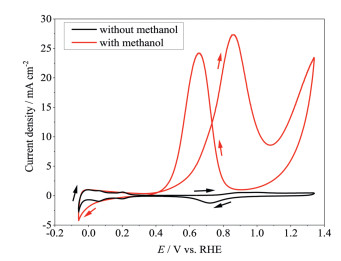
Figure 1.
Cyclic voltammograms of Pt electrode in H2SO4 solutions (0.5 mol/dm3) with or without CH3OH (1 mol/dm3) at room temperature in the cell using Pt as counter electrode.
Degradation of platinum electrocatalysts for methanol oxidation by lead contamination
Zihao Li , Xianggui Zhou , Subhash Singh , Xiaoming Wei , Chunlei Guo , Weishan Li

Many technologies have been explored for replacing fossil fuel [1, 2], among which hydrogen fuel cell is one of the most efficient and sustainable [3, 4]. However, challenges remain for hydrogen generation, storage and transportation. Therefore, searching for alternatives to hydrogen fuel cells becomes one of the hottest topics in the field of fuel cells [5, 6]. Direct methanol fuel cell (DMFC) is an ideal candidate, because methanol is easily available and can be used directly without transformation, and DMFC shares almost the same configuration as well as electrode materials with hydrogen fuel cell [7, 8]. Platinum (Pt) is the most active electrocatalyst for methanol oxidation but its activity is affected by the intermediates during methanol oxidation [9, 10]. Lead (Pb) is detrimental to many chemical reactions [11], but can help Pt electrocatalyst resist the poisoning from the intermediates. This benefit brought by Pb was demonstrated using Pt electrode with Pb atoms gained from under-potential-deposition (UPD) in Pb(NO3)2 solution (10−4–10−1 mol/m3) [12]. Metal Pb might mitigate the electrocatalytic activity when its usage is in a large amount, which can be ascribed to the physical coverage of Pt [13]. While massive investigations were made to modify Pt by architecting microstructures or introducing secondary elements or compounds to enhance the electrocatalytic activity of Pt, less attention was paid to the effect of fuel cell environment such as the compositions possibly dissolved in the solution.
In this communication, we report an abnormal phenomenon that the electrocatalytic activity of Pt toward methanol oxidation is largely lost in a Pb-contaminated environment resembling the working conditions of DFMCs. The activity loss of Pt is demonstrated in a three-electrode cell with H2SO4 solutions (0.5 mol/dm3) with or without methanol (1 mol/dm3). It is found that the methanol oxidation current that is obviously recorded on Pt working electrode in the cell using Pt counter electrode decreases significantly when the Pt counter electrode is replaced by Pb. The possible mechanism on the activity loss of Pt by Pb contamination is given and confirmed by simulations.
Cyclic voltammetry (CV) and chronoamperometry (CA) were performed to present direct evidences. CV was conducted on CHI1040C electrochemical workstation (CH Instruments Ins., USA) with a scan rate of 50 mV/s and the cyclic voltammograms at the 10th cycle were presented. A three-electrode cell with Pt disk electrode of 0.3 cm in diameter (0.07 cm2) as working electrode, Hg/HgSO4 in saturated K2SO4 solution as reference electrode and metal Pt or Pb used as counter electrode. To mitigate the effect of Ohmic resistance, the counter electrode with much larger area (1 cm2) than the working electrode was used. The electrolytes were H2SO4 solutions (0.5 mol/dm3) with or without methanol (1 mol/dm3). To further confirm the effect of Pb2+ ions, CA was conducted in H2SO4 (0.5 mol/dm3) and CH3OH (1 mol/dm3) solutions with or without Pb2+ ions, using Pt as counter electrode under a potential where methanol oxidation takes place. The Pb-containing solution was prepared by adding excess Pb(NO3)2 in H2SO4 solution. Before all the measurements, nitrogen gas was bubbled into the electrolytes to purge the dissolved oxygen. The current densities reported in this work are referred to the geometric surface area of the working electrode. All potentials are reported with respect to standard hydrogen electrode (RHE).
Simulations were carried out to explain the mechanism on the activity loss of platinum by Pb2+ ions. The ionic structures were optimized with density functional theory (DFT) using the Gaussian 09 package and B3LYP hybrid exchange-correlation function. The calculations for oxygen and metal ions were performed based on the standard 6–311++G(d) and SDD basis sets, respectively. In consideration of the solvent environment effect, a dielectric constant of water via polarized continuum model was used simultaneously. Vibrational frequency was employed to confirm all the minimum energy point of each structure.
In order to provide a reference, Pt was used as both working and counter electrodes to carry out CV. Fig. 1 presents the cyclic voltammograms of Pt electrode in H2SO4 solutions (0.5 mol/dm3) with or without methanol (1 mol/dm3) in the electrolytic cell using Pt as counter electrode. In the solution without methanol (black curve in Fig. 1), hydrogen adsorption/desorption currents on/from Pt can be observed in the potential range between 0 and 0.3 V, while the oxidation of Pt starts at 0.6 V, forming platinum oxide with low valence of Pt such as PtO [14]. The current is stable after 0.9 V in forward sweeping, suggesting that the Pt on the surface has been fully turned into platinum oxide that might contain high valence Pt such as PtO2. In backward sweeping, the reduction peak of platinum oxide could be found at around 0.7 V.
When switching to the solution with methanol (red curve in Fig. 1), the hydrogen absorption/desorption current is still observed but the curve shape changes slightly, suggesting that the methanol oxidation process affects the rearrangement of the surface Pt atoms. At the potentials higher than 0.4 V in forward sweeping, the recorded currents in the methanol-containing solution are larger than those without methanol, suggesting that the methanol oxidation happens at these potentials. Especially, the methanol oxidation current increases significantly at the potential for the formation of surface platinum oxide, indicating that the presence of partial surface platinum oxide plays an important role in the electrocatalytic methanol oxidation on Pt [15]. When the potential reaches 0.85 V, the rising of current stops, which is a sign that the Pt surface has been completely turned into platinum oxide. In backward sweeping, the oxidation peak emerges again at 0.65 V, which can be explained by the reduction of platinum oxide and the recovery of the Pt electrocatalytic activity toward methanol oxidation due to the co-existence of metal Pt atom and platinum oxide [6].
When substituting Pb for Pt as counter electrode but keeping the same Pt working electrode, different results were obtained, as shown in Fig. 2. In the solution without methanol (black curve in Fig. 2), the Pt electrode behaves similar to that in the cell using Pt counter electrode (black curve in Fig. 1): The currents for hydrogen adsorption/desorption and formation/reduction of platinum oxide can be identified, suggesting that the introduction of Pb counter electrode does not change the surface properties of Pt. In the solution with methanol, however, the cyclic voltammogram (red curve in Fig. 2) is remarkably different from that with Pt as counter electrode (red curve in Fig. 1). The methanol oxidation peak current decreases significantly when Pt is replaced by Pb, with the forward sweeping methanol oxidation peak reduced from 27.6 mA/cm2 to 6.5 mA/cm2, suggesting that the electrocatalytic activity of Pt toward methanol oxidation is inhibited due to the introduction of Pb counter electrode.
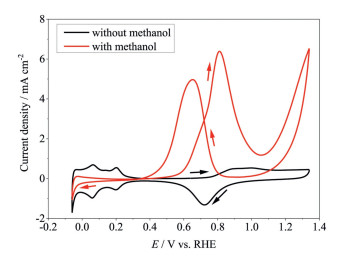
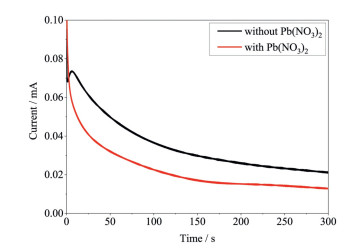
Since Pb2+ is difficult to dissolve in H2SO4 solution and no oxidation and reduction currents related to Pb can be recorded (the same shapes of the black curves in Figs. 1 and 2), it can be inferred that the activity loss of Pt toward methanol oxidation does not result from the deposition of Pb as metal. To confirm that the activity loss of Pt is not from the metal Pb, CA was conducted in H2SO4 and CH3OH solutions with or without Pb2+ ions, using Pt as counter electrode under a potential where methanol oxidation takes place. Fig. 3 presents the chronoamperograms of Pt in H2SO4 and CH3OH solutions with and without Pb2+ ions after the potential is stepped from open circuit potential to 0.74 V. A significant reduction in methanol oxidation currents can be identified when the Pb-free solution is replaced by the solution that contains Pb2+. Since the potential bias was applied positively, Pb2+ ions in the solution will not be reduced to metal Pb and the reduced oxidation currents result from Pb2+ ions rather than metal Pb. It can be noted that there is a sharp drop and then increase of current during the initial CA for the solution without Pb2+ ions compared to the monotonic drop for the solution with Pb2+ ions. This difference indicates the electrocatalytic mechanism of Pt toward methanol oxidation. The sharp drop of current results from adsorption of methanol oxidation intermediates and double layer charging, while the increase of current is attributed to the further oxidation of the intermediates due to the available platinum oxide. It was reported that the trace metal Pb from UPD on Pt was beneficial for the activity improvement of methanol oxidation [12]. Apparently, there is an abnormal mechanism on the activity loss of platinum when Pb is used as counter electrode or Pb2+ ions are present in the solution.
The possible mechanism of platinum degradation can be explained as following. Pt exhibits electrocatalytic activity toward methanol oxidation because there is an appropriate ratio of Pt atom to platinum oxide on the surface of Pt electrode. The involved electrocatalytic mechanism is illustrated in Fig. 4a. The methanol oxidation results in intermediates CHxO (x < 4), especially CO. These intermediates are strongly adsorbed on Pt and hinder the subsequent methanol oxidation [16]. When a neighboring platinum oxide is available, it plays a role of spilling over proton or oxygen, which assists the further oxidation of the intermediates, providing the Pt electrode with an electrocatalytic activity toward methanol oxidation. Due to the steric hindrance from the intermediates and the weak spillover effect from PtO, a pure Pt electrode does not exhibit a satisfactory electrocatalytic activity toward the methanol oxidation. This is why it is effective for improving the activity of Pt by delicately constructing the surface microstructures or by introducing the secondary elements or compounds [5]. In the case that the electrocatalytic activity of Pt toward the methanol oxidation was improved by the trace metal Pb from UPD [12], the deposited Pb atoms provided appropriate space that facilitates further oxidation of the intermediates.
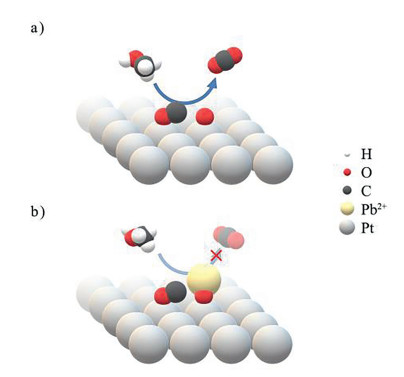
Based on the role of PtO, we propose the mechanism on the activity loss of Pt by Pb contamination, as shown in Fig. 4b. The trace Pb2+ ions dissolved in the solution from Pb counter electrode are adsorbed on PtO via the combination of Pb2+ and O2− ions, suppressing the spillover effect of PtO, which is important to eliminate adsorbed intermediates, especially (CO)ad, deteriorating the electrocatalytic activity of Pt. To confirm this assumption, the geometric structures of O2−, Pb2+ and Pt2+ were optimized and the binding energies of O2− with Pb2+ and Pt2+ were calculated based on density functional theory. The obtained binding energy of Pt2+-O2− and Pb2+-O2− is −564.77 and −1196.15 kJ/mol, respectively. The far lower binding energy of Pb2+-O2− suggests that there is a strong interaction between Pb2+and O2−. It is this strong interaction that induces a strong adsorption of Pb2+ ions on PtO and suppresses the spillover effect of PtO, leading to the activity loss of Pt towards methanol oxidation.
To conclude, we have demonstrated that the electrocatalytic activity of Pt toward methanol oxidation will be severely lost in a slightly Pb-contaminated environment. The trace Pb2+ ions present in the solution are easily adsorbed on PtO due to the strong interaction between Pb2+ and O2− ions. It is this strong interaction that suppresses the spillover effect of PtO, which is important to eliminate adsorbed intermediates from methanol oxidation, especially (CO)ad, and deteriorates the electrocatalytic activity of Pt toward methanol oxidation. The stronger interaction between Pb2+ and O2− ions has been demonstrated by the far lower binding energy of Pb2+ with O2− than that of Pt2+ with O2−. Since Pb might be present in various components in the fuel cell systems or even in air, attention should be paid to reliably design and efficiently manage direct methanol fuel cells.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work is financially supported by Bill & Melinda Gates Foundation (No. OPP1119542). The author W. Li would like to thank South China Normal University for the financial support for his visit to University of Rochester.
I.E.L. Stephens, J. Rossmeisl, Science 354 (2016) 1378. doi: 10.1126/science.aal3303
Q. Xu, J. Lin, C. Ye, et al., Adv. Energy Mater. 10 (2019) 1903292.
K. Wang, N. Li, Y. Yang, et al., Chin. Chem. Lett. 32 (2021) 3159–3163. doi: 10.1016/j.cclet.2021.02.045
C. Pak, S.W. Lee, C. Baik, et al., Chin. Chem. Lett. 30 (2019) 1186–1189. doi: 10.1016/j.cclet.2019.02.020
M.E. Scofield, C. Koenigsmann, L. Wang, H. Liu, S.S. Wong, Energy Environ. Sci. 8 (2015) 350. doi: 10.1039/C4EE02162B
W. Wu, X. Xiang, Y. Lin, W. Li, Int. J. Hydrog. Energy 38 (2013) 11080. doi: 10.1016/j.ijhydene.2013.02.123
X. Zhu, Z. Hu, M. Huang, et al., Chin. Chem. Lett. 32 (2021) 2033–2037. doi: 10.1016/j.cclet.2020.11.071
X. Liu, Y. Zhang, S. Deng, et al., Chin. Chem. Lett. 31 (2020) 299–304. doi: 10.1016/j.cclet.2019.03.053
M. Li, K. Kuttiyiel, F. Lu, O. Gang, R.R. Adzic, J. Electrochem. Soc. 166 (2019) F3300. doi: 10.1149/2.0321907jes
J. Lu, J. Du, W. Li, J. Fu, Chin. Chem. Lett. 15 (2004) 703–706.
L. Bonin, V. Vitry, F. Delaunois, SM&T 23 (2020) 00130.
B. Beden, F. Kadirgan, C. Lamy, J.M. Leger, J. Electroanal. Chem. 127 (1981) 75. doi: 10.1016/S0022-0728(81)80469-X
A.F. Diwell, B. Harrison, Platinum Met. Rev. 25 (1981) 142–151.
Z. Fu, W. Li, W. Zhang, et al., Int. J. Hydrog. Energy 35 (2010) 8101. doi: 10.1016/j.ijhydene.2010.01.024
X. Xiang, W. Li, Z. Zhou, et al., J. Solid State Electrochem. 14 (2010) 903. doi: 10.1007/s10008-009-0988-x
W. Li, L. Tian, Q. Huang, et al., J. Power Sources 104 (2002) 281. doi: 10.1016/S0378-7753(01)00961-2

Figure 1 Cyclic voltammograms of Pt electrode in H2SO4 solutions (0.5 mol/dm3) with or without CH3OH (1 mol/dm3) at room temperature in the cell using Pt as counter electrode.

Figure 2 Cyclic voltammograms of Pt electrode in H2SO4 solutions (0.5 mol/dm3) with or without CH3OH (1 mol/dm3) at room temperature in the cell using Pb as counter electrode.

Figure 3 Chronoamperograms of Pt electrode in H2SO4 (0.5 mol/dm3) and CH3OH (1 mol/dm3) solutions with and without Pb(NO3)2 under 0.74 V at room temperature in the cell using Pt as counter electrode.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: