46, 53-55]. Dinitrogen extrusion of diazenes that well planned out is practical because these methods have been successfully applied in the complex natural product synthesis. We envisage that trapping the biradical generated from diazenes using various radicals could be insightful for new method development.
Despite being a promising strategy in organic synthesis, some limitations on the transformation of diazenes are yet to be addressed. Cyclopropane can be produced efficiently from the corresponding 1, 2-diazene while the synthesis of cyclobutane is in its infancy (Scheme 2). Moreover, photoirradiation of 8-membered 1, 2-diazene gave cyclohexane as the desired product while hex-1-ene was observed as a side product [56]. Instead of cyclization via radical recombination, the distant linear 1, 6-biradical resulting from dinitrogen extrusion may undergo other competing radical pathways, such as radical abstraction that led to the formation of alkene. Besides, the synthesis of medium-sized carbocycles (i.e., 8-11 membered) from dinitrogen extrusion of 1, 2-diazene is rarely reported. This may be related to the higher kinetic and thermodynamic barriers associated with their synthesis compared to other rings sizes [57]. On the other hand, the necessity of a benzyl fragment or a tertiary alkyl connected to a nitrogen atom to stabilize the biradical generated from dinitrogen extrusion of 1, 1-diazene appears as a major limitation of the method. We suggest that other radical stabilizing functionalities such as O, N atoms, cyano or ketone group installed at the adjacent position of the radical center(s) [58] may facilitate the formation of biradical species and eventually expand the substrate scope of the reaction.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
C. Xu is grateful to Fuzhou University for the funding support (No. GXRC21051) and greatly acknowledges the Award Program for Minjiang Scholar Professorship.
K.R. Campos, P.J. Coleman, J.C. Alvarez, et al., Science 363 (2019) eaat0805. doi: 10.1126/science.aat0805
G.M. Whitesides, Angew. Chem. Int. Ed. 54 (2015) 3196-3209. doi: 10.1002/anie.201410884
X. Jiang, L. Shi, H. Liu, A.H. Khan, J.S. Chen, Org. Biomol. Chem. 10 (2012) 8383-8392. doi: 10.1039/c2ob26152a
H.Y. Lee, Acc. Chem. Res. 48 (2015) 2308-2319. doi: 10.1021/acs.accounts.5b00178
J. Kim, M. Movassaghi, Acc. Chem. Res. 48 (2015) 1159-1171. doi: 10.1021/ar500454v
P.S. Engel, Chem. Rev. 80 (1980) 99-150. doi: 10.1021/cr60324a001
Y. Ren, S. Zhu, Q. Zhou, Org. Biomol. Chem. 16 (2018) 3087-3094. doi: 10.1039/c8ob00473k
B.D. Bergstrom, L.A. Nickersom, J.T. Shaw, L.W. Souza, Angew. Chem. Int. Ed. 60 (2021) 6864-6878. doi: 10.1002/anie.202007001
H.M.L. Davies, R.E.J. Beckwith, Chem. Rev. 103 (2003) 2861-2904.
L. Dubinsky, B.P. Krom, M.M. Meijler, Bioorg. Med. Chem. 20 (2012) 554-570.
M.P. Doyle, J. Tauton, S. Oon, et al., Tetrahedron Lett. 29 (1988) 5863-5866. doi: 10.1016/S0040-4039(00)82210-8
C. Xu, V.U.B. Rao, F. Chen, Green. Synth. Catal. 3 (2022) 1-3. doi: 10.1016/j.gresc.2021.12.001
C. Zippel, J. Seibert, S. Bräse, Angew. Chem. Int. Ed. 60 (2021) 19522-19524. doi: 10.1002/anie.202107490
Z. Wang, J. Liu, Beilstein J. Org. Chem. 16 (2020) 3015–3031. doi: 10.3762/bjoc.16.251
G. Koga, J.C. Warner, J.P. Anselme, Radical reactions of azo, hydrazo and azoxy compounds, in: S. Patai, (Eds. ), The Chemistry of the Hydrazo, Azo and Azoxy Groups. John Wiley & Sons, 1997.
J. Liu, Y. Yang, K. Ouyang, W. Zhang, Green Synth. Catal. 2 (2021) 87-122. doi: 10.1117/12.2604794
H. Yi, G. Zhang, H. Wang, et al., Chem. Rev. 117 (2017) 9016-9085. doi: 10.1021/acs.chemrev.6b00620
Y. Fan, X. Gao, J. Yue, Sci. China Chem. 59 (2016) 1126-1141. doi: 10.1007/s11426-016-0233-1
B. Zhen, X. Suo, J. Dang, et al., Chin. Chem. Lett. 32 (2021) 2338-2341.
Z. Meng, F. Mi, F. Lu, et al., Chin. Chem. Lett. 31 (2020) 1903-1905.
J. Li, K. Gao, M. Bian, H. Ding, Org. Chem. Front. 7 (2020) 136-154.
S. Poplata, A. Tröster, Y. Zou, T. Bach, Chem. Rev. 116 (2016) 9748-9815. doi: 10.1021/acs.chemrev.5b00723
Y. Xu, M.L. Conner, M.K. Brown, Angew. Chem. Int. Ed. 54 (2015) 11918-11928. doi: 10.1002/anie.201502815
E.N. Hancock, J.M. Wahl, M.K. Brown, Nat. Prod. Rep. 36 (2019) 1383-1393. doi: 10.1039/c8np00083b
C. Ebner, E.M. Carreira, Chem. Rev. 117 (2017) 11651-11679. doi: 10.1021/acs.chemrev.6b00798
M. Mato, A. Franchino, C. García-Morales, A.M. Echavarren, Chem. Rev. 121 (2021) 8613-8684. doi: 10.1021/acs.chemrev.0c00697
A. Padwa, A. Rodriguez, Tetrahedron Lett. 22 (1981) 187-190.
M.S. Kirillova, M.E. Muratore, R. Dorel, A.M. Echavarren, J. Am. Chem. Soc. 138 (2016) 3671-3674. doi: 10.1021/jacs.6b01428
P.A. Wender, J.M. Kee, J.M. Warrington, Science 320 (2008) 649-652. doi: 10.1126/science.1154690
V. Mascitti, E.J. Corey, J. Am. Chem. Soc. 126 (2004) 15664-15665. doi: 10.1021/ja044089a
M. Movassaghi, O.K. Ahmad, S.P. Lathrop, J. Am. Chem. Soc. 133 (2011) 13002-13005. doi: 10.1021/ja2057852
S. P Lathrop, M. Pompeo, W.T.T. Chang, M. Movassaghi, J. Am. Chem. Soc. 138 (2016) 7763-7769. doi: 10.1021/jacs.6b04072
M.M. Pompeo, J.H. Cheah, M. Movassaghi, J. Am. Chem. Soc. 141 (2019) 14411-14420. doi: 10.1021/jacs.9b07397
P. Lindovska, M. Movassaghi, J. Am. Chem. Soc. 139 (2017) 17590-17596. doi: 10.1021/jacs.7b09929
B.M. Trost, Angew. Chem. Int. Ed. 25 (1986) 1-20.
J.A. Berson, C.D. Duncan, L.R. Corwin, J. Am. Chem. Soc. 96 (1974) 6175-6177. doi: 10.1021/ja00826a035
R.D. Little, G.W. Muller, J. Am. Chem. Soc. 101 (1979) 7129-7130. doi: 10.1021/ja00517a086
R.D. Little, G.W. Muller, J. Am. Chem. Soc. 103 (1981) 2744-2749. doi: 10.1021/ja00400a043
T. Kang, W.Y. Kim, Y. Yoon, B. G. Kim, H.Y. Lee, J. Am. Chem. Soc. 133 (2011) 18050-18053. doi: 10.1021/ja207591e
T. Kang, S.B. Song, W.Y. Kim, B.G. Kim, H.Y. Lee, J. Am. Chem. Soc. 136 (2014) 10274-10276. doi: 10.1021/ja5054412
H. Lee, T. Kang, H.Y. Lee, Angew. Chem. Int. Ed. 56 (2017) 8254-8257. doi: 10.1002/anie.201704492
W.D. Hinsberg III, P.B. Dervan, J. Am. Chem. Soc. 100 (1978) 1608-1610. doi: 10.1021/ja00473a051
P.G. Schultz, P.B. Dervan, J. Am. Chem. Soc. 102 (1980) 878-880. doi: 10.1021/ja00522a090
P.G. Schultz, P.B. Dervan, J. Am. Chem. Soc. 103 (1981) 1563-1564. doi: 10.1021/ja00396a048
P.G. Schultz, P.B. Dervan, J. Am. Chem. Soc. 104 (1982) 6660-6668. doi: 10.1021/ja00388a031
S.H. Kennedy, B.D. Dherange, K.J. Berger, M.D. Levin, Nature 593 (2021) 223-227. doi: 10.1038/s41586-021-03448-9
H, Qin, W. Cai, S. Wang, et al. Angew. Chem. Int. Ed. 60 (2021) 20678-20683.
X. Zou, J. Zou, L. Yang, G. Li, H. Lu, J. Org. Chem. 82 (2017) 4677-4688. doi: 10.1021/acs.joc.7b00308
C. Hui, L. Brieger, C. Strohmann, A.P. Antonchick, J. Am. Chem. Soc. 143 (2021) 18864-18870. doi: 10.1021/jacs.1c10175
L.G. O'Neil, J.F. Bower, Angew. Chem. Int. Ed. 60 (2020) 25640-25666.
M. Zenzola, R. Doran, L. Degennaro, R. Luisi, J.A. Bull, Angew. Chem. Int. Ed. 55 (2016) 7203-7207. doi: 10.1002/anie.201602320
T. Glachet, H. Marzag, N.S. Rosa, et al. J. Am. Chem. Soc. 141 (2019) 13689-13696. doi: 10.1021/jacs.9b07035
B.D. Dherange, P.Q. Kelly, J.P. Liles, M.S. Sigman, M.D. Levin, J. Am. Chem. Soc. 143 (2021) 11337-11344. doi: 10.1021/jacs.1c06287
D. Ma, B.S. Martin, K.S. Gallagher, T. Saito, M. Dai, J. Am. Chem. Soc. 143 (2021) 16383-16387. doi: 10.1021/jacs.1c08626
A.M. Szpilman, E.M. Carreira, Angew. Chem. Int. Ed., 49 (2010) 9592-9628. doi: 10.1002/anie.200904761
W. Adam, T. Oppenländer, Can. J. Chem. 63 (1985) 1829-1832. doi: 10.1139/v85-303
A.K. Clarke, W.P. Unsworth, Chem. Sci. 11 (2020) 2876-2881. doi: 10.1039/d0sc00568a
M.D.E. Forbes, Carbon-Centered Free Radicals and Radical Cations: Structure, Reactivity, and Dynamics, John Wiley & Sons, 2009.

Scheme 1 Candidates for nitrogen extrusion and various modes of decomposition of diazene.

Scheme 2 Small carbocycle formation from cyclic 1, 2-diazene via dinitrogen extrusion.

Scheme 3 Fragmentation of linear 1, 2-diazene in homo or heterodimerization in organic synthesis.

Scheme 5 Cycloaddition with TMM-diyl generated from 1, 2-diazene and its application in organic synthesis.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: