Citation:
Kun Chen, Wei Zhang, Yu Bai, Wanbing Gong, Ning Zhang, Ran Long, Yujie Xiong. Boosting electrochemical hydrogen evolution by coupling anodically oxidative dehydrogenation of benzylamine to benzonitrile[J]. Chinese Chemical Letters,
2023, 34(3): 107319.
doi:
10.1016/j.cclet.2022.03.042
Boosting electrochemical hydrogen evolution by coupling anodically oxidative dehydrogenation of benzylamine to benzonitrile
English
Boosting electrochemical hydrogen evolution by coupling anodically oxidative dehydrogenation of benzylamine to benzonitrile
School of Chemistry and Materials Science, Frontiers Science Center for Planetary Exploration and Emerging Technologies, National Synchrotron Radiation Laboratory, and Experimental Center of Engineering and Material Sciences, University of Science and Technology of China, Hefei 230026, China
b.
Key Laboratory of Magnetic Materials at Universities of Inner Mongolia Autonomous Region, Baotou Teachers' College, Baotou 014030, China
c.
Department of Applied Physics, The Hong Kong Polytechnic University, Hong Kong 999077, China
d.
Institute of Energy, Hefei Comprehensive National Science Center, Hefei 230031, China
Received Date:
26 January 2022 Accepted Date:
10 March 2022 Revised Date:
28 February 2022 Available Online:
15 March 2023
Abstract:
The electricity-driven water splitting acts as a promising pathway for renewable energy conversion and storage, yet anodic oxygen evolution reaction (OER) largely hinders its efficiency. Seeking the alternatives to OER exhibits the competitive advance to address this predicament. In this work, we show a more thermodynamically and kinetically favorable reaction, electrochemical oxidative dehydrogenation (EODH) of benzylamine to replace the conventional OER, catalyzed by a cobalt cyclotetraphosphate (Co2P4O12) nanorods catalyst grown on nickel foam. This anodic reaction lowers the electricity input of 317 mV toward the desired current density of 100 mA/cm2, together with a highly selective benzonitrile product of more than 97%. More specifically, when coupling it with cathodic hydrogen evolution reaction (HER), the proposed HER||benzylamine-EODH configuration only requires a cell voltage of 1.47 V@100 mA/cm2, exhibiting an energy-saving up to 17% relative to conventional water splitting, as well as the near unit selectivity toward cathodic H2 and anodic benzonitrile products.
In order to achieve the goal of Carbon Neutrality in 2060s, it is highly anticipated to seek the effective routes for renewable energy conversion and storage. Specifically, it is firmly believed as an appealing approach to convert the intermittent electricity derived from sustainable solar/wind energies (so called "green electricity") into the value-added hydrogen feedstock through water splitting [1, 2]. In the electrolyzer, this well-studied water splitting process is commonly divided into two-half electrochemical reactions, namely, cathodic hydrogen evolution (HER) and anodic oxygen evolution (OER). Despite the great potential, its energy conversion efficiency is still largely hindered by OER due to the high thermodynamic barrier as well as sluggish reaction kinetics [3-6]. For instance, a voltage of more than 1.9 V is required for overall water splitting over the advanced cathodic-Pt/C||anodic-IrOx/C electrocatalyst to reach the benchmark current density of 100 mA/cm2 [7, 8]. Furthermore, the anodically generated O2 product is a relatively valueless product, giving rise to the further wasting of positive charge in anode.
Alternatively, it is a compelling concept to replace OER by other more favorable molecular oxidative reactions, such as nitrogen-based molecules (e.g., hydrazine [9, 10] and urea [11]) and biomass organics (e.g., alcohols [12, 13], glucose [14] and 5-hydroxymethylfurfural [15, 16]). When coupling these oxidative reactions with cathodic HER, the required energy input can be substantially reduced to achieve the energy saving. However, given the robust oxidation ability in anode, especially for the highly operated potentials, their products may be uncontrollable and further overoxidized to undesired molecules. Taking biomass ethanol as an example, the direct electrochemical oxidation usually results in the thorough CO2 production, giving the additional carbon emission. Taken together, it is still challenging to explore the suitable oxidative reaction instead of OER with excellent energy saving and value-added products. Among various candidates, electrochemical oxidative dehydrogenation (EODH) of amine (X-CH2NH2) to nitrile (X-C≡N) receives our specific attention owing to its several intrinsic merits. First of all, nitrile group is a vitally important class in chemistry with the widespread applications in fine chemicals and pharmaceuticals [17, 18]. Unfortunately, its traditional synthesis usually uses toxic oxidants under rigorous conditions (i.e., high temperature and pressure), giving rise to severe energy consumption and environmental pollution [19]. Instead, the electrochemical route offers a renewable choice with "green electricity" as energy input under mild conditions (Fig. 1a). In addition, amine EODH is not only a favorable reaction in terms of both thermodynamics and kinetics, but also gives hydrophobic product of nitrile to self-separate from the electrolyte, thereby avoiding its overoxidation for high selectivity [20-22].
Figure 1
Figure 1.
(a) Merits of renewable electro-route for amine oxidative dehydrogenation to nitrile relative to traditional thermo-route. (b) Schematic illustration of coupling HER with BA EODH in a two-electrode configuration using "green electricity" as energy input.
Herein, we demonstrate an earth-abundant electrocatalyst, cobalt cyclotetraphosphate nanorods grown on nickel foam (Co2P4O12/NF) as an attractive anode to furnish the benzylamine (BA) EODH to the desired benzonitrile (BN), which reduces the potential of 317 mV to reach the current density of 100 mA/cm2 relative to OER and shows a small Tafel slope of 76 mV/dec. Post-catalyst characterizations uncover that the high activity originates from the kinetically favorable nucleophile dehydrogenation reaction with Co3+/Co2+ redox active species [13, 20]. Moreover, profiting from the remarkable activity of Co2P4O12/NF catalyst for HER, we further assemble a two-electrode configuration through coupling HER with BA EODH using our Co2P4O12/NF as bifunctional electrocatalyst for simultaneous H2 generation and BA-to-BN conversion, as schemed in Fig. 1b. As a result, this advanced HER||BA-EODH system exerts an overall energy-saving up to 17% with the desired current density ranging from 50 mA/cm2 to 100 mA/cm2 in comparison with conventional water splitting, together with good durability as well as high selectivity toward both cathodic H2 and anodic BN products.
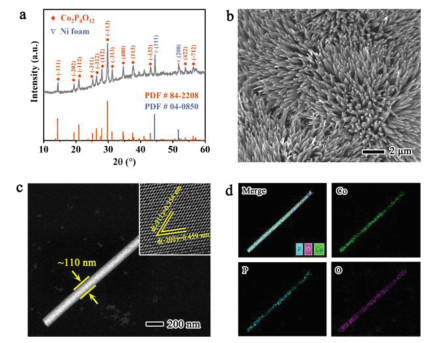
The electrocatalyst of Co2P4O12 nanorods anchored on nickel foam substrate (Fig. S1 in Supporting information) was synthesized by a typical two-step procedure [23]. A Co(OH)F precursor was firstly prepared by a hydrothermal method [24], which then underwent a phosphorization process using NaH2PO2 as phosphorus source to obtain our proposed Co2P4O12 nanorods electrocatalyst (see experimental details in Supporting information). X-ray diffraction (XRD) pattern of our as-obtained sample (Fig. 2a) can be well indicative of the crystal phase of monoclinic Co2P4O12 with the space group of C2/c (PDF#84–2208). Scanning electron microscopy (SEM) images (Fig. 2b and Fig. S2 in Supporting information) demonstrate the one-dimension nanorod structure of Co2P4O12 perpendicularly grown on NF substrate, which is further affirmed by dark-field transmission electron microscopy (TEM) with the width of ca. 110 nm (Fig. 2c). The high-resolution TEM (HRTEM) image (insert in Fig. 2c) exhibits the clear lattice fringes with the spacings of 0.459 and 0.354 nm, well matching the (202) and (311) planes in Co2P4O12 lattice matrix, respectively. The spatially elemental distribution was further examined by energy-dispersive X-ray spectroscopy (EDS) mapping. As displayed in Fig. 2d, the Co, P and O elements are uniformly dispersed in the lattice matrix, suggesting the well synthesis of Co2P4O12. The atomic ratio of P and Co elements was determined to be 1.9 by inductively coupled plasma mass spectrometry (ICP-MS), in good agreement with the stoichiometric ratio of 2.
Figure 2
Figure 2.
Structure characterizations of Co2P4O12/NF. (a) XRD pattern. (b) SEM image. (c) Dark-field TEM image. Insert is the HRTEM image. (d) The relative EDS mapping images with Co, P and O elements.
To investigate the chemical states of various elements, we conducted X-ray photoelectron spectroscopy (XPS) measurement. The deconvoluted Co 2p core-level spectra (Fig. S3a in Supporting information) demonstrate the characteristic peaks at the binding energy (BE) of 782.4 and 798.6 eV, together with the observable satellites at 786.2 and 804.0 eV, respectively, suggesting the dominant existence of Co2+ species in Co2P4O12 [25, 26]. Meanwhile, the high-resolution P 2p and O 1s XPS spectra (Figs. S3b and c in Supporting information) ascertain the P-O bond (i.e., BEs at 134.3 eV for P 2p spectra and 532.4 eV for O 1s spectra, respectively) in cyclotetraphosphate building block [25, 26].
Given the as-characterized crystalline and electronic structure, we then investigated the electrochemical activity for EODH of BA over Co2P4O12/NF anode in a standard H-type three-electrode cell using Ar-saturated 1 mol/L KOH as electrolyte (see the experimental details in Supporting information). Fig. 3a exhibits the linear sweep voltammetry (LSV) polarization curves with and without adding BA substrate into the electrolyte, respectively. In the absence of BA, the conventional OER is triggered only after the pre-oxidation of Co active species (Co2+/Co3+ redox couple at ~1.4 V vs. reversible hydrogen electrode (RHE) [27, 28]). A high potential of 1.659 V vs. RHE is observed to reach the current density of 100 mA/cm2. To be sharply different, after introducing 1 mmol of BA into electrolyte, BA EODH apparently occurs prior to catalyst pre-oxidation, and the required potential for current density of 100 mA/cm2 is dramatically reduced to as low as 1.342 V vs. RHE (i.e., a potential decrease of 317 mV relative to OER). Moreover, we also determined the Tafel slopes of these two oxidative reactions. As shown in Fig. 3b, the Tafel slope for BA EODH is considerably reduced to 78 mV/dec relative to OER (193 mV/dec). Such operated potential as well as Tafel slope decrease manifest the substantially more favorable BA EODH over conventional OER at anode both thermodynamically and kinetically. We also performed the same BA EODH test using bare NF as anode, whereas the poor catalytic performance (Fig. S4 in Supporting information) further consolidates the intrinsic catalytic activity for BA EODH originating from our Co2P4O12 electrocatalyst.
Figure 3
Figure 3.
Activity evaluation for BA EODH over Co2P4O12/NF anode in 1 mol/L KOH electrolyte with 1-mmol BA. (a) LSV polarization curves. (b) The determined Tafel slope. (c) The determined FEs of BN product under different applied potentials from 1.25 V to 1.55 V vs. RHE. (d) The calculated yields and FEs of BN product in consecutive cycling tests. (e) High-resolution Co 2p XPS spectra of Co2P4O12/NF after BA EODH and OER, respectively. (f) Mechanistic illustration of nucleophile dehydrogenation reaction to reduce Co3+ into Co2+ species on electrochemically reconstructed CoOxHy surface.
After confirming the superiority of BA EODH in terms of energy-saving, we then focused on the selectivity of the products. We performed the chronoamperometry tests with the charge of about 385 C (a theoretical charge for complete BA-to-BN conversion) under the different applied potentials (Fig. S5 in Supporting information). The observed products were quantitively analyzed by gas chromatography-mass spectrometry (GC–MS, Fig. S6 in Supporting information). No other possible by-products can be recognized from GC except for BN. The Faradaic efficiencies (FEs) of desired BN product were determined to be more than 97% at the measured potentials in the range from 1.25 V to 1.45 V vs. RHE (Fig. 3c), demonstrating that this BA EODH route can yield BN product with excellent selectivity. The slight charge loss (2%~3%) may be ascribed to the background current as well as inevitable charge consumption in electrochemical cell system, which has also been widely reported [15, 20, 22]. Higher operation potential (> 1.5 V vs. RHE) will reduce the FEs owing to the competing OER with O2 bubbles. For instance, when applied with a potential of 1.55 V vs. RHE, the FE of BN decreases to only 79.3%. Next, we evaluated the durability over our Co2P4O12/NF catalyst for BA EODH with five cyclic chronoamperometry tests at the potential of 1.4 V vs. RHE. As depicted in Fig. 3d, both FE and yield of BN product can be well retained without recognizable decay, illustrating the good durability. Finally, we selected different amine substrates (including aromatic and aliphatic amines) to carry out EODH evaluations under the same conditions. The observed LSV polarization curves (Fig. S7 in Supporting information) show that Co2P4O12/NF catalyst can well turn on the EODH for all of amines with comparable activity to benzylamine, underlining the universality of Co2P4O12/NF catalyst toward amine molecules.
To recognize the intrinsic activity origin, we conducted the comprehensive characterizations for the Co2P4O12 electrocatalyst after catalyzing the reaction. SEM images (Fig. S8 in Supporting information) indicate the irreversible reconstruction of catalyst surface to generate sheet-like active species. EDS mapping (Fig. S9 in Supporting information) demonstrates the largely leaching of P element with the dominate existence of Co and O elements in the electrochemically reconstructed surface. This argument about P leaching is further consolidated by the ICP-MS measurement, whereas the P-to-Co atomic ratio is reduced from 1.9 to 1.6 after the electrocatalysis. Given that the observed XRD pattern (Fig. S10 in Supporting information) shows the well-retained crystal phase of Co2P4O12 without other peaks, we can regard the sheet-like species to be amorphous cobalt (oxy)hydroxide (CoOxHy) containing oxidized Co species as catalytically active sites, which is usually formed during electrocatalytic oxidative reactions.
To understand the critical role of Co sites, we specifically checked the electronic evolution of Co species after BA EODH and OER, respectively. As displayed in the deconvoluted Co 2p XPS spectra (Fig. 3e), both Co2+ and Co3+ species are observable in the surface CoOxHy. For the sample after OER, Co3+ species dominantly exists with the Co3+-to-Co2+ ratio of 1.13 (based on the integrated area in 2p3/2 region). This result coincides with the fact that OER is triggered after electrochemical oxidation of Co species (see violet line in Fig. 3a). Interestingly, the percentage of Co3+ species dramatically decreases in the sample after BA EODH, whereas the Co3+-to-Co2+ ratio is determined as low as 0.43 along with the increased signals of characteristic satellites for Co2+ species (Fig. 3e) [29], even though the applied potential is higher than the potential of Co oxidation (i.e., 1.45 V vs. RHE). Considering that BA EODH can be readily triggered at the same conditions, we propose a reaction mechanism as illustrated in Fig. 3f. In detail, Co active species is firstly oxidized to high-valence Co3+ species, generating electrophilic Co3+-O motif with high oxidation ability. Given the nucleophilic feature of amino group (-NH2) with active hydrogen as reactant, the Co3+-O active species can favorably seize the hydrogen in -NH2 to furnish the dehydrogenation process, forming a Co2+-OH state with low Co valence. In comparison with OER, this nucleophile dehydrogenation reaction is much kinetically faster, thereby contributing to the high activity toward BA EODH reaction [13, 20].
It is well reported that cobalt-based compounds can serve as the promising electrocatalyst alternatives toward HER [30-36]. As expected, our Co2P4O12/NF catalyst can well catalyze HER under alkaline conditions (Fig. S11 in Supporting information), requiring an overpotential of 70 mV to reach the current density of 10 mA/cm2, only 48 mV higher than benchmark 20% Pt/C catalyst (i.e., 28 mV). We further determined the electrochemically active surface area (ECSA) up to 595 cm2 (Fig. S12 in Supporting information). Such high ECSA originates from the surface reconstruction of Co2P4O12/NF catalyst, beneficial to the exposure of catalytically active sites [37]. The specific activity was also normalized by ECSA, in which the current density is 0.11 mA/cmECSA2 at the overpotential of 150 mV, further underlining the superior intrinsic HER activity (Fig. S12c).
Given the good HER activity, we then attempted to couple HER with BA EODH into a full electrolyzer with two-electrode configuration. One specific matter is whether BA molecule can impact on the cathodic HER activity or not. To address the issue, we carried out HER evaluation in the presence of BA using our Co2P4O12/NF catalyst as cathode. As a result, the LSV polarization curves in the presence and absence of BA nearly overlap with each other (Fig. 4a) and the determined Tafel slopes are also comparable to about 50 mV/dec (insert in Fig. 4a). In addition, we also conducted a chronoamperometry test at the current density of 10 mA/cm2 with 1 mmol of BA, which only increases the required potential of less than 35 mV for a long-term 27-h test (Fig. S13 in Supporting information). The observed HER activity exhibits the good tolerance of our Co2P4O12/NF electrocatalyst toward BA molecule.
Figure 4
Figure 4.
Coupling HER with BA EODH over Co2P4O12/NF bifunctional catalyst. (a) LSV polarization curves for individual cathodic HER test. Inset is the determined Tafel slopes. (b) LSV polarization curves for the integrated HER||BA-EODH electrochemical system containing 1 mmol of BA. (c) The comparison of the applied voltages to reach the desired current density between HER||BA-EODH and conventional water splitting. (d) The determined FEs of cathodic H2 and anodic BN products in consecutive cycling tests.
Given the excellent activity of Co2P4O12/NF catalyst toward BA EODH as well as HER in the alkaline electrolyte, we further assembled a two-electrode electrolyzer through coupling cathodic HER with anodic BA EODH (i.e., HER||BA-EODH) to achieve the simultaneous H2 generation and BN electrosynthesis for sustainable energy-saving. The anodic electrolyte contains 1 mol/L KOH with 1 mmol of BA. Conventional water splitting (i.e., HER coupled with OER) was also tested in the same electrolyzer for activity comparison. As depicted in Fig. 4b, an obviously reduced cell voltage is required for HER||BA-EODH system in comparison with conventional water splitting. Specifically, to afford the current densities of 50, 70 and 100 mA/cm2, the cell voltages of 1.442, 1.455 and 1.472 V are only required for HER||BA-EODH, respectively (Fig. 4c), which can offer the energy-saving (i.e., (Uw.s. − UH.E.)/Uw.s. × 100%, where Uw.s. and UH.E. represent the required cell voltage for water splitting and HER||BA-EODH system, respectively) of 14%~17% relative to water splitting (Fig. S14 in Supporting information). To be appealing, our proposed Co2P4O12/NF bifunctional electrocatalyst exhibits superior activity to some recent reports for coupled HER||amine-EODH systems, such as CoP||NiSe (1.49 V for 20 mA/cm2 for BA) [20], CoS2−MoS2||Ni(OH)2 (~1.55 V for 50 mA/cm2 for propylamine) [21], and Ni2P/NF||Ni2P/NF (~1.55 V for 100 mA/cm2 for BA) [22]. To further quantify the observed products (i.e., cathodic H2 and anodic BN), we implemented the consecutive cycling tests through chronopotentiometry with the passed charge for total BA consumption at an applied voltage of 1.5 V. As shown in Fig. 4d, the calculated FE of anodic BN product can reach up to 97%, well matching with the cathodic H2 product (near 100%). Furthermore, this high selectivity can well be retained during five consecutive tests without decay, further underlining the promising potential for practicality of this integrated HER||BA-EODH configuration.
In summary, we have demonstrated an integrated electrochemical configuration through coupling cathodic HER with anodic BA EODH to apparently lower the electricity consumption, using Co2P4O12/NF as a bifunctional electrocatalyst. Owing to the thermodynamically and kinetically favorable nature of BA EODH, this proposed configuration only requires a cell voltage as low as 1.47 V to afford the current density of 100 mA/cm2 with an energy saving up to 17% relative to conventional water splitting, together with the excellent catalytic durability. Moreover, the anode simultaneously offers the value-added BN as product with a unit Faradaic efficiency of 97% toward cathodic H2 product of near 100%, further enhancing the conversion value of renewable electric energy. Our work offers an attractive and effective choice for electrochemical hydrogen evolution with lower energy consumption as well as sustainable nitrile synthesis from amine.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported in part by National Key R&D Program of China (No. 2020YFA0406103), National Natural Science Foundation of China (NSFC, Nos. 21725102, 22122506, 22075267, U1832156, 91961106, 51902311), DNL Cooperation Fund, CAS (No. DNL201922), Strategic Priority Research Program of the CAS (No. XDPB14), Anhui Provincial Natural Science Foundation (No. 2008085J05), Youth Innovation Promotion Association of CAS (No. 2019444), and Open Funding Project of National Key Laboratory of Human Factors Engineering (No. SYFD062010K). XPS measurements were performed at the beamline BL11U in the NSRL. The authors thank the support from USTC Center for Micro- and Nanoscale Research and Fabrication.
Supplementary materials
Supplementary material associated with this article can be
found, in the online version, at doi:10.1016/j.cclet.2022.03.042.
[1]
I. Roger, M.A. Shipman, M.D. Symes, Nat. Rev. Chem. 1 (2017) 0003. doi: 10.1038/s41570-016-0003
K. Xu, H. Cheng, H. Lv, et al., Adv. Mater. 30 (2018) 1703322. doi: 10.1002/adma.201703322
[31]
L. Su, X. Cui, T. He, et al., Chem. Sci. 10 (2019) 2019–2024. doi: 10.1039/c8sc04589e
[32]
X. Li, B. Lv, X.P. Zhang, et al., Angew. Chem. Int. Ed. 61 (2022) e202114310.
[33]
L. Xie, J. Tian, Y. Ouyang, et al., Angew. Chem. Int. Ed. 59 (2020) 15844–15848. doi: 10.1002/anie.202003836
[34]
X. Li, H. Lei, J. Liu, et al., Angew. Chem. Int. Ed. 57 (2018) 15070–15075. doi: 10.1002/anie.201807996
[35]
X. Gao, Y. Chen, T. Sun, et al., Energy Environ. Sci. 13 (2020) 174–182. doi: 10.1039/c9ee02380a
[36]
H. Li, X. Li, H. Lei, et al., ChemSusChem 12 (2019) 801–806. doi: 10.1002/cssc.201802765
[37]
X. Shang, K.-.L. Yan, Y. Rao, et al., Nanoscale 9 (2017) 12353–12363. doi: 10.1039/C7NR02867A
Figure 1
(a) Merits of renewable electro-route for amine oxidative dehydrogenation to nitrile relative to traditional thermo-route. (b) Schematic illustration of coupling HER with BA EODH in a two-electrode configuration using "green electricity" as energy input.
Figure 2
Structure characterizations of Co2P4O12/NF. (a) XRD pattern. (b) SEM image. (c) Dark-field TEM image. Insert is the HRTEM image. (d) The relative EDS mapping images with Co, P and O elements.
Figure 3
Activity evaluation for BA EODH over Co2P4O12/NF anode in 1 mol/L KOH electrolyte with 1-mmol BA. (a) LSV polarization curves. (b) The determined Tafel slope. (c) The determined FEs of BN product under different applied potentials from 1.25 V to 1.55 V vs. RHE. (d) The calculated yields and FEs of BN product in consecutive cycling tests. (e) High-resolution Co 2p XPS spectra of Co2P4O12/NF after BA EODH and OER, respectively. (f) Mechanistic illustration of nucleophile dehydrogenation reaction to reduce Co3+ into Co2+ species on electrochemically reconstructed CoOxHy surface.
Figure 4
Coupling HER with BA EODH over Co2P4O12/NF bifunctional catalyst. (a) LSV polarization curves for individual cathodic HER test. Inset is the determined Tafel slopes. (b) LSV polarization curves for the integrated HER||BA-EODH electrochemical system containing 1 mmol of BA. (c) The comparison of the applied voltages to reach the desired current density between HER||BA-EODH and conventional water splitting. (d) The determined FEs of cathodic H2 and anodic BN products in consecutive cycling tests.


 DownLoad:
DownLoad:



 下载:
下载:





 下载:
下载:
