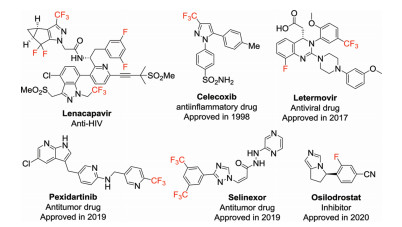
Figure 1.
Examples of fluorine or fluoroalkyl-containing drugs.

Fluorine is a halogen element with atomic number 9 in group 17 of the periodic table. As the most reactive chemical element, fluorine possesses many unique properties, such as high electronegativity (4.0 in Pauling scale), small atomic radius (rv = 1.47 Å) and strong C−F bond energy (~116 kcal/mol) [1]. The introduction of fluorine atom(s) or fluoroalkyl groups (e.g., trifluoromethyl, -CF3) into organic molecules can greatly impact their physicochemical, pharmacokinetic, and pharmacodynamic properties. Consequently, organofluorine compounds has been widely applied in medicine, pesticides, and materials science [2, 3]. For instance, many important drugs, such as lenacapavir [4], mavacoxib [5], celecoxib [6], letermovir [7], pexidartinib [8], selinexor [3] and osilodrostat [9] containing at least one fluorine atom or/and fluoroalkyl groups, have recently been approved for effective treatment of various diseases (Fig. 1). It has been reported that more than 20% of the marketed pharmaceuticals and up to 40% of agrochemicals contain at least one fluorine atom [10, 11]. As a consequence, the development of effective methods to incorporate fluorine atom(s) or fluoroalkyl groups into organic molecules has attracted much interest from organic and medicinal chemists for drug development [12-14].
Various fluorinating reagents such as the Ruppert-Prakash reagent and its analogues [15, 16], Chen's reagent [17], the Togni reagents [18], the Umemoto reagent [19], sulfur-based fluorination and fluoroalkylation reagents [20, 21], [(phen)CuCF3] [22], fluoroform-derived CuCF3 [23], ((CF3SO2)2Zn [24], the Langlois reagent (CF3SO2Na) [25], [(phen)2Cu](O2CRF) [26, 27], and building blocks such as trifluorodiazoethane [28], methyl perfluoroalk-2-ynoates [29], 2,2,2-trifluoroacetohydrazide [30] and 2–bromo-3,3,3-trifluoropropene [31] have been used for the preparation of fluoroalkylated compounds. However, some of these reagents are expensive, or require multistep synthesis, and thus widespread synthetic applications are restricted.
Trifluoroacetic anhydride (TFAA) is an inexpensive and atom-efficient CF3-source with commercial availability on ton-scale. These advantages and the enormous potential of trifluoromethyl group in fluorine chemistry make TFAA attractive in synthetic chemistry. In addition, TFAA also plays important roles in organic synthesis [32, 33], and polymer chemistry [34, 35]. However, despite this promising precedence for the application of TFAA in organic synthesis, the use of TFAA in trifluor-omethylation reactions has only scarcely been studied.
This tutorial review summarizes the recent advances in the synthesis of fluoroalkylated compounds using TFAA and its analogues [(RFCO)2O, RF = CF2H, CF2Cl, CnF2n+1]. The available literature reports of these procudures were classified into four significant sections based on the reaction with the fragment of the fluoroalkyl anhydrides (Scheme 1): (ⅰ) coupling reactions of fluoroalkyl groups (RF), (ⅱ) coupling reactions of fluoroalkanoyl groups (RFCO), (ⅲ) cyclization reactions with fluoroalkyl groups (RF), (ⅳ) cyclization reactions with fluoroalkanoyl groups (RFCO). We hope it will provide a reference for the development of new strategies for the synthesis of fluoroalkylated compounds, and the large-scale application of these reagents in synthetic chemistry in the future.
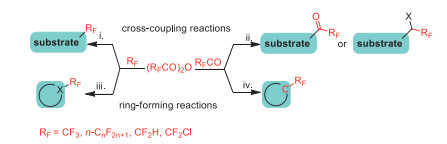
The coupling reaction of substrate with fluoroalkyl reagents is one of the most effective ways to access the fluoroalkylated compounds. Fluorine-containing anhydrides have been extensively employed in the synthesis of fluorine-containing organic compounds through coupling reaction strategies by many research groups in recent years. These reactions include coupling reactions of fluoroalkl groups and fluoroalkanoyl groups.
It has been well established that bis(fluoroalkanoyl) peroxides generated from reaction of (RFCO)2O with peroxides can furnish perfluoroalkyl radicals. In 1982, Jiang and co-workers reported the synthesis and thermal decomposition of some perfluoro- and polyfluorodiacyl peroxides [36]. The major decomposition products were characterized as the coupling products of the corresponding radicals (RF−RF).
In 1985 and 1986, Kobayashi and co-workers reported the reaction of heteroaryl compounds and electron-rich olefins with bis(heptafluorobutyryl) peroxide to give corresponding perfluoropropyl products (Scheme 2) [37, 38].
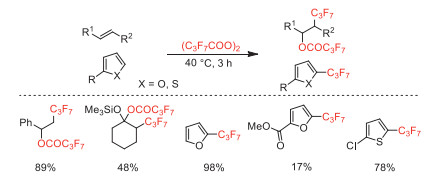
Two years later, Yoshida and co-workers reported the heptafluoropropylation of thiophenes with this reagent and further explored the synthesis of 3-heptafluoropropylated thiophenes (Scheme 3) [39].

After this, this group also reported the trifluoromethylation of (hetero)aromatic compounds with bis(trifluoroacetyl) peroxide to provide correspounding trifluoromethylated products in moderate to high yields, including trifluoromethylated benzenes, furans, thiophenes, nitrogen-containing heteroaromatics and benzo derivatives [34, 40-42]. From 1992 to 1994, this group synthesized bis(chlorodifluoroacetyl) peroxide reagent using hydrogen peroxide and CDFAA in Freon, and reported the chlorodifluoromethylation of electron-rich aromatics and olefins with this reagent to give corresponding chlorodifluoromethylated products that can be further transfered into other difluoromethylene-functionalized compounds by hydrogenation or reaction with Tin reagents under radical conditions (Scheme 3) [43, 44].
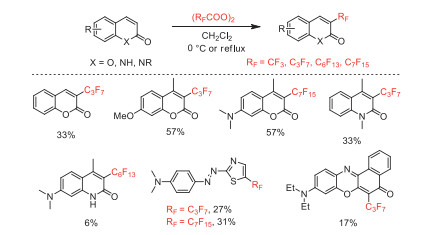
In 1992 and 1995, Matsui and co-workers reported the perfluoroalkylation of coumarins, azo compounds, quinolinones and phenoxazones with bis(perfluoroalkanoyl) peroxides (Scheme 4). The fluorescence intensities and photostabilities of corresponding perfluoroalkylated products with non-fluorine substituted products were compared [45-48].
In 2015, Bräse and co-workers developed a metal-free perfluoroalkylation of (hetero)arenes with perfluoroalkyl radicals generated in situ from perfluoro carboxylic anhydrides in the presence of urea–hydrogen peroxide (UHP) [49]. This method provided an access to perfluoroalkylated (hetero)arenes, including benzene derivatives, furans, thiophenes, pyrroles, and highly functionalized compounds. This strategy effectively avoided explosion hazard of bis(perfluoroalkanoyl) peroxides and use of environmentally hazardous Freon (Scheme 5).
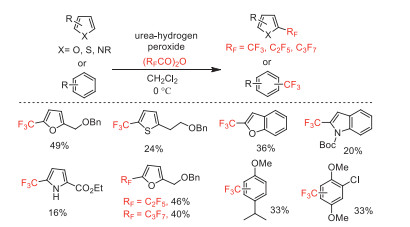
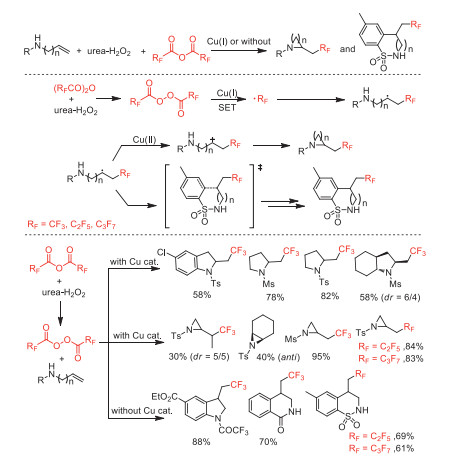
In 2016, Sodeoka and co-wokers developed an efficient perfluoroalkylation of unactivated alkenes with perfluoro acid anhydrides as the perfluoroalkyl source [50]. In this process, the use of Cu(Ⅰ) catalyst was crucial for achieving perfluoroalkylation reaction. The authors proposed that single electron transfer (SET) between the bis(fluoroalkanoyl) peroxide and the Cu(Ⅰ) catalyst initiates the generation of perfluoroalkyl radical via decarboxylation. In addition, the carboperfluoroalkylation of alkenes under metal-free conditions afforded perfluoroalkyl-substituted polycycle or heterocycle compounds (Scheme 6).

In 2018, the same group reported a Cu(Ⅱ) catalyzed or metal-free chlorodifluoromethylation of alkenes and N-heterocycles using chlorodifluoroacetic anhydride (CDFAA) as a chlorodifluoromethyl radical source [51]. The reaction was optimized through variation of copper salt catalysts with addition of pyridine as additive. Base on the fact that the relatively fast thermal decomposition of bis(chlorodifluoroacetyl) peroxide (BCDFAP) would generate an excess amount of CF2Cl radicals without the SET process with Cu(Ⅰ), the authors employed Cu(Ⅱ) salt as a pre-catalyst instead of Cu(Ⅰ) salt to improve the yield of desired chlorodifluoromethylated products (Scheme 7).
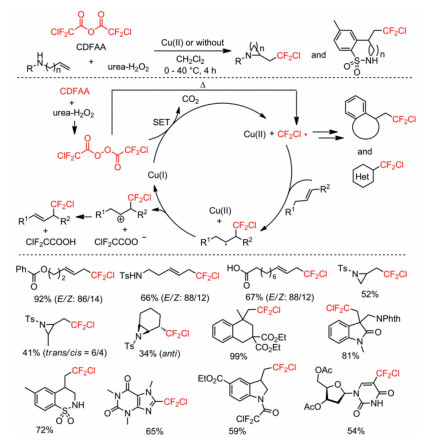
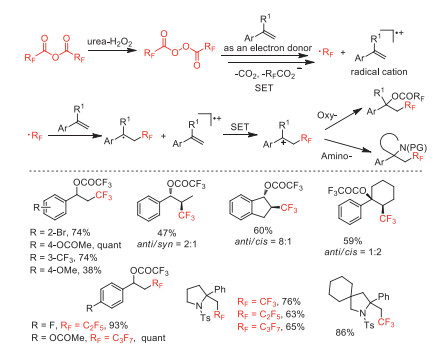
In the same year, this group also reported a metal-free oxy- and amino-perfluoroalkylations of alkenes with (RFCO)2O (RF = CF3, C2F5, n-C3F7) to access oxy-perfluoroalkylation products and amino-perfluoroalkylation cyclization products via carbocation intermediates formation [52]. The authors proposed that the styrene substrate could serve as an electron donor to accelerate generation of the perfluoroalkyl radical through decomposition of the diacyl peroxide by SET (Scheme 8).
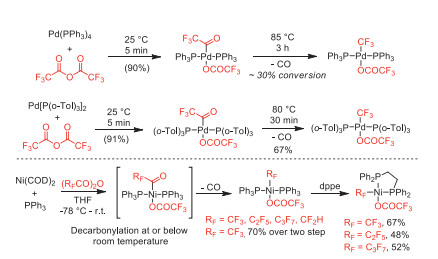
In 2014, Sanford and co-worker reported a Pd-mediated decarbonylative trifluoromethylation of diarylzincs using trifluoroacetic esters as CF3 sources [53]. The perfluoroacyl complexes were porposed as the key intermediates, which could be alternatively synthesized from oxidative addition of Pd[P(o-Tol)3]2 with perfluoro acid anhydrides (Scheme 9).
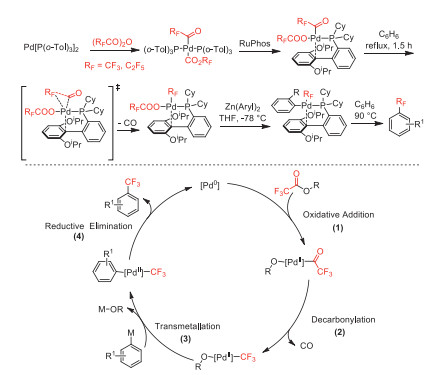
In the same year, this group further reported the synthesis of fluoroalkyl palladium(Ⅱ) and nickel(Ⅱ) complexes through decarbonylation of the perfluoroacyl-metal species [54]. The higher decarbonylation reactivity of perfluoroacyl-Ni species over Pd species was proposed as that nickel could undergo decarbonylation without ligand dissociation via a pentacoordinate transition state (Scheme 10).
In 2015, Stephenson and co-workers described a radical trifluoromethylation of (hetero)arenes with TFAA under photoredox catalysis conditions [55]. Pyridine N-oxide (Py-O) was used as the reagent for the formation of a reducible TFAA adduct, which readily underwent N–O bond cleavage and CO2 extrusion to form the trifluoromethyl radical. A variety of arenes, heterocycles, and alkenes could be trifluoromethylated in moderate yields (Scheme 11).
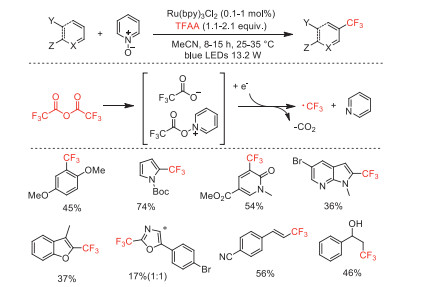
In 2016, Stephenson and co-workers further reported an improved perfluoroalkylation of (hetero)arenes with (RFCO)2O (RF = CF3, C2F5, n-C3F7) in combination with continuous flow on a kilogram scale using pyridine N-oxide derivatives (Scheme 12) [56]. This protocol overcame the limitations associated with small-scale synthesis and using superstoichiometric copper salt by lowing catalyst loadings and utilizing visible light. The mechanistic studies revealed that the conjugation and electrophilicity of the pyridinium ring system promotes the formation of photoactive electron donor-acceptor complexes. This work was highlighted by Reiser as "the underlying methodology are of great relevance in medicinal chemistry" [57].

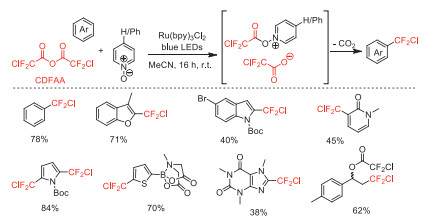
Afterwards, in 2018, Stephenson and co-worker reported a decarboxylative radical chlorodifluoromethylation of (hetero)arenes with CDFAA as a chlorodifluoromethyl radical source by using (4-phenyl)pyridine N-oxide as a redox trigger under visible light irradiation conditions (Scheme 13) [58]. This reaction runs successfully in both batch and flow processing with high regioselectivity and broad substrate scope compatibility.
In 2018, Duan and co-workers reported the synthesis of a recyclable ruthenium tris[4,4′-bis(dinonylmethyl)−2,2′-bipyridine] (Ru[(DNM)2bpy]
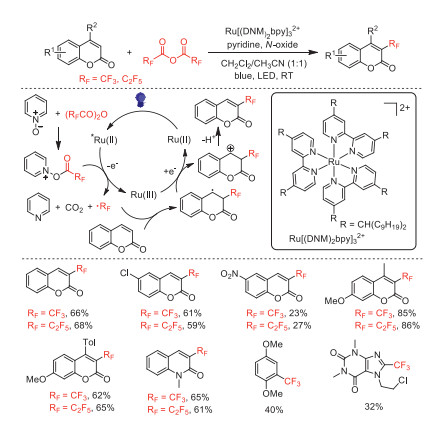
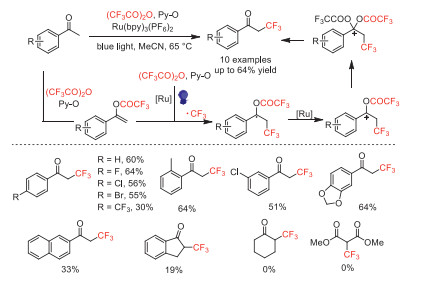
In 2019, Schaub and co-workers developed a direct α-trifluoromethylation of acetophenone derivatives by using TFAA as the CF3 source, pyridine N-oxide (Py-O) as activator and oxidant, and tris-(2,2′-bipyridine)ruthenium(Ⅱ) hexafluorophosphate [Ru(bpy)3(PF6)2] as the photocatalyst under blue light irradiation [60]. The acetophenone derivatives substituted with different functional groups could be converted to the corresponding α-CF3 derivatives in moderate yields with high regioselectivity. However, cyclohexanone or dimethyl malonate were not suitable for these conditions. Based on in-situ monitoring experiments and existing literature reports [55], the mechanism was proposed and showed in Scheme 15. The authors proposed that vinyl trifluoroacetate was the key intermediate for this transformation.
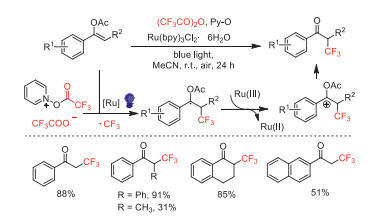
In the same year, Singh and Garg reported a visible light-mediated radical trifluoromethylation of enol acetates using TFAA (Scheme 16) [61]. Compared with Schaub's work, this method afforded the corresponding α-trifluoromethylated acetophenones with a higher yield and wider substrate scope.
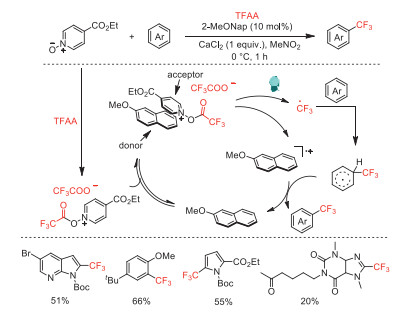
In 2020, Stephenson and co-workers demonstrated that the electron donor-acceptor (EDA) complexes from the combination of 2-methoxynaphthalene donor and acylated ethyl isonicotinate N-oxide acceptor can controllably generate trifluoromethyl radical from TFAA through photoexcitation (Scheme 17) [62]. This strategy solves the problem of fast deactivation of photocatalytic species originated from back electron transfer. The authors reported the synthetic utility by visible light-driven radical trifluoromethylation and Minisci alkylation reactions. A multigram-scale trifluoromethylation of methyl N-Boc pyrrole-2-carboxylate by the EDA platform in a continuous flow manifold was also demonstrated.
Fluoroalkanoyl groups, such as trifluoroacetyl (CF3CO), can be found in some biologically active molecules [63]. Perfluoroketones are useful building blocks for the synthesis of more complex structures and these compounds are also important intermediates in medicinal chemistry.
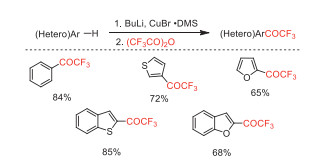
In 1991, Kerdesky and co-worker reported a nucleophilic reaction of TFAA with (hetero)arylcopper reagents to prepare (hetero)aryl trifluoromethyl ketones (Scheme 18) [64].
From 1992 to 1995, Zard and co-workers reported a series of methods for the synthesis of trifluoromethyl ketones and their further trifluoromethylated heterocycles from reaction of carboxylic acids or its chlorides with TFAA in the present of pyridine (Scheme 19). Trifluoromethyl acyl ketenes were used as versatile precursors for the synthesis of various trifluoromethylated heterocycles [65-67].
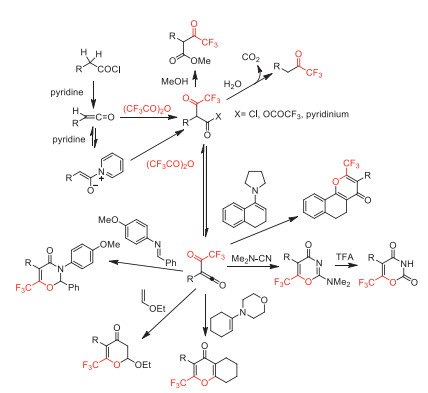
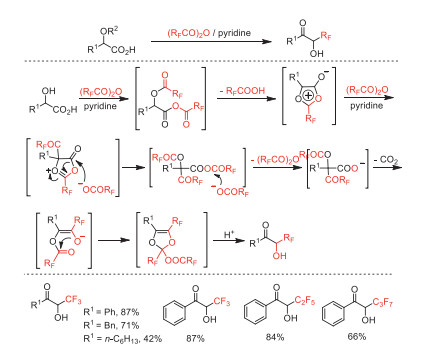
In 1994, Kawase and co-workers reported a transformation of α–hydroxy acids to α-perfluoroalkylated acyloins by using perfluoroalkylcarboxylic anhydrides in the presence of pyridine (Scheme 20) [68]. These α-perfluoroalkylated acyloins serve as a class of perfluoroalkyl-containing building blocks for further transformation.
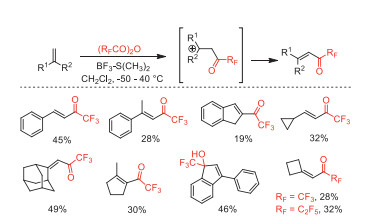
In the same year, Balenkova and co-workers developed a new class of perfluoroacetylation reagents, (RFCO)2O-BF3·SMe2. The perfluoroacetylating reactivity of these reagents with different types of alkenes were investigated (Scheme 21) [69].
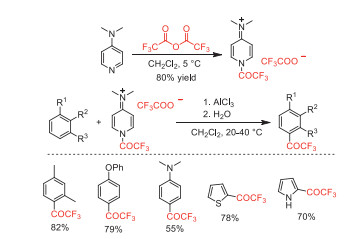
Two years later, Simchen and co-worker synthesized a stable and easy to handle trifluoroacetylating agent, 4-dimethylamino-1-trifluoroacetylpyridinium trifluoroacetate [70]. This reagent was used in AlCl3-mediated trifluoroacetylation of aromatic compounds to obtain aryl trifluormethyl ketones in good yields (Scheme 22).
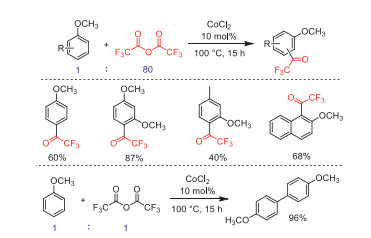
In the same year, Gilbert and co-workers developed a CoCl2-catalyzed trifluoroacetylation and dimerization of methoxyaromatics with neat TFAA under fixed ratio conditions (anisole/TFAA 1:80 or 1:1, respectively) (Scheme 23) [71].
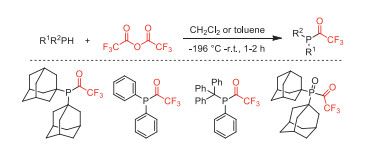
In 1996, Schmutzler and co-workers reported the C−P bond coupling reaction of secondary phosphines (1-Ad)2PH and Trt(Ph)PH with TFAA to yield the trifluoroacetylphosphines, and the secondary phosphines oxide (1-Ad)2P(O)H also reacted with TFAA to furnish the corresponding trifluoroacetylated product (Scheme 24) [72].
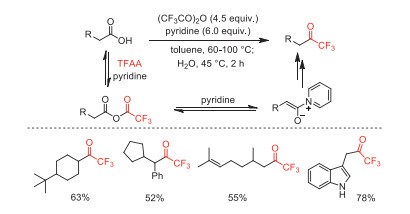
In 2007, Reeves and co-workers greatly improved the previous procedure for the transformation of carboxylic acid chlorides to trifluoromethyl ketones reported by Zard and co-workers [65, 66]. They developed a direct one-pot conversion of primary and secondary carboxylic acids with TFAA to the corresponding trifluoromethyl ketones (Scheme 25) [73].
In the same year, Porco, Jr. and co-workers reported a Lewis acid-catalyzed nucleophilic addition to N-phosphinylimines in the presence of TFAA [74]. Asymmetric trifluoroacetamide products are obtained in this novel process which likely involved succeeding acylation/hydration procedure of N-phosphinylimine substrates followed immediately nucleophilic addition (Scheme 26).
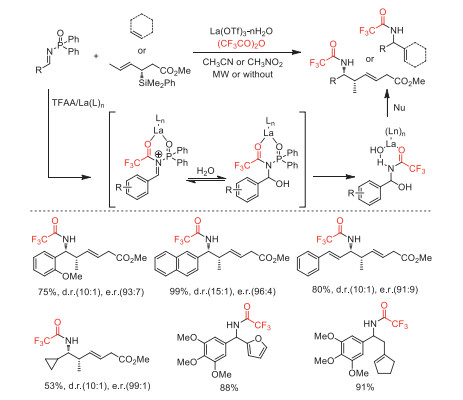
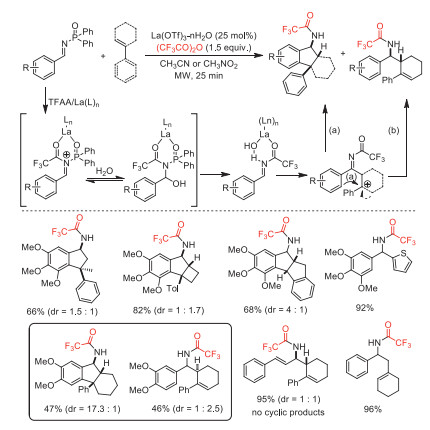
In 2010, the same group reported a diverse nucleophilic addition of N-diphenylphosphinylimines, including aza-Prins/intramolecular Friedel-Crafts annulations, employed La(OTf)3·nH2O as a catalyst and TFAA as an activator to obtain N-trifluoroacetylated products [75]. This method provided a series of complex polycyclic scaffolds containing diarylmethane pharmacophore. The control experiments and DFT calculation revealed that thermodynamic equilibration of stereoisomeric cationic intermediates and steric hindrance of Friedel-Crafts cyclization determines the distribution of acyclic-cyclic products and stereochemistries (Scheme 27).
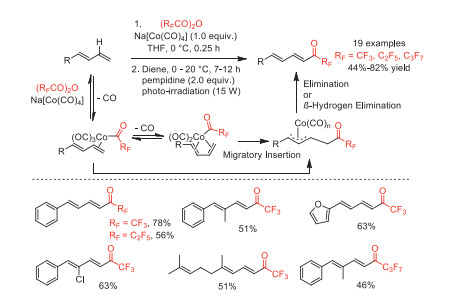
In 2014, Rovis and co-workers reported a cobalt-mediated direct perfluoroacylation of 1,3-dienes with (RFCO)2O (RF = CF3, C2F5, n-C3F7) to afford useful perfluoroketones under mild conditions [76]. A series of mechanism experiments were performed to demonstrate the formation of key π-allylcobalt intermediate, and the reaction pathway is shown in Scheme 28.
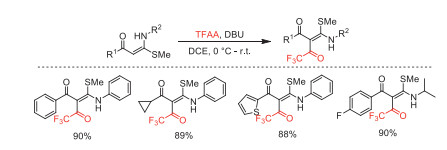
In 2017, Bhagat and co-workers reported an electrophilic trifluoroacetylation of enaminones with TFAA by using DBU as a base (Scheme 29) [77]. Various enaminones were obtained in excellent yields. This methodology features some advantages, such as maintained E stereoselectivity of products, broad functional group tolerance, and products extensibility.
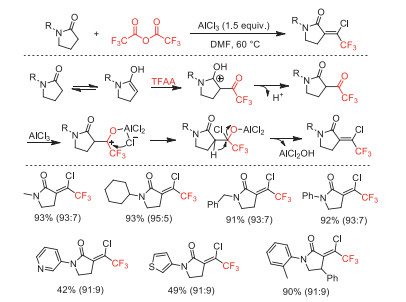
In 2018, our group reported an AlCl3-mediated one-pot cascade reaction of pyrrolidones with TFAA through cascaded electrophilic trifluoroacetylation, nucleophilic chlorination, and elimination, achieving the synthesis of various 1–chloro-2,2,2-trifluoroethylidene-substituted pyrrolidones in moderate to high yields under simple and mild conditions (Scheme 30) [78]. The obtained products with a wide range of substituents can be applied to further functionalization as trifluoromethyl-containing building blocks and some compounds showed fungicidal activity against cucumber downy mildew (CDM).
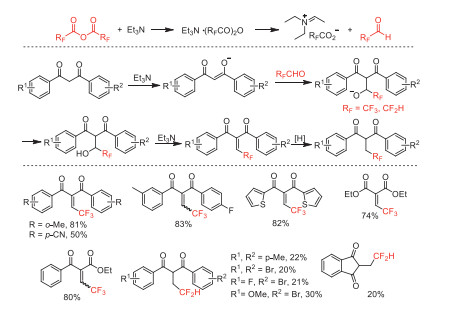
Next year, our group reported a Knoevenagel-type reaction of 1,3-diketones with TFAA or difluoroacetic anhydride to access 2-(2,2,2-trifluoroethylidene) and (2,2-difluoroethyl)−1,3-dicarbonyl compounds, respectively (Scheme 31) [79]. This reaction process promoted by trifluoroacetaldehyde/difluoroacetaldehyde generated in situ from trifluoroacetic/difluoroacetic anhydride and Et3N [80] shows broad substrate applicability and excellent chemoselectivity under metal-free conditions.
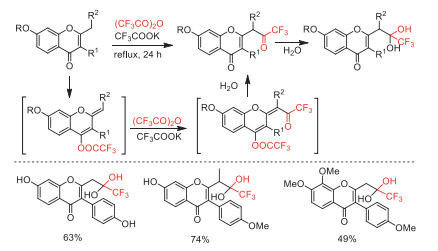
In 2019, Frasinyuk and co-workers developed an efficient trifluoroacetylation of 2-methyl- and 2-ethylchromones with TFAA to access 2-(3,3,3-trifluoroacetonyl) chromones and isoflavones [81]. The reaction mechanism was proposed with involving the generation of the vinyl ethers from substrates followed by trifluoroacetylation with TFAA in the presence of CF3COOK (Scheme 32).
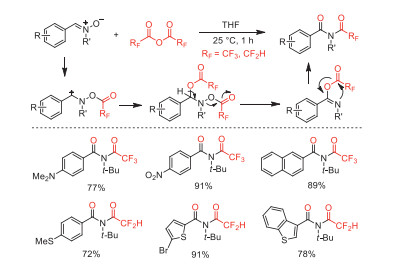
In the same year, our group reported an efficient N-trifluoroacetylation of nitrones to synthesize N-trifluoroacetyl amides in good to excellent yields under mild conditions [82]. Based on existing literature reported by Drabowicz and co-workers in 1997 [83], we proposed that this method involved electrophilic trifluoroacetylation, nucleophilic addition, elimination, and subsequent intramolecular substitution cascade process to afford corresponding amide products (Scheme 33).
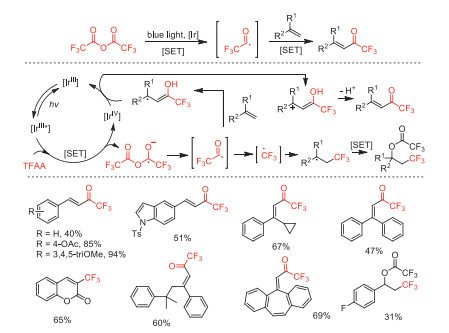
In 2021, Katayev and co-workers reported a visible-light-promoted radical trifluoroacylation strategy of alkenes with TFAA to afford a variety of α, β-unsaturated trifluoromethyl ketone derivatives [84]. Furthermore, the strategy can be turned into a trifluoromethylation reaction of alkenes and aromatics by simply changing the reaction condition parameters. For trifluoroacylation, mechanistic studies showed that the generation of a fleeting trifluoroacyl radical (CF3CO·) promoted by a Ir photocatalyst triggered a C—O bond fragmentation, revealing fundamentally different from conventional methodologies (Scheme 34).
Cyclization reactions can deliver initial cyclic compounds, structurally diverse scaffolds and biologically active fragments. Some compounds with uncommon heterocycles or sterically hindered substituents, which are not easily available through coupling reaction, are synthesized through the cyclization process.
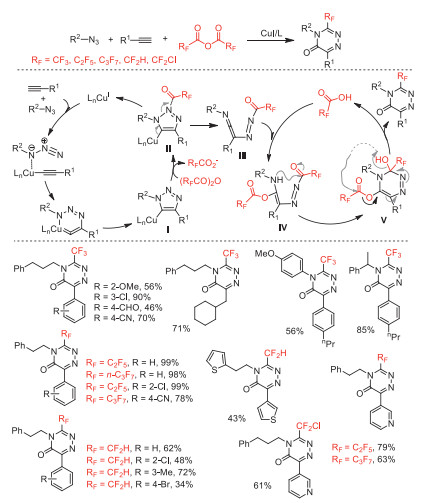
After had reported the perfluoroalkylation coupling reaction and cyclization reaction of unactivated alkenes with acid anhydrides [50], in 2017, Sodeoka and co-workers reported an efficient metal-free or copper-catalyzed N-heterocycle-forming amino/carboperfluoroalkylations of aminoalkenes by using (RFCO)2O (RF = CF3, C2F5, n-C3F7) in conbination with urea·H2O2 as perfluoroalkyl radical sources to provide diversified perfluoroalkylated amines (Scheme 35) [85]. A wide variety of perfluoroalkylated N-heterocycles including complex molecules were synthesized through this method, confirming its potential synthetic utility. Mechanism investigations suggested that the Cu(Ⅰ) catalyst could accelerate perfluoroalkyl radical (·RF) formation by promoting the decomposition of perfluoro diacyl peroxide, while the Cu(Ⅱ) controled the product selectivity. Subsequently, this group reported chlorodifluoromethylation coupling reaction and cyclization reaction of unactivated alkenes with chlorodifluoroacetic anhydride [51].
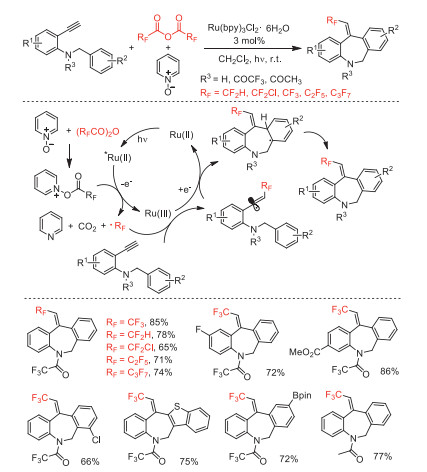
In 2019, Zhong and co-workers reported a one-pot ring-forming approach to afford various fluoroalkylated dibenz[b, e]azepines by visible-light catalysis and using (RFCO)2O (RF = CF2H, CF2Cl, CF3, C2F5, n-C3F7) as a reactive fluoroalkyl radicals (·RF) source [86]. The combination of Ru(bpy)3Cl2·6H2O and Py-O was employed as an efficient photoinduced catalytic system. Based on radical inhibitor and literature, the authors proposed the reaction mechanism involving the fluoroalkylation of substrates followed by 7-ortho radical cyclization and oxidation to obtain the desired products (Scheme 36).
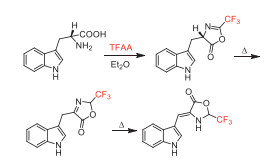
In 1989, Bergman and co-worker reported a quickly ring-formation reaction of tryptophan with TFAA to obtain corresponding oxazolone (Scheme 37), which was used as a starting material for the synthesis of tryptophan containing peptides [87].
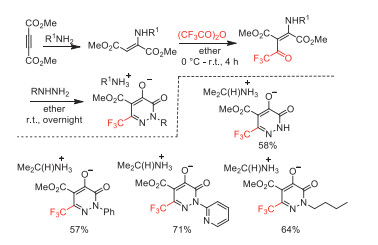
In 1993, Hegde and co-worker reported the cyclization reaction of hydrazines and 2-(alkylamino)-3-(trifluoroacetyl)butenedioates starting from TFAA, primary amine and dimethyl acetylenedicarboxylate to provide pyridazinone-containing heterocyclic compounds (Scheme 38) [88].
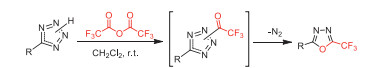
In 2007, Kizhnyaev and co-workers reported the ring formation of 5-trifluoromethyl-1,3,4-oxadiazoles through the reaction of tetrazoles with TFAA under milder conditions (at 20–25 ℃) comparing with non-fluorinated anhydride (at 100–120 ℃) (Scheme 39) [89].
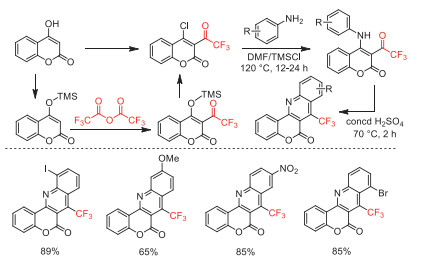
In 2011, Iaroshenko and co-workers reported a straightforward and high-yield method for the synthesis of 7-(trifluoromethyl)−6H-chromeno[4,3-b]quinolin-6-ones by the reaction of anilines with 4–chloro-3-(trifluoroacetyl)coumarin, which was synthesized via TMSCl-mediated trifluoroacetylation of 4-hydroxycoumarin with TFAA (Scheme 40) [90].
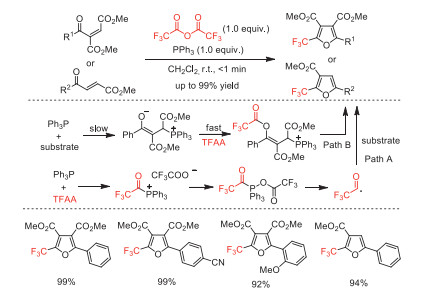
In the same year, Xu and co-workers developed a cascade cyclization reaction to synthesize 2-trifluoromethylfurans [91]. This cascade reaction was extremely efficient because the raw materials were completely converted within 1 min and the desired products were obtained in good to excellent yields (87%~99%) with wide substrate generality. Based on the results of control experiments and 19F NMR analysis, the authors proposed two possible pathways shown in Scheme 41.
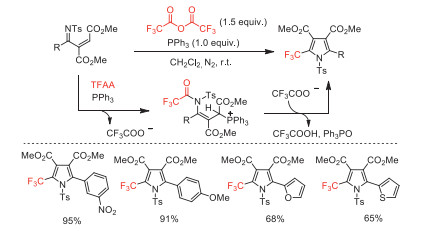
Two years later, Xu and co-workers reported a PPh3-mediated cascade cyclization reaction to synthesize 2-trifluoromethylpyrroles (Scheme 42) [92]. This method still maintains the advantages of high yields (up to 99%), mild reaction conditions (at room temperature), simple operation, broad functional group tolerance and super-short reaction time (within 1 min).
In 2017, Schäfer and co-workers reports a scalable synthesis of trifluoromethylated imidazo-fused N-heterocycles using TFAA as CF3 reagent and dehydrating agent [93]. This reaction proceeded via N-trifluoroacetylation of benzylamines followed by dehydrative cyclization to the imidazo-fused polycyclic products in the present of TFAA (Scheme 43).
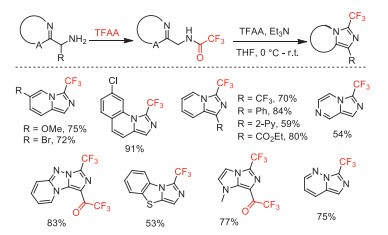
In the same year, our group reported an efficient CuI-catalyzed interrupted click reaction in the presence of TFAA for the synthesis of 3-trifluoromethyl-substituted 1,2,4-triazinones (Scheme 44) [94]. The discovery of this process by using trifluoroacetyl group (CF3CO) from TFAA to accelerate the ring-opening of the 5-Cu(Ⅰ) triazolide species had allowed us to extend the scope of classical copper-catalyzed azide-alkyne cycloaddition (CuAAC) 1,2,3-triazole products to 1,2,4-triazinone derivatives. Under the optimized reaction conditions, a broad range of azides and alkynes with electron-withdrawing and electron-donating groups could participate in this transformation, affording corresponding products in moderate to excellent yields. By finely optimizing reaction conditions, this method allowed facile synthesis of structurally diverse 3-fluoroalkyl-substituted 1,2,4-triazinones with other fluoroalkyl anhydrides, including (C2F5CO)2O, (n-C3F7CO)2O [95], (CF2ClCO)2O, and (CF2HCO)2O [96].
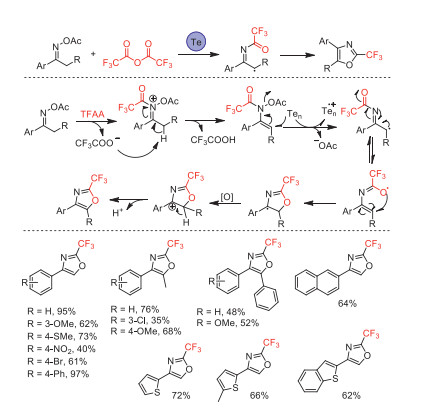
In 2018, our group reported a tellurium-mediated tandem ring-forming reaction for the synthesis of 2-(trifluoromethyl)oxazoles using acetophenone oxime acetates as substrate and TFAA as CF3 source [97]. This new cyclization reaction proceeded in good to excellent yields under extremely simple and easy-to-operate conditions. The reaction mechanism involving a SET reduction followed by a 5-endo-trig tandem cyclization was proposed based on a series of mechanistic control experiments and radical trapping (Scheme 45). The biological activity studies suggested that some of 2-(trifluoromethyl)oxazoles displayed potent fungicidal and insecticidal activities.
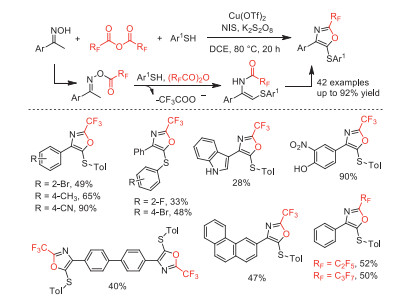
Subsequently, in 2019, Deng and co-workers reported a copper-catalyzed oxidative three-component domino cyclization of oxime, arylthiol, and TFAA to selectively synthesize 4-aryl-5-(arythio)−2-(trifluoromethyl)oxazoles (Scheme 46) [98]. This reaction was proposed to involve N-O bond cleavage, new C-N and C-S bond formation, C-H functionalization, single-electron oxidation and intramolecular 5-endo cyclization.
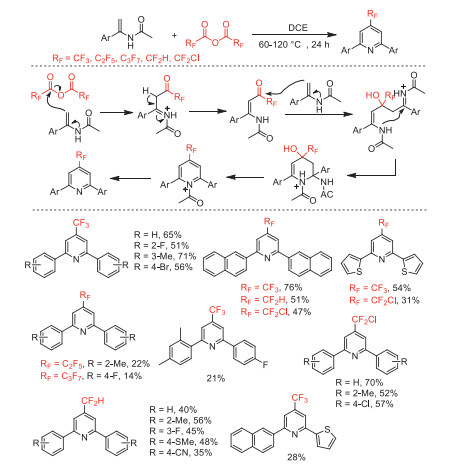
In 2019, our group reported a (RFCO)2O (RF = CF2H, CF2Cl, CF3, C2F5, n-C3F7) triggered cyclization to access 2,6-diaryl-4-fluoroalkylpyridines under metal-free conditions through a double nucleophilic addition, 6-endo cyclization, and subsequent elimination tandem processes (Scheme 47) [99]. This process provides a simple method to construct 4-perfluoroalkylpyridines from easily available and inexpensive starting materials.
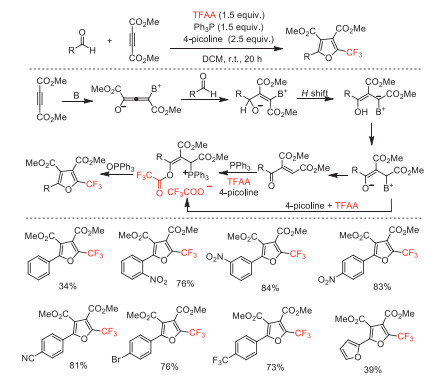
In the same year, Saito and co-workers reported an efficient method to access polysubstituted 2-trifluoromethylfurans in moderate to excellent yields under metal-free conditions [100]. The combination of 4-picoline and Ph3P has been employed to promote a formal [2 + 2 + 1] ring-forming for the synthesis of 2-trifluoromethylfurans from TFAA, aldehydes and alkynoates via Michael-type addition, 1,2-H-shift, 1,4-H-shift and subsequent O-trifluoroacylation of the enone intermediates and intramolecular Wittig reaction cascade procedure (Scheme 48). Compared with the report in 2011 by Xu's group [91], it is clear that an advantage of this method is the direct use of α, β-enone precursors, and a disadvantage is the relatively low yield of the products.
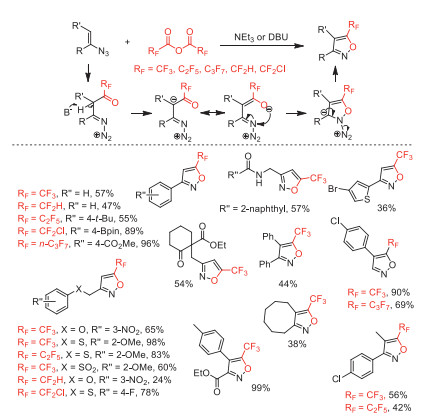
In 2020, our group developed an operationally convenient and efficient denitrogenative cyclization of vinyl azides with (RFCO)2O (RF = CF2H, CF2Cl, CF3, C2F5, n-C3F7) to synthesize structurally diverse 5-(fluoroalkyl)isoxazoles under metal-free and mild conditions [101, 102]. This ring-forming reaction with broad functional group tolerance was involved fluoroalkanoyl of initial substrates, deprotonation and subsequent cyclization, dinitrogen elimination procedures (Scheme 49).
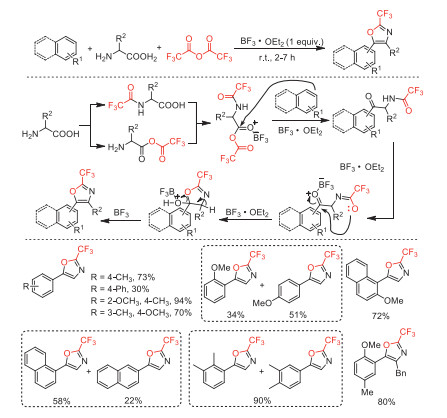
In 2020, Ilangovan and co-worker reported a TFAA-BF3·OEt2 mediated one-pot three-component cyclization of TFAA, aromatics and amino acid to access 2-trifluoromethyl oxazoles under mild conditions [103]. This tandem procedures included amidation (the formation of C-N bond), anhydride formation (the formation of C-O bond), Friedel-Crafts acylation (the formation of C-C bond), Robinson-Gabriel annulation, and subsequently dehydrative aromatization (Scheme 50).
Fluoroalkyl-containing compounds, owing to their unique physicochemical properties, have been allowed to be used across an array of fields. TFAA and its anhydride analogues used as a class of high-efficiency and low-cost fluoroalkyl reagents have attracted great attention for the numerous synthesis of fluoroalkyl-containing compounds. Recently, diversified strategies for high value-added utilization of easily available TFAA and its anhydride analogs have been developed. Herein, the review summarized the advances in the development of synthetic methods of fluoroalkyl-containing compounds using fluoroalkyl anhydrides as fluoroalkyl reagents, including (CF3CO)2O, (CF2HCO)2O, (CF2ClCO)2O, (CnF2n+1CO)2O. We hope that this review can provide a reference for subsequent application and help chemists to develope new methodologies for the synthesis of fluoroalkylated compounds. Despite the tremendous efforts and substantial achievements, some noteworthy issues still exist in this field:
1. More economical methods should be developed. Fluoroalkyl reagents derived from fluoroalkyl anhydrides participated in coupling reactions that mainly focused on the use of bis(fluoroalkanoyl) peroxides and the photocatalytic reactions of noble metal (e.g., Ru, Ir), and only a few examples of cyclization reactions of these reagents were provided. The methods using low-cost metal catalysis and organic catalysis as sustainable processes and the cyclization strategies of fluoroalkyl reagents should be explored.
2. The coupling reactions initiated by fluoroalkanoyl groups mainly focus on fluoroalkanoylation of electron-rich aromatics and olefins. Undoubtedly, expanding the substrate scope will broaden the application of trifluoromethyl ketone compounds and trifluoromethyl-containing building blocks.
3. A library synthesis of fluoroalkylated heterocyclic compounds should be established. The cyclization reaction induced by fluoroalkanoyl groups conveniently constructes structurally diverse heterocyclic compounds in one-pot, which were difficultly synthesized by coupling reactions.
4. The asymmetric synthesis of chiral fluoroalkylated compounds originated from fluoroalkyl anhydrides remains limited. Bsed on the inportance of chiral molecules, fluoroalkyl and fluoroalkanoyl groups, more attention needs to be devoted to this direction.
5. While some progress has been made, kilogram scale utilizing of low-consumption fluoroalkyl anhydrides and late-stage fluoroalkylating of polyfunctional compounds are less reported and still in infancy.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 21772022, and 22171124), Minjiang University (No. MJY21041), and Fuzhou University.
B.E. Smart, J. Fluorine Chem. 109 (2001) 3-11. doi: 10.1016/S0022-1139(01)00375-X
P.K. Mykhailiuk, Chem. Rev. 121 (2021) 1670-1715. doi: 10.1021/acs.chemrev.0c01015
M. Inoue, Y. Sumii, N. Shibata, ACS Omega 5 (2020) 10633-10640. doi: 10.1021/acsomega.0c00830
J.O. Link, M.S. Rhee, W.C. Tse, et al., Nature 584 (2020) 614-618. doi: 10.1038/s41586-020-2443-1
P. Lees, L. Pelligand, J. Elliott, et al., J. Vet. Pharmacol. Ther. 38 (2015) 1-14. doi: 10.1111/jvp.12185
S. Chen, W. Cao, P. Yue, et al., Cancer Res. 71 (2011) 6270. doi: 10.1158/0008-5472.CAN-11-0838
R.F. Chemaly, A.J. Ullmann, S. Stoelben, et al., New Engl. J. Med. 370 (2014) 1781-1789. doi: 10.1056/NEJMoa1309533
L.H. Boal, J. Glod, M. Spencer, et al., Clin. Cancer Res. 26 (2020) 6112. doi: 10.1158/1078-0432.CCR-20-1696
L. Bessiène, F. Bonnet, F. Tenenbaum, et al., Eur. J. Endocrinol. 184 (2021) L13-L15. doi: 10.1530/EJE-21-0147
J. Choi, Y. Wang David, S. Kundu, et al., Science 332 (2011) 1545-1548. doi: 10.1126/science.1200514
F. Leroux, M. Ford, S. Pazenok, Org. Process Res. Dev. 18 (2014) 980-980. doi: 10.1021/op500216s
C. Zhu, M.M. Sun, K. Chen, H. Liu, C. Feng, Angew. Chem. Int. Ed. 60 (2021) 20237-20242. doi: 10.1002/anie.202106531
J.R. Box, A.P. Atkins, A.J.J. Lennox, Chem. Sci. 12 (2021) 10252-10258. doi: 10.1039/D1SC01574E
J. Sang, L. Feng, R. Hu, et al., Org. Lett. 23 (2021) 7666-7671. doi: 10.1021/acs.orglett.1c02932
G.K.S. Prakash, A.K. Yudin, Chem. Rev. 97 (1997) 757-786. doi: 10.1021/cr9408991
X. Liu, C. Xu, M. Wang, Q. Liu, Chem. Rev. 115 (2015) 683-730. doi: 10.1021/cr400473a
Q. Xie, J. Hu, Chin. J. Chem. 38 (2020) 202-212. doi: 10.1002/cjoc.201900424
J. Charpentier, N. Früh, A. Togni, Chem. Rev. 115 (2015) 650-682. doi: 10.1021/cr500223h
T. Umemoto, S. Ishihara, J. Am. Chem. Soc. 115 (1993) 2156-2164. doi: 10.1021/ja00059a009
X.H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115 (2015) 731-764. doi: 10.1021/cr500193b
C. Ni, M. Hu, J. Hu, Chem. Rev. 115 (2015) 765-825. doi: 10.1021/cr5002386
H. Morimoto, T. Tsubogo, N.D. Litvinas, J.F. Hartwig, Angew. Chem. Int. Ed. 50 (2011) 3793-3798. doi: 10.1002/anie.201100633
A. Zanardi, M.A. Novikov, E. Martin, J. Benet-Buchholz, V.V. Grushin, J. Am. Chem. Soc. 133 (2011) 20901-20913. doi: 10.1021/ja2081026
Y. Ji, T. Brueckl, R.D. Baxter, et al., Proc. Natl. Acad. Sci. 108 (2011) 14411-14415. doi: 10.1073/pnas.1109059108
M. Tordeux, B. Langlois, C. Wakselman, J. Org. Chem. 54 (1989) 2452-2453. doi: 10.1021/jo00271a041
X. Lin, C. Hou, H. Li, Z. Weng, Chem. Eur. J. 22 (2016) 2075-2084. doi: 10.1002/chem.201504306
Y. Huang, M.J. Ajitha, K.W. Huang, Z. Zhang, Z. Weng, Dalton Trans. 45 (2016) 8468-8474. doi: 10.1039/C6DT00277C
Y.J. Chen, F.G. Zhang, J.A. Ma, Org. Lett. 23 (2021) 6062-6066. doi: 10.1021/acs.orglett.1c02139
D. Shen, J. Han, J. Chen, et al., Org. Lett. 17 (2015) 3283-3285. doi: 10.1021/acs.orglett.5b01479
Z. Yuan, S. Chen, Z. Weng, Org. Chem. Front. 7 (2020) 482-486. doi: 10.1039/C9QO01131E
W. Wu, B. Luo, Y. You, Z. Weng, Org. Chem. Front. 8 (2021) 1997-2001. doi: 10.1039/D1QO00157D
H.C. Lo, H. Han, L.J. D'Souza, S.C. Sinha, E. Keinan, J. Am. Chem. Soc. 129 (2007) 1246-1253. doi: 10.1021/ja0668668
P. Nussbaumer, K. Baumann, T. Dechat, M. Harasek, Tetrahedron 47 (1991) 4591-4602. doi: 10.1016/S0040-4020(01)86465-6
M. Mitani, H. Sawada, M. Nakayama, M. Yoshida, N. Kamigata, Polym. J. 22 (1990) 623-627. doi: 10.1295/polymj.22.623
Y. Hayakawa, N. Terasawa, H. Sawada, Polymer (Guildf) 42 (2001) 4081-4086. doi: 10.1016/S0032-3861(00)00673-X
C. Zhao, R. Zhou, H. Pan, et al., J. Org. Chem. 47 (1982) 2009-2013. doi: 10.1021/jo00132a007
M. Yoshida, K. Moriya, H. Sawada, M. Kobayashi, Chem. Lett. 14 (1985) 755-758. doi: 10.1246/cl.1985.755
H. Sawada, M. Yoshida, H. Hagii, K. Aoshima, M. Kobayashi, Bull. Chem. Soc. Jpn. 59 (1986) 215-219. doi: 10.1246/bcsj.59.215
M. Yoshida, T. Yoshida, N. Kamigata, M. Kobayashi, Bull. Chem. Soc. Jpn. 61 (1988) 3549-3552. doi: 10.1246/bcsj.61.3549
H. Sawada, M. Nakayama, M. Yoshida, T. Yoshida, N. Kamigata, J. Fluorine Chem. 46 (1990) 423-431. doi: 10.1016/S0022-1139(00)82927-9
M. Yoshida, N. Kamigata, H. Sawada, M. Nakayama, J. Fluorine Chem. 49 (1990) 1-20. doi: 10.1016/S0022-1139(00)80359-0
M. Yoshida, T. Yoshida, M. Kobayashi, N. Kamigata, J. Chem. Soc., Perkin Trans. 1 (1989) 909-914.
M. Yoshida, Y. Morinaga, M. Ueda, N. Kamigata, M. Iyoda, Chem. Lett. 21 (1992) 227-230. doi: 10.1246/cl.1992.227
M. Yoshida, Y. Morinaga, M. Iyoda, J. Fluorine Chem. 68 (1994) 33-38. doi: 10.1016/0022-1139(93)02985-N
M. Matsui, K. Shibata, H. Muramatsu, H. Sawada, M. Nakayama, Chem. Ber. 125 (1992) 467-471. doi: 10.1002/cber.19921250226
M. Matsui, H. Ishikawa, K. Hiramatsu, K. Shibata, H. Muramatsu, Dyes Pigments 27 (1995) 143-151. doi: 10.1016/0143-7208(94)00049-8
M. Matsui, S. Kawamura, K. Shibata, et al., J. Fluorine Chem. 57 (1992) 209-217. doi: 10.1016/S0022-1139(00)82833-X
M. Matsui, K. Shibata, H. Muramatsu, H. Sawada, M. Nakayama, Synlett 1991 (1991) 113-114. doi: 10.1055/s-1991-20647
S. Zhong, A. Hafner, C. Hussal, M. Nieger, S. Bräse, RSC Adv. 5 (2015) 6255-6258. doi: 10.1039/C4RA13430C
S. Kawamura, M. Sodeoka, Angew. Chem. Int. Ed. 55 (2016) 8740-8743. doi: 10.1002/anie.201604127
S. Kawamura, C.J. Henderson, Y. Aoki, et al., Chem. Commun. 54 (2018) 11276-11279. doi: 10.1039/C8CC05905E
E. Valverde, S. Kawamura, D. Sekine, M. Sodeoka, Chem. Sci. 9 (2018) 7115-7121. doi: 10.1039/C8SC02547A
A. Maleckis, M.S. Sanford, Organometallics 33 (2014) 2653-2660. doi: 10.1021/om500398z
A. Maleckis, M.S. Sanford, Organometallics 33 (2014) 3831-3839. doi: 10.1021/om500535x
J.W. Beatty, J.J. Douglas, K.P. Cole, C.R.J. Stephenson, Nat. Commun. 6 (2015) 7919. doi: 10.1038/ncomms8919
Joel W. Beatty, James J. Douglas, R. Miller, et al., Chem 1 (2016) 456-472. doi: 10.1016/j.chempr.2016.08.002
O. Reiser, Chem 1 (2016) 344-345. doi: 10.1016/j.chempr.2016.08.005
R.C. McAtee, J.W. Beatty, C.C. McAtee, C.R.J. Stephenson, Org. Lett. 20 (2018) 3491-3495. doi: 10.1021/acs.orglett.8b01249
X. Zhang, Y. Li, X. Hao, et al., Tetrahedron 74 (2018) 1742-1748. doi: 10.1016/j.tet.2018.01.040
S. Das, A.S.K. Hashmi, T. Schaub, Adv. Synth. Catal. 361 (2019) 720-724. doi: 10.1002/adsc.201801305
P. Garg, A. Singh, Asian J. Org. Chem. 8 (2019) 849-852. doi: 10.1002/ajoc.201900181
E.J. McClain, T.M. Monos, M. Mori, J.W. Beatty, C.R.J. Stephenson, ACS Catal. 10 (2020) 12636-12641. doi: 10.1021/acscatal.0c03837
W. Wu, Z. Weng, Synthesis 50 (2018) 1958-1964. doi: 10.1055/s-0036-1591971
F.A.J. Kerdesky, A. Basha, Tetrahedron Lett. 32 (1991) 2003-2004. doi: 10.1016/S0040-4039(00)78892-7
J. Boivin, L.E. Kaim, S.Z. Zard, Tetrahedron Lett. 33 (1992) 1285-1288. doi: 10.1016/S0040-4039(00)91602-2
J. Boivin, L.E. Kaim, S.Z. Zard, Tetrahedron 51 (1995) 2573-2584. doi: 10.1016/0040-4020(95)00006-T
J. Boivin, L. El Kaim, S.Z. Zard, Tetrahedron 51 (1995) 2585-2592. doi: 10.1016/0040-4020(95)00007-U
M. Kawase, T. Kurihara, Tetrahedron Lett. 35 (1994) 8209-8212. doi: 10.1016/0040-4039(94)88284-3
V.G. Nenajdenko, I.D. Gridnev, E.S. Balenkova, Tetrahedron 50 (1994) 11023-11038. doi: 10.1016/S0040-4020(01)85712-4
G. Simchen, A. Schmidt, Synthesis (Mass) 1996 (1996) 1093-1094. doi: 10.1055/s-1996-4444
J. Ruiz, D. Astruc, L. Gilbert, Tetrahedron Lett. 37 (1996) 4511-4514. doi: 10.1016/0040-4039(96)00870-2
J.R. Goerlich, V. Plack, R. Schmutzler, J. Fluorine Chem. 76 (1996) 29-35. doi: 10.1016/0022-1139(95)03288-6
J.T. Reeves, F. Gallou, J.J. Song, et al., Tetrahedron Lett. 48 (2007) 189-192. doi: 10.1016/j.tetlet.2006.11.068
W.W. Ong, A.B. Beeler, S. Kesavan, J.S. Panek, J.A. Porco Jr, Angew. Chem. Int. Ed. 46 (2007) 7470-7472. doi: 10.1002/anie.200700694
H. Kinoshita, O.J. Ingham, W.W. Ong, A.B. Beeler, J.A. Porco, J. Am. Chem. Soc. 132 (2010) 6412-6418. doi: 10.1021/ja100346w
B.L. Kohn, T. Rovis, Chem. Sci. 5 (2014) 2889-2892. doi: 10.1039/C4SC00743C
P. Kumari, N. Sharma, A. Kumar, S.C. Mohapatra, S. Bhagat, Synlett 28 (2017) 2008-2013. doi: 10.1055/s-0036-1588865
Z. Wang, Z. Yuan, X. Han, Z. Weng, Adv. Synth. Catal. 360 (2018) 2178-2182. doi: 10.1002/adsc.201800275
Y. Chen, Y. You, Z. Weng, Org. Chem. Front. 6 (2019) 213-217. doi: 10.1039/C8QO01118D
S.L. Schreiber, Tetrahedron Lett. 21 (1980) 1027-1030. doi: 10.1016/S0040-4039(00)78830-7
G.P. Mrug, I.M. Biletska, S.P. Bondarenko, V.M. Sviripa, M.S. Frasinyuk, ChemistrySelect 4 (2019) 11506-11510. doi: 10.1002/slct.201903629
J. Wang, Z. Weng, Eur. J. Org. Chem. 2019 (2019) 1330-1334. doi: 10.1002/ejoc.201801662
J. Drabowicz, A. Kotyński, Z.H. Kudzin, R.B. Nazarski, M.K. Tasz, Tetrahedron 53 (1997) 14169-14178. doi: 10.1016/S0040-4020(97)00918-6
K. Zhang, D. Rombach, N.Y. Nötel, G. Jeschke, D. Katayev, Angew. Chem. Int. Ed. 60 (2021) 22487-22495. doi: 10.1002/anie.202109235
S. Kawamura, K. Dosei, E. Valverde, K. Ushida, M. Sodeoka, J. Org. Chem. 82 (2017) 12539-12553. doi: 10.1021/acs.joc.7b02307
X.K. Qi, H. Zhang, Z.T. Pan, et al., Chem. Commun. 55 (2019) 10848-10851. doi: 10.1039/C9CC04977K
J. Bergman, G. Lidgren, Tetrahedron Lett. 30 (1989) 4597-4600. doi: 10.1016/S0040-4039(01)80754-1
S.G. Hegde, C.R. Jones, J. Heterocycl. Chem. 30 (1993) 1501-1508. doi: 10.1002/jhet.5570300607
L.I. Vereshchagin, O.N. Verkhozina, F.A. Pokatilov, et al., Russ. J. Org. Chem. 43 (2007) 1710-1714. doi: 10.1134/S1070428007110218
V.O. Iaroshenko, S. Ali, T.M. Babar, et al., Tetrahedron Lett. 52 (2011) 373-376. doi: 10.1016/j.tetlet.2010.11.028
Y. Wang, Y.C. Luo, X.Q. Hu, P.F. Xu, Org. Lett. 13 (2011) 5346-5349. doi: 10.1021/ol2022092
D.D. Tang, Y. Wang, J.R. Wang, P.F. Xu, Tetrahedron Lett. 55 (2014) 4133-4135. doi: 10.1016/j.tetlet.2014.05.127
G. Schäfer, M. Ahmetovic, S. Abele, Org. Lett. 19 (2017) 6578-6581. doi: 10.1021/acs.orglett.7b03291
W. Wu, J. Wang, Y. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 10476-10480. doi: 10.1002/anie.201705620
B. Lin, W. Wu, Z. Weng, Tetrahedron 75 (2019) 2843-2847. doi: 10.1016/j.tet.2019.04.005
W. Wu, C. Xu, Z. Weng, J. Fluorine Chem. 226 (2019) 109359. doi: 10.1016/j.jfluchem.2019.109359
B. Luo, Z. Weng, Chem. Commun. 54 (2018) 10750-10753. doi: 10.1039/C8CC05670F
F. Xiao, S. Yuan, H. Huang, F. Zhang, G.J. Deng, Org. Lett. 21 (2019) 8533-8536. doi: 10.1021/acs.orglett.9b02934
Z. Wang, C. You, C. Wang, Z. Weng, J. Org. Chem. 84 (2019) 14926-14935. doi: 10.1021/acs.joc.9b02254
K. Tateishi, Y. Matsumoto, A. Saito, Eur. J. Org. Chem. 2019 (2019) 5603-5609. doi: 10.1002/ejoc.201900806
W. Wu, Q. Chen, Y. Tian, et al., Org. Chem. Front. 7 (2020) 1878-1883. doi: 10.1039/D0QO00243G
J. Lin, W. Wu, Z. Weng, Tetrahedron Lett. 61 (2020) 152672. doi: 10.1016/j.tetlet.2020.152672
V. Karuppusamy, A. Ilangovan, Org. Lett. 22 (2020) 7147-7151. doi: 10.1021/acs.orglett.0c02484

Scheme 4 Perfluoroalkylation of coumarins, azo compounds, quinolinones and phenoxazones.

Scheme 6 Plausible mechanism and substrate scope for the perfluoroalkylation and the intramolecular carboperfluoroalkylation.

Scheme 8 Proposed mechanisms and substrate scope of the oxy- and amino-perfluoroalkylations.

Scheme 11 Mechanistic proposal and substrate scope for Ru-catalyzed perfluoroalkylation of (hetero)arenes.

Scheme 12 Proposed mechanism and substrate scope of perfluoroalkylation of (hetero)arenes.

Scheme 15 Proposed mechanism and substrate scope for the photoassisted α-trifluoromethylation of acetophenones with TFAA.

Scheme 16 Proposed mechanism and substrate scope of radical trifluoromethylation of enol acetates.

Scheme 17 Plausible mechanism for the trifluoromethylation with EDA complex catalysis.

Scheme 19 The synthesis of trifluoromethyl ketones and their trifluoromethylated heterocycles from TFAA.

Scheme 20 Proposed mechanism and substrate scope of the transformation of α–hydroxy acids to α-perfluoroalkylated acyloins.

Scheme 22 The synthesis and application of 4-dimethylamino-1-trifluoroacetylpyridinium trifluoroacetate.

Scheme 23 The comparatively CoCl2-catalyzed trifluoroacetylation and dimerization of methoxyaromatics.

Scheme 25 The one-pot conversion of primary and secondary carboxylic acids to trifluoromethyl ketones.

Scheme 26 Proposed mechanism and substrate scope for asymmetric crotylation of N-phosphinylimines.

Scheme 27 Proposed mechanism and substrate scope for La-catalyzed nucleophilic addition of N-diphenylphosphinylimines.

Scheme 28 Proposed mechanism and substrate scope for the cobalt-mediated direct fluoroacylation of 1,3-dienes.

Scheme 30 Proposed mechanism and substrate scope for the formation of 1–chloro-2,2,2-trifluoroethylidene-substituted pyrrolidones.

Scheme 31 Proposed mechanism and substrate scope of the Knoevenagel-type reaction of 1,3-diketones.

Scheme 32 Reaction procedure and substrate scope of the trifluoroacetylation of 2-methyl-and 2-ethylchromones.

Scheme 33 Proposed reaction pathway and substrate scope for the synthsis of N-trifluoroacetyl amides.

Scheme 34 Proposed mechanism and substrate scope of the alkene radical trifluoroacylation and trifluoromethylation.

Scheme 35 Proposed mechanism and substrate scope of the amino- and carboperfluoroalkylations.

Scheme 36 Proposed reaction pathway and substrate scope for the synthsis of fluoroalkylated dibenz[b, e]azepines.

Scheme 40 The reaction pathway and substrate scope for the synthesis of 7-(trifluoromethyl)−6H-chromeno[4,3-b]quinolin-6-ones.

Scheme 41 Possible mechanistic pathways and substrate scope for the synthesis of 2-trifluoromethylfurans.

Scheme 42 Possible reaction pathways and substrate scope of PPh3-mediated cascade cyclization reaction.

Scheme 43 The reaction precedure and substrate scope of trifluoromethylated imidazo-fused N-heterocycles.

Scheme 44 Possible mechanistic pathways and substrate scope for the coppercatalyzed synthesis of trifluoromethyl 1,2,4-triazinones.

Scheme 45 Possible mechanistic pathways and substrate scope of the telluriummediated tandem ring-forming reaction.

Scheme 46 Tentative reaction mechanism and substrate scope of copper-catalyzed oxidative three-component domino cyclization.

Scheme 47 Possible mechanistic processes and substrate scope for the synthesis of 4-perfluoroalkylpyridines.

Scheme 48 Possible mechanistic processes and substrate scope for the synthesis of 2-trifluoromethylfurans.

Scheme 49 Possible mechanistic processes and substrate scope for the synthesis of 5-(fluoroalkyl)isoxazoles.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

















 下载:
下载: