
Figure 1.
Top and side views of optimized structures of (a) Fe-N3P-C and (b) Fe-N4–C catalysts. The C, N, P and Fe atoms are presented with gray, blue, pink and orange circles, respectively.
The triggering of catalysis via structural engineering at atomic level: Direct propane dehydrogenation on Fe-N3P-C
Wenyi Bian , Xueli Shen , Huang Tan , Xing Fan , Yunxia Liu , Haiping Lin , Youyong Li

Propylene is an important raw material in vast downstream productions in the chemical industry [1]. Traditional approaches for propylene production, such as stream cracking of naphtha and fluid catalytic cracking of oil fractions have been suffered from high energy consumption and low propylene selectivity [2]. Correspondingly, intense researches are stimulated in propylene production from selective dehydrogenations of cheap propane [2-6]. In comparison with oxidative approaches, direct propane dehydrogenations (PDH) is characterized with high propylene selectivity [7], and therefore has been industrialized with two primary technologies, namely UOP Oleflex and CB & I Lummus processes [8]. Despite of progresses in PDH technologies, challenges in cost, activity, selectivity and stability still need to be met. Mechanistically, the propane conversion is dominated by thermal dynamics. As a result, highly active catalysts (noble metal-based materials) are often employed to drive reactions at high temperatures (> 500 ℃) in order to achieve sufficient propane conversion efficiencies [9, 10]. This inevitably increases production cost and promote undesired side reactions [1, 11]. The catalytic selectivity, however, is dependent on reaction kinetics, i.e., whether the produced propylene can desorb from active sites before over-dehydrogenations and C–C bond cracking [11-13]. In this context, catalysts should not only promote sp3 C–H bond scission, suppress C–C bond breaking, but also facilitate propylene desorption.
The prevalent industrial PDH catalyst is Pt-Sn alloy, in which Pt is regarded as active catalyst and Sn play a positive role in Pt dispersion and propylene desorption [14]. Yang et al. studied possible microscopic active sites in Pt-Sn catalysts by means of first-principles calculations and reported that the catalytic selectivity of Pt-Sn was originated from the weak adsorption of propylene on the Pt2Sn site [15]. In recent years, researches on new PDH catalysts have been motivated by current drawbacks of Pt-Sn including: high cost, fast coke formation and using ecologically harmful Cl2 or Cl containing compounds. Reported catalysts including metal oxides such as ZrO2 [16, 17], TiO2 [18, 19] and Al2O3 with coordinative unsaturated species [20, 21], Pt based bimetallic catalysts [22, 23], and metal-free carbonaceous materials [24, 25]. In the past decade, large number of single-atom catalysts (SACs) have been produced and reported to exhibit extraordinary activity in a variety of catalysis, due to distinctive merits of electronic and geometric structures and 100% atom utilization efficiency [26-32]. Very recently, Niu et al. and Kong et al. reported that isolated metal sites in SACs could exhibit desired PDH activity and propylene selectivity [33, 34]. Nevertheless, the performance dependency of local chemical environments of SAC to PDH has not yet been addressed. In the present work, Fe embedded N-doped carbon (Fe-N4–C) site is selected as a model system to study the impact of local chemical environment of SAC on PDH, due to following reasons: (ⅰ) Fe-N4–C sites have been synthesized experimentally [35-38]; (ⅱ) Fe-N4–C sites are reported to exhibit outstanding thermal stability (ⅲ) the local environment of Fe-N4–C can be tuned by replacing a N atom with P, leading to buckled N and P dual-coordinated Fe SAC (Fe-N3P-C) site [39]. Herein, catalytic performance of Fe-N4–C and Fe-N3P-C sites toward PDH are studied systematically with density functional theory (DFT) calculations. Compared with the in-plane Fe-N4–C site, the buckled Fe-N3P-C exhibits much higher activity in C–H bond activations due to suitable H affinity. Subsequent calculations show that the out-of-plane Fe-N3P-C site may also promote propylene selectivity and therefore is a fascinating SAC for PDH. By contrast, the Fe-N4–C is an inactive site towards PDH chiefly due to geometric effects. The present work has broadened the design strategy of PDH catalysts, where the performance of SACs can be tuned by adjusting local geometric structure of active sites. Moreover, the prediction of catalytic activities in C–H bond cleavage with H affinity can be applied not only to O-terminated catalysts but also to main-group element-metal complexes [40].
Computational details and methods are discussed in Supporting information. The optimized atomic configuration and stability of the Fe-N3P-C site are investigated before PDH calculations. As shown in Fig. 1a, the single Fe atom is coordinated with three N atoms and one P atom. The average lengths of Fe-N bonds are 1.92 Å and 2.15 Å for the Fe-P bond, respectively. Since the atomic radius of P is larger than N, it is noteworthy that the P atom in Fe-N3P-C protrudes outward, and the Fe atom is slightly pulled out of the carbon matrix. By contrast, the Fe-N4–C adopts a planar configuration, in which Fe-N bonds (1.90 Å) are slightly shorter than that in Fe-N3P-C (Fig. 1b) [41, 42]. The binding energies of Fe atoms on Fe-N3P-C and Fe-N4–C are both larger than the cohesive energy of Fe crystal [43], indicating isolated Fe atom can be firmly anchored on these sites (Table S1 in Supporting information), consisting well with previous experimental observations [39, 42]. As industrial PDH processes operate at high temperatures around 900 K [44], ab initio molecular dynamics (AIMD) simulations are performed to evaluate the thermal stability of Fe-N3P-C at 1000 K. As shown in Fig. S1 (Supporting information), the atomic structure of Fe-N3P-C is well retained without significant structural changes. In addition, the climbing image-nudged elastic band (CI-NEB) calculations show that the Fe migration away from N3P-C center needs to overcome a high energy barrier of 4.85 eV, indicating that Fe-N3P-C exhibits outstanding stability at high temperatures (Fig. S2 in Supporting information).
The electronic structures of Fe-N3P-C and Fe-N4–C are investigated with projected density of states (PDOS), partial charge densities, and charge density differences. As shown in Figs. 2a and b, PDOS plots reveal that the front orbitals are dominated by Fe 3d states in both sites. Partial charge density analysis shows that Fe 3d states are significantly hybridized with N 2p states in Fe-N4–C, resulting in conjugation-like filled and empty states (Fig. 2b). Such bonding characteristics can also be seen in charge density differences upon Fe incorporation (Fig. 2d). This bonding mode often implies stable electronic properties at neutral conditions. By contrast, in the Fe-N3P-C site, both filled and empty states protrude outward, with electrons accumulated on top of Fe and P atoms (Figs. 2a and c), suggesting activity to form covalent and/or coordination bonds.

The activation of first C−H bond is often regarded as the rate-limiting step in PDH since the C–H bond of propane molecule has a high bonding energy of 409 kJ/mol [45]. As shown in Table 1, reaction energies to cleavage C–H bonds on Fe-N3P-C are 0.79 and 0.73 eV, which is less than that on the Pt2-Sn site (0.82 eV) [15]. By contrast, reaction energies on the Fe-N4–C site are up to 1.91 eV and 1.87 eV (Fig. S3 in supporting information), implying that Fe-N3P-C is much more active than Fe-N4–C to drive C–H bond scissions. Subsequently, the catalytic selectivity of these two sites are estimated via propylene binding energies: weak propylene adsorption results in high propylene selectivity [15]. The binding energies are calculated with the following equation: Eads = E(*C3H6) – E(*) – E(C3H6), where the active site is represented with *. As seen in Table S2 (Supporting information), the adsorption energies of propylene on Fe-N3P-C (0.09 eV) and Fe-N4–C (1.18 eV) are positive, revealing fast propylene desorption, and therefore high catalytic selectivity. The aforementioned calculations show that both Fe-N3P-C and Fe-N4–C exhibit propylene selectivity but only Fe-N3P-C demonstrates reasonable PDH activity. Subsequent studies are thus focused on the energy profiles of PDH on Fe-N3P-C.
 DownLoad:
CSV
DownLoad:
CSV

|
The possible elementary steps during propane dehydrogenations and their reaction energies and activation barriers summarized in Scheme 1 and Table 1, respectively. Before PDH, the propane molecule is physiosorbed on Fe-N3P-C, with an adsorption energy of −0.36 eV. As shown in Scheme 1, PDH may proceed via three possible pathways: dehydrogenation on the terminal methyl group (reaction 1), dehydrogenation on the methylene group (reaction 2), and demethylation to produce a methyl group and an ethyl group (reaction 3). Reaction energies and possible reaction configurations of dehydrogenation paths are shown in Fig. S4 (Supporting information). As can be seen, dissociated H atoms resulted from the first C–H bond scission tend to bind on the P atom rather than C or N atoms. To be specific, reaction energies of dehydrogenations on the methyl and methylene groups are 0.73 eV (reaction 1) and 0.79 eV (reaction 2), respectively (Table 1). The corresponding activation barriers are 1.80 eV (reaction 1) and 1.81 eV (reaction 2), respectively. These activation barriers are similar to that on Pt2Sn (1.22 eV), implying feasible propane conversion [15]. The C–C bond scission (reaction 3), however, is kinetically inhibited by a very high energy barrier of 3.93 eV (Table 1)

Starting from 1-propyl (CH3CH2CH2*) and 2-propyl (CH3CHCH3*), as can be seen in Scheme 1, PDH can proceed along three pathways, including two kinds of dehydrogenations (reactions 4 and 5, reactions 7 and 8) and one C–C bond cleavage (reactions 6 and 9). Reaction 4 and reaction 7 are desired paths to produce propylene, while the others are side reactions. Fig. S5 (Supporting information) shows that H atoms dissociated from CH3CH2CH2* and CH3CHCH3* prefer to bind with the Fe atom with reaction energies being 0.89 (reaction 4) and 0.95 eV (reaction 7), respectively. The activation barriers of reaction 4 and reaction 7 are lower than those in reaction 1 and reaction 2, indicating that the second desired C–H bond scissions are faster than the first ones (Table 1). Of importance, as shown in Fig. 3, the second C–H bond scissions are accompanied with propylene desorption, suggesting intrinsic impression of undesired deep dehydrogenations. The reaction energies of side dehydrogenation paths (reactions 5 and 8), as shown in Table 1, are 1.52 eV and 1.71 eV. They are clearly higher than activation barriers of reaction 4 (1.13 eV) and reaction 7 (1.22 eV), indicating that their activation barriers should be even higher and these undesired second hydrogenations are very slow processes. Similar situation is also observed in C–C bond cleavages (reactions 6 and 9), whose reaction energies (1.92 eV and 2.25 eV) are much higher than energy barriers to produce propylene (reactions 4 and 7). The calculations shown above indicate that propylene can be selectively produced over Fe-N3P-C.

The regeneration of active sites requires remove of dissociated H atoms. The first reaction path that has been considered is that the H atom dissociated in the second C–H bond scission bind directly with the one dissociated in the first C–H cleavage and produce a H2 molecule. However, this pathway can be ruled out due to high activation barriers (1.99 and 2.60 eV, Fig. S6 in Supporting information). The second reaction path is the Tafel process, where the H2 molecule is generated by two adsorbed H atoms. Our calculations show that the first dissociated H atom is anchored on the P atom due to large diffusion barriers (Fig. S7 in Supporting information), and the second dissociated H atom binds with the Fe atom (Fig. 3a). The combination of these two adsorbed H atoms and regenerate the clean Fe-N3P-C site can be feasibly achieved by crossing over an activation barrier of 0.97 eV (Table 1 and Fig. 3). Please note that this barrier is higher than that of propylene desorption (0.33 eV), suggesting that regenerations of active sites are slower than product desorption and further enable satisfied propylene selectivity [16]. These calculations clearly indicate that the PDH can proceed on Fe-N3P-C with propylene being the major product.
The contributions of temperature and entropy in PDH is investigated with Gibbs free energies (T = 873.15 K) under ambient pressure. The energy profiles of potential energies and Gibbs free energies are represented in Fig. 3b. As can be seen, the increase of temperature will not bring adverse effects on the activity and selectivity of PDH on Fe-N3P-C. In addition, as listed in Table 1, the stability of products in reaction 10 increases dramatically at 873.15 K, consisting well with van't Hoff equation that chemical equilibrium prefers propylene production at elevated temperatures.
As can be seen in our calculations, replacing just one N atom with a P atom in Fe-N4–C results in significant catalytic differences in PDH. This clearly illustrates that local chemical environments of active sites can bring vital impacts on catalysis. The origin of enhanced catalytic activity resulted from N-P dual coordination is then analyzed Fig. 4. shows that the P atom in Fe-N3P-C exhibits much higher H affinity than N atoms in Fe-N4–C, while binding interactions of propyl groups on Fe atoms of these two sites do not differ significantly. This indicates that the poor activity of Fe-N4–C towards C–H bond scission can be ascribed to the low H affinity on N atoms.

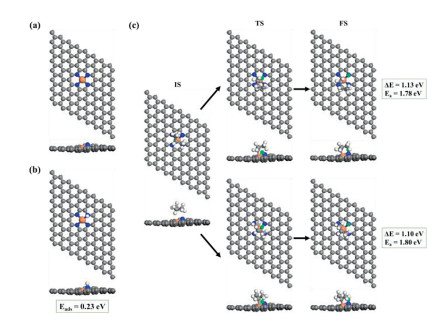
In this context, an interesting question is that if the improved H affinity upon P substitution is originated from electronic modification or geometric effect. We thus designed an artificial Fe-N4–C site by replacing the P atom in the optimized Fe-N3P-C site with an N atom (Fig. 5a). As shown in Fig. 5b, the N atom protruding from the plane shows a much higher H affinity (Eads = 0.23 eV) than the planarized Fe-N4 center (Eads = 0.95 eV). In addition, as shown in Fig. 5c, by calculating reaction energies and energy barriers of the first C–H bond scission of propane, which is the decisive step in PDH activity, we find that compared with the C–H bond scission on the planar Fe-N4 site (∆E = 1.87 eV, Ea = 1.92 eV, Fig. S3), the reaction energy of propane C–H bond activation on the protruding Fe-N4 is reduced to about 1.10 eV. The energy barrier is also reduced from the original 1.92 eV to 1.80 eV, which is quite similar to that on Fe-N3P-C. Therefore, it can be concluded that enhanced activity upon P substitution is chiefly a geometric effect: the polar configuration can break conjugation-like electronic states near Fermi level and provide moderate H affinity which is not only important for C–H bond scissions, but also exhibit desired H diffusion rate that ensures high propylene selectivity [46, 47].
In summary, first-principles calculations have been performed to reveal the intrinsic coordinating effect of Fe single-atom catalysts in PDH. Through systematic theoretical investigations, we report that compared with the in-plane Fe-N4–C site, the N and P dual-coordinated single Fe (Fe-N3P-C) site exhibits outstanding performance for PDH with desired activity and selectivity at industrial PDH temperatures. Subsequent calculations reveal that the catalytic activity and selectivity of Fe-N3P-C resulted from P substitution in Fe-N4–C can be attributed to the geometric effect, where the out-of-plane structure of Fe-N3P-C can break conjugation-like electronic states near Fermi level and exhibits suitable H affinity, which not only effectively promotes the C–H bond scission but also provides the appropriate H diffusion rate that ensures both the high selectivity of propylene and the regeneration of catalysts. The current work not only reports an active SAC for PDH but also reveals a novel mechanism of triggering the activity of PDH by tuning the local environment with atomic precision.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank support from National Science Foundation of China (Nos. 21771134, 22173067), National Key R&D Program of China (No. 2017YFA0204800), Science and Technology Project of Jiangsu Province (No. BZ2020011), Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the 111 Project, the Science and Technology Development Fund, Macau SAR (FDCT No. 0052/2021/A).
Supplementary material associated with this article can be
found, in the online version, at doi:
J.J. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez, et al., Chem. Rev. 114 (2014) 10613–10653. doi: 10.1021/cr5002436
S. Saerens, M.K. Sabbe, V.V. Galvita, et al., ACS Catal. 7 (2017) 7495–7508. doi: 10.1021/acscatal.7b01584
Q. Wang, R. Li, Renewable Sustainable Energy Rev. 74 (2017) 715–720. doi: 10.1016/j.rser.2017.03.007
P.C. Bruijnincx, B.M. Weckhuysen, Angew. Chem. Int. Ed. 52 (2013) 11980–11987. doi: 10.1002/anie.201305058
Z. Nawaz, Rev. Chem. Eng. 31 (2015) 413–436.
M.M. Bhasin, J.H. McCain, B.V. Vora, et al., Appl. Catal. A 221 (2001) 397–419. doi: 10.1016/S0926-860X(01)00816-X
T. Saelee, S. Namuangruk, N. Kungwan, et al., J. Phys. Chem. C 122 (2018) 14678–14690. doi: 10.1021/acs.jpcc.8b03939
J.J.H.B. Sattler, I.D. Gonzalez-Jimenez, L. Luo, et al., Angew. Chem. Int. Ed. 53 (2014) 9251–9256. doi: 10.1002/anie.201404460
J.A. Labinger, J.E. Bercaw, Nature 417 (2002) 507–514. doi: 10.1038/417507a
S. Vajda, M.J. Pellin, J.P. Greeley, et al., Nat. Mater. 8 (2009) 213–216. doi: 10.1038/nmat2384
L. Nykänen, K. Honkala, ACS Catal. 3 (2013) 3026–3030. doi: 10.1021/cs400566y
Z.J. Zhao, C.C. Chiu, J. Gong, Chem. Sci. 6 (2015) 4403–4425. doi: 10.1039/C5SC01227A
S. Zha, G. Sun, T. Wu, et al., Chem. Sci. 9 (2018) 3925–3931. doi: 10.1039/C8SC00802G
N. Kaylor, R.J. Davis, J. Catal. 367 (2018) 181–193. doi: 10.1016/j.jcat.2018.09.006
M.L. Yang, Y.A. Zhu, X.G. Zhou, et al., ACS Catal. 2 (2012) 1247–1258. doi: 10.1021/cs300031d
Y. Zhang, Y. Zhao, T. Otroshchenko, et al., Nat. Commun. 9 (2018) 3794. doi: 10.1038/s41467-018-06174-5
Y. Zhang, Y. Zhao, T. Otroshchenko, et al., ACS Catal. 10 (2020) 6377–6388. doi: 10.1021/acscatal.0c01455
Z. Xie, T. Yu, W. Song, et al., ACS Catal. 10 (2020) 14678–14693. doi: 10.1021/acscatal.0c02825
C.F. Li, X. Guo, Q.H. Shang, et al., Ind. Eng. Chem. Res. 59 (2020) 4377–4387. doi: 10.1021/acs.iecr.9b06759
Z. Lu, R.W. Tracy, M.L. Abrams, et al., ACS Catal. 10 (2020) 13957–13967. doi: 10.1021/acscatal.0c03391
P. Wang, Z. Xu, T. Wang, et al., Catal. Sci. Technol. 10 (2020) 3537–3541. doi: 10.1039/d0cy00779j
Y. Wang, Y. Suo, X. Lv, et al., J. Colloid Interface Sci. 593 (2021) 304–314. doi: 10.1016/j.jcis.2021.02.129
S.W. Han, H. Park, J. Han, et al., ACS Catal. 11 (2021) 9233–9241. doi: 10.1021/acscatal.1c01808
Y. Song, G. Liu, Z.Y. Yuan, RSC Adv. 6 (2016) 94636–94642. doi: 10.1039/C6RA20726J
S.F. Pan, J.L. Yin, X.L. Zhu, et al., Carbon 152 (2019) 855–864. doi: 10.1016/j.carbon.2019.06.106
X.F. Yang, A. Wang, B. Qiao, et al., Acc. Chem. Res. 46 (2013) 1740–1748. doi: 10.1021/ar300361m
B. Qiao, A. Wang, X. Yang, et al., Nat. Chem. 3 (2011) 634–641. doi: 10.1038/nchem.1095
P. Liu, Y. Zhao, R. Qin, et al., Science 352 (2016) 797–801. doi: 10.1126/science.aaf5251
X. Huang, Y. Xia, Y. Cao, et al., Nano Res. 10 (2017) 1302–1312. doi: 10.1007/s12274-016-1416-z
M. Zhang, Y.G. Wang, W. Chen, et al., J. Am. Chem. Soc. 139 (2017) 10976–10979. doi: 10.1021/jacs.7b05372
H. Yang, C. He, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3202–3206. doi: 10.1016/j.cclet.2021.03.038
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J. 414 (2021) 128857. doi: 10.1016/j.cej.2021.128857
K. Niu, Z. Qi, Y. Li, et al., J. Phys. Chem. C 123 (2019) 4969–4976. doi: 10.1021/acs.jpcc.8b12274
N. Kong, X. Fan, F. Liu, et al., ACS Nano 14 (2020) 5772–5779. doi: 10.1021/acsnano.0c00659
Y. Zhu, W. Sun, J. Luo, et al., Nat. Commun. 9 (2018) 3861. doi: 10.1038/s41467-018-06296-w
J. Li, H. Zhang, W. Samarakoon, et al., Angew. Chem. Int. Ed. 58 (2019) 18971–18980. doi: 10.1002/anie.201909312
M. Lefevre, E. Proietti, F. Jaouen, et al., Science 324 (2009) 71–74. doi: 10.1126/science.1170051
X. Cui, H. Li, Y. Wang, et al., Chem 4 (2018) 1902–1910. doi: 10.1016/j.chempr.2018.05.006
K. Yuan, D. Lutzenkirchen-Hecht, L. Li, et al., J. Am. Chem. Soc. 142 (2020) 2404–2412. doi: 10.1021/jacs.9b11852
A.A. Latimer, A.R. Kulkarni, H. Aljama, et al., Nat. Mater. 16 (2017) 225–229. doi: 10.1038/nmat4760
X. Long, Z. Li, G. Gao, et al., Nat. Commun. 11 (2020) 4074. doi: 10.1038/s41467-020-17903-0
H. Fei, J. Dong, Y. Feng, et al., Nat. Catal. 1 (2018) 63–72. doi: 10.1038/s41929-017-0008-y
P.H.T. Philipsen, E.J. Baerends, Phys. Rev. B 54 (1996) 5326–5333. doi: 10.1103/PhysRevB.54.5326
Y. Wang, Y. Wang, S. Wang, et al., Catal. Lett. 132 (2009) 472–479. doi: 10.1007/s10562-009-0119-4
X. Sun, B. Li, D. Su, Chem. Commun 50 (2014) 11016–11019. doi: 10.1039/C4CC02841D
X. Sun, J. Phys. Chem. Lett. 11 (2020) 9224–9229. doi: 10.1021/acs.jpclett.0c02400
R. Wang, C. He, W. Chen, et al., Chin. Chem. Lett. 32 (2021) 3821–3824. doi: 10.1016/j.cclet.2021.05.024

Figure 1 Top and side views of optimized structures of (a) Fe-N3P-C and (b) Fe-N4–C catalysts. The C, N, P and Fe atoms are presented with gray, blue, pink and orange circles, respectively.

Figure 2 Projected density of states (PDOS) of (a) Fe-N3P and (b) Fe-N4 sites with Fermi levels set as 0.00 eV. The inset figures in (a) and (b) show partial charge densities in the energy ranges of −8.00 eV to 0.00 eV and 0.00 eV to 6.00 eV, the values of isosurface contours are 0.04 |e|/Å3 for bonding orbitals and 0.005 |e|/Å3 for antibonding orbitals, respectively. Charge density differences of (c) Fe-N3P and (d) Fe-N4 before and after Fe anchored into framework, respectively. Cyan and yellow regions represent charge depletion and accumulation, respectively, and the isosurface value is 0.004 |e|/Å3.

Scheme 1 Possible reaction paths in propane dehydrogenations. Pathways to form propylene are marked in red. C–C bond scissions and undesired dehydrogenation reactions are marked with yellow and blue lines, respectively.

Figure 3 (a) Corresponding reaction pathways of the synthesis of propylene. H atoms in the adsorbed molecules are represented by white circles. C, N, P, Fe and H atoms adsorbed on the active site are represented with gray, dark blue, pink, orange and green circles, respectively. (b) Energy profiles of propane dehydrogenations to propylene on Fe-N3P-C. The potential and Gibbs free energies are represented by solid and dashed lines, respectively.

Figure 4 (a) Atomic structures and adsorption energies (Eads) of adsorbed H atoms on active sites of Fe-N4–C and Fe-N3P-C. (b) The reaction energies (∆E) of the first C–H bond activation of propane on Fe-N4–C and Fe-N3P-C, respectively.

Figure 5 (a) The top and side views of the artificial Fe-N4–C structure. (b) The atomic structures and adsorption energies (Eads) of the adsorbed H atom on the protruding Fe-N4 site. (c) Two pathways of the first dehydrogenation on the artificial Fe-N4–C to get 1-propyl (CH3CH2CH2*) and 2-propyl (CH3CHCH3*), respectively. IS, TS and FS represent the initial state, transition state and final state, respectively. Green circles represent dissociated H atoms.
Table 1. Changes of potential energies (ΔE), Gibbs free energies (ΔG), and corresponding energy barriers (Ea and Ga) of each elementary reaction on Fe-N3P-C.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: