Received Date:
27 September 2021 Accepted Date:
26 October 2021 Revised Date:
25 October 2021 Available Online:
15 April 2022
Abstract:
Platinum-based anticancer agents such as cisplatin and its analogues are widely used for treating multiple cancers. However, due to the inferior water-solubility, chemoresistance and consequent adverse side effects, their clinical applications are limited. Herein, cholesPt(IV), a lipophilic platinum(IV) prodrug was synthesized for manufacture of CholesPt(IV)-Liposomes aiming to resolve the predefined obstacles encountered by platinum drugs. Following systematic screening, CholesPt(IV)-Liposomes showed a small particle size (105.6 nm), the rapid release of platinum (Pt) ions, and notable apoptosis of cancer cells. In addition, according to the fluidity and safety results of animal experiments in mice, CholesPt(IV)-Liposomes also showed better therapeutic effect, which significantly inhibited the growth of patient-derived xenograft tumors of hepatocellular carcinoma with an inhibition ratio of 80.7%, and effectively alleviated the drug toxicity brought by traditional platinum drugs. Overall, this study provides a promising route to enhance the therapeutic efficiency of platinum drugs in cancer treatment.
Recent years, cancer has become a formidable medical hot issue [1, 2]. Platinum-based anticancer agents such as cisplatin and its analogues are widely used to treat multiple cancers [3-7]. However, the inferior water-solubility, chemoresistance and consequent adverse side effects limit their clinical applications [8–10]. Therefore, it is of great clinical significance to develop a novel strategy to improve platinum efficiency and reduce side effects.
Nanotechnology-based drug delivery systems have been shown to improve the therapeutic efficiency of chemotherapeutics and reduce system toxicity due to their enhanced permeability and retention effect, which are emerging as the next-generation anticancer agents [11–20]. As a tempting modality of nanomedicine, liposomes characterized with spherical nano-scaled vesicles from phospholipids (and other lipids), have been intensively explored as drug delivery vehicles with respect to its excellent biocompatibility, capable of avoiding detection by the body's immune system, specifically, the cells of reticuloendothelial system [21–23]. Besides, liposomes are available to facile post chemistry functionalization, such as PEG surface coating (inert in the body) onto liposomes, which entitle prolonged blood retention for improved bioavailability to the targeted tumor [24–26]. For example, He and co-workers reported that non-PEGylated formulations were quickly cleared, while non-PEGylated prodrug nanoparticles (PTX-S-S-OA/TPGS2k NPs) exhibited persistently high concentrations of PTX in plasma by evading the identification and phagocytosis of reticulo-endothelial system (RES) [27]. Nevertheless, the poor molecular affinity of platinum drugs to the lipids imposes difficulty to direct incorporation of platinum drugs into the liposomes. To tackle this drawback, we attempted to couple the lipophilic cholesterol to the platinum drugs. Of note, pioneering research advocated the importance of construction of appropriate Pt(IV) prodrugs as rationale alternative to Pt(II) [28–33]. In contrast to Pt(II) susceptible to ligand substitution in the extracellular milieu to induce detoxification of Pt(II), the Pt(IV) prodrugs haven been elucidated to impart improved inertness to ligand substitution, consequently lower the extent of platinum sequestration and deactivation en route to the tumor cells. In this respect, we are encouraged to engineer a cisPt(IV) lipophilic prodrug by attaching cisPt(IV) at the terminus of cholesterol [referred as cholesPt(IV) hereafter] for manufacture of liposomes.
Herein, cholesPt(IV) was synthesized according to a ligation reaction between cisPt(IV) and cholesterol (Scheme S1 in Supporting information). The yielded product was confirmed by exhaustive chemistry characterizations including FT-IR, 1H NMR, ESI-MS, HPLC and elemental analysis (Figs. S1–S4 and Table S1 in Supporting information). The obtained cholesPt(IV) molecule was used as supplementary component to integrate with phospholipids of HSPC and PEGylated lipid for preparation of liposome (Fig. 1a). A film hydration and membrane extrusion method was adopted to create liposome [34]. Aiming for identification of an optimal condition for construction of cholesPt(IV)-functionalized liposomes, the liposome was prepared from constant quantity of phospholipids of HSPC and PEGylated lipid, but varied quantities of cholesPt(IV). The feed ratio of cholesPt(IV) and lipids was varied from 0.2:1 to 1:1. The integration efficiency of cholesPt(IV) into liposomes was quantified by ICP-MS measurement (Fig. 1b). Apparently, high integration percentage of cholesPt(IV) could be achieved at relative low feed ratio of cholesPt(IV)/lipids. The total integration quantity calculated by the quantified integration percentage and the total feed quantity of cholesPt(IV), led to identification of an optimal formulation at cholesPt(IV)/lipid feed ratio of 0.4:1 to possess maximal cholesPt(IV) integration.
Figure 1
Figure 1.
Schematic illustration and characterizations of cholesPt(IV)-functionalized liposome. Liposomes integrated by cholesPt(IV) were prepared by sequential film hydration and extrusion approach. (a) Anchor of cholesPt(IV) into the lipid bilayer of liposome to yield CholesPt(IV)-Liposome. (b) Integration percentage of cholesPt(IV) as a function of varying feed ratios of cholesPt(IV)/lipids. (c) Representative DLS result of cholesPt(IV) integrated liposome at the identified feed ratio of cholesPt(IV)/lipid of 0.4:1. (d) Colloidal stability of CholesPt(IV)-Liposome (at the identified feed ratio of cholesPt(IV)/lipid ratio of 0.4:1). The size derivation of the formulated liposome was monitored by DLS measurement.
The formulated structures were further transferred to dynamic light scattering (DLS) and zeta potential measurement. No marked difference in the size and surface charge was observed for the class of formulated liposomes irrespective of feed ratio of cholesPt(IV)/lipid, as evidenced by comparable diameters (ranging from 100 nm to 140 nm) and zeta potentials (ranging from −22 mV to −27 mV) (Table S2 in Supporting information). The identified liposome at cholesPt(IV)/lipid feed ratio of 0.4:1 was determined to possess an average diameter of 105.6 nm with unimodal polydispersity index of 0.07 by DLS measurement (Fig. 1c). TEM of the liposome (0.4:1) also showed the similar formation as DLS (Fig. S5 in Supporting information). Furthermore, excellent colloidal stability of the formulated liposome was confirmed from the observation of constant DLS size over extended incubation (over 10 days) in PBS (Fig. 1d).
Prior to the drug potency test, it is important to demonstrate the proposed CholesPt(IV)-Liposome capable of releasing the active Pt(II) drugs inside the cells. Here, we evaluated the drug release rate from liposomes under diverse stimuli (presented inside the cells). Given that the cell interior is characterized as a distinctive reductive compartment (abundant existence of glutathione and ascorbic acids), the conversion of Pt(IV) derivatives to Pt(II) is presumed to be facilitated by means of redox-triggered release of two ligands and regeneration of the active Pt(II). In addition, nano-scaled structures are envisaged to internalize the cell through endocytosis pathway, followed by transfer to acidic endosome [35–37]. In view of readily hydrolysis of Pt(IV) particularly in acidic milieu, it could be conjectured that the release of drug from liposome is facilitated in the subcellular endosome compartment. The subsequent investigations approved our speculation that both reductive treatment (ascorbate acid resembling the cell interior) and acidic treatment (pH 5 resembling the endosome pH) could stimulate the drug release. As opposed to negligible drug release at pH 7.4, liposome incubated at pH 5 exhibited markedly accelerated drug release profile. To our interest, liposome under treatment of ascorbate (10 mmol/L) exhibited most appreciable drug release profile (Fig. S6 in Supporting information). These results approved appealing characters of the proposed liposome that managed to preserve the drug payload in the formulation in the extracellular milieu but allow selective intracellular drug release of drug, which apparently is favorable in pursuit of minimized non-specific toxicity and premature drug detoxification.
To validate the feasibility of the proposed CholesPt(IV)-Liposome in circumventing the drug resistance, a comparative study was conducted on the drug potency of diverse platinum derivatives to two types of human ovarian cancer cells, A2780 (cisplatin susceptible cell line) and A2780 DDP (cisplatin resistant cell line). Cisplatin and cisPt(IV) were included as controls [cholesPt(IV) was not included in this study due to its poor solubility). Herein, the cells were treated by platinum drug derivatives at varying concentration for 72 h incubation (Figs. 2a and b). The drug potency was assessed by measuring the cell viability relative to the control blank sample. Pertaining to the cisplatin-susceptible cell line of A2780, cisplatin was observed to exert most pronounced potency (IC50: 2.7 µmol/L), whose IC50 is markedly lower than that of cisPt(IV) (IC50: 20.7 µmol/L) and CholesPt(IV)-Liposome (IC50: 10.2 µmol/L) (Fig. 2c). A plausible reason for the high potency of cisplatin [Pt(II)] over Pt(IV) derivatives should be attributed to the chemistry of Pt(IV) derivatives requiring additional step for activation of Pt(IV). Noteworthy was the significantly elevated drug potency of CholesPt(IV)-Liposome as compared to cisPt(IV). In respect to poor affinity of cisPt(IV) to the lipophilic cell membrane, most likely, the increased drug potency of CholesPt(IV)-Liposome could be accredited to its superior affinity to the cell membrane for promoted cellular uptake. The subsequent quantification of intracellular platinum by ICP-MS for the samples of cell lysis affirmed promoted internalization of CholesPt(IV)-Liposome as compared to cisPt(IV) (Fig. 3a).
Figure 2
Figure 2.In vitro toxicity of diverse platinum formulations of cisPt(IV), cisplatin and CholesPt(IV)-Liposome in A2780 and A2780 DDP cells. Cell viability of A2780 (a) and A2780 DDP (b) under treatment of cisPt(IV), cisplatin and cholesPt-NPs for 72 h. The IC50 of cisPt(IV), cisplatin and CholesPt(IV)-Liposome on A2780 and A2780 DDP cells at 72 h were shown in (c). (d) The drug resistance fold of each platinum drug formulation was calculated by the ratio of IC50 at A2780 DDP to IC50 at A2780.
Figure 3.
Impact of drug resistance on cellular uptake of diverse platinum derivatives. Quantification of intracellular platinum in A2780 (a) and A2780 DDP (b) after 2 and 6 h drug incubation with cisplatin, cisPt(IV) and CholesPt(IV)-Liposome. The uptake-reducing index was summarized in (c). The uptake-reducing was defined as the ratio of platinum uptake at A2780 to platinum uptake at A2780 DDP. (d) Cellular uptake activity of CholesPt(IV)-Liposome at 37 and 4 ℃.
Pertaining to cisplatin-resistant cell line of A2780 DDP, CholesPt(IV)-Liposome mediated notable potency, possessing an IC50 of 12.6 µmol/L even exceeding the potency of cisplatin (IC50 18.6 µmol/L) (Fig. 2c). Close observation of the IC50 values of cisplatin in A2780 and A2780 DDP identified the drug resistance characters of A2780 DDP substantially curtailed the drug potency of cisplatin, whose IC50 was subjected to pronounced rise from 2.7 µmol/L in A2780 to 18.6 µmol/L in A2780 DDP (Fig. 2c). The reduced drug potency of cisplatin may be attributable to the characteristic impeded drug internalization implicated in the drug resist cells. Of note, cisplatin is elucidated to translocate to the cell interior either through passive diffusion or active transportation with aids of cell membrane protein (copper transporter 1:Ctr1) [38]. Molecular insight identified the reduced expression of Ctr1 protein in the platinum-resistant cells, which hampers the efficiency of the Ctr1-dependent cisplatin internalization. In accordance, quantification of intracellular platinum affirmed remarkably reduced internalization of cisplatin [also cisPt(IV)] at drug-resistant cell line. Particular noteworthy was reduced intracellular cisplatin at 6 h post incubation as compared to that at 2 h (Figs. 3a–c), implying the critical impact of platinum-resistant cells, not only feasible of impeding internalization but also capable of prompting efflux of molecular platinum drugs. On the contrary, nanoparticles have been documented to steer an alternative endocytosis pathway rather than Ctr1-mediated transportation. Hence, the cell internalization of CholesPt(IV)-Liposome is envisioned to be unperturbed by the reduced expression of Ctr1 in the drug resistant cells, thereby capable of overcoming the impeded cellular uptake presented by drug resistant cells. Subsequent investigations approved CholesPt(IV)-Liposome capable of overcoming drug resistance, as evidenced by comparable drug potency of CholesPt(IV)-Liposome to A2780 and A2780 DDP, approximate 9.7 µmol/L for A2780 and 11.4 µmol/L for A2780 DDP, respectively (Figs. 2c and d). In accordance to comparable IC50 value of CholesPt(IV)-Liposome in A2780 and A2780 DDP, CholesPt(IV)-Liposome mediated similar level of cell uptake efficiency, which is in stark contrast to aforementioned cisplatin whose cell uptake efficiency was substantially lowered. These results suggest CholesPt(IV)-Liposome adopted an alternative pathway for cell internalization. CLSM observation for intracellular distribution of internalized CholesPt(IV)-Liposome captured colocalization of CholesPt(IV)-Liposome and endosome, confirmed the proposed CholesPt(IV)-Liposome internalized cell through endocytosis (Fig. S7 in Supporting information). Still, marked reduced cell uptake of CholesPt(IV)-Liposome at 4 ℃ as compared to 37 ℃ (Fig. 3d), again affirmed CholesPt(IV)-Liposome internalized cells through an energy-dependent endocytosis pathway. Moreover, as opposed to aforementioned distinctive efflux character of A2780 DDP for cisplatin, the intracellular platinum was observed to progressive increase (Fig. 3b), suggesting utilization of liposome by means of integration of lipophilic cholesPt(IV) prodrug in liposome could address the impeded cell internalization drawbacks of the molecular drugs encountered in drug resistant cells.
For a quantitative evaluation of the capacity of proposed system in overcoming the drug resistance (impeded cellular uptake), the uptake-reducing index was calculated by quantification of the ratios of diverse platinum derivatives uptake level at A2780 to the uptake level at A2780 DDP. Higher value of uptake-reducing index stands for the crucial impact of drug resistance on impeding drug internalization. In agreement of drug potency evaluation, cisplatin showed highest uptake-resistance index (Fig. 2d), suggesting its drug potency was markedly dwindled by the characters of drug resistant A2780. The molecular drug of cisPt(IV) also showed considerable high uptake resistance index, consistent with the previous claim that the drug resistance is versatile to a wide spectrum of molecular drugs. On the contrary, the uptake reducing index of CholesPt(IV)-Liposome is in proximity of 1, which approved appreciable strategy of manufacture of liposome as drug delivery vehicle to manage the impeded cell uptake schemed by drug resistant cells.
Similarly, based on IC50 values of diverse platinum drugs for A2780 and A2780 DDP (Fig. 2c), the drug resistance index [defined by the ratio of IC50 obtained for platinum-resistance cell line (A2780 DDP) to the IC50 obtained for platinum-susceptible cell line (A2780)] (Fig. 2d) was calculated to characterize the capacity of diverse platinum drugs in overcoming drug resistance. In principal, lower drug resistance index, particularly approximate to 1 (similar drug potency to platinum-susceptible cell line and platinum-resistant cell line), stands for higher capacity in overcoming the drug resistance. Overall, distinctive drug resistance was observed for cisplatin, as evidenced by high drug resistance index of 5 (Fig. 2d). Considerable drug resistance was also observed for Pt(IV), despite reduced drug resistance index of 2.3 (Fig. 2d). To our interest, CholesPt(IV)-Liposome afforded appreciable capacity in overcoming the drug resistance, whose drug resistance index is 1.2 (Fig. 2d).
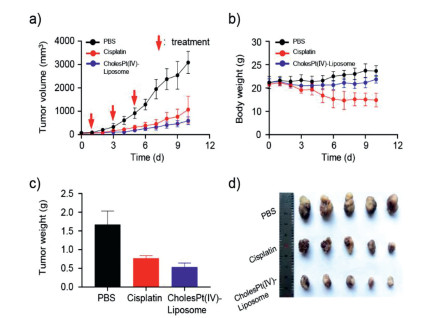
CholesPt(IV)-Liposome displayed stronger cytotoxicity than cisplatin and platinum(IV) at the same concentration (Fig. S8 in Supporting information). Inspired by the results of cytotoxicity in vitro, the antitumor effects of CholesPt(IV)-Liposome were further investigated in vivo. We established a patient-derived xenograft (PDX) model of hepatocellular carcinoma (HCC) on BALB/c nude mice to reproduce the histological and microenvironmental features of tumor. Although cisplatin showed significant inhibition of tumor growth, the CholesPt(IV)-Liposome exhibited the best antitumor efficacy, with an inhibitory rate of 80.7% compared to the saline group (Figs. 4a and d). Notably, the mice treated with cisplatin were observed apparent toxic side effects. In detail, the reduced weights of the mice failed to return to the normal level at the end of treatment. The body weights of CholesPt(IV)-Liposome-treated mice, however, were no apparent changes (Fig. 4b). Mice treated with CholesPt(IV)-Liposome had smaller tumors (0.53 ± 0.11 g), which was 1/3 that of tumors from mice received PBS treatment (1.67 ± 0.36 g) (Fig. 4c). These results indicate that CholesPt(IV)-Liposome had stronger antitumor efficacy but with lower systemic toxicity.
Figure 4
Figure 4.In vivo antitumor effects of CholesPt(IV)-Liposome. (a) Tumor growth curves of HCC PDX-bearing mice during the treatment process. (b) Body weight curves of mice. Weight (c) and image (d) of the tumors harvested from mice on day 10.
***P < 0.001.
Hematoxylin and eosin (H & E), TdT-mediated dUTP Nick-End Labeling (TUNEL)-staining, and immunofluorescence analysis of tumor tissues results revealed that CholesPt(IV)-Liposome could cause more necrosis and apoptosis, as well as damaged DNA strands (Fig. 5a), which demonstrated that CholesPt(IV)-Liposome had superior antitumor effects. In addition, there was no obvious histological toxicity in main organs (heart, liver, spleen, lung, and kidney) and blood biochemistry, which indicated the excellent biocompatibility of CholesPt(IV)-Liposome (Fig. 5b and Fig. S9 in Supporting information).
Figure 5
Figure 5.
Therapeutic efficacy and biocompatibility evaluation of CholesPt(IV)- Liposome on HCC PDX models. (a) H&E staining, immunohistochemistry of TUNEL (brown), and γ -H2A.X (green) analyses of tumor sections. Scale bar: 50 μm. (b) Biochemical analysis of serum after the treatments on 10 d.
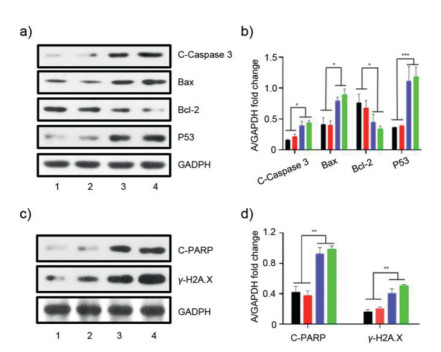
We further explored the antitumor mechanism of CholesPt(IV)-Liposome by western blotting. Compared with cisplatin or platinum(IV), the CholesPt(IV)-Liposome-treated cells exhibited marked up-regulation of apoptosis-related proteins, such as cleaved caspase 3 (abbreviated as C-Caspase 3), P53, Bax, as well as decreased anti-apoptosis protein Bcl-2 (Figs. 6a and b). Compared with cisplatin and platinum(IV), the CholesPt(IV)-Liposome could lead to increased expression of cleaved PARP (abbreviated as C-PARP) and γ-H2A.X, which suggested that CholesPt(IV)-Liposome could effectively cause DNA damages (Figs. 6c and d).
Figure 6
Figure 6.
Antitumor mechanism investigation of CholesPt(IV)-Liposome. Western blot analysis (a) and quantification (b) of antitumor-associated proteins in HepG2 cells after various treatments. Western blot analysis (c) and quantification (d) of DNA damage related proteins. *P < 0.05, **P < 0.01, ***P < 0.001.
To this end, we demonstrated a valid strategy of coupling Pt(IV) prodrug to a lipophilic cholesterol for fabrication of CholesPt(IV)-Liposome. The subsequent investigations explicated the proposed formulation capable of promoting cell internalization of platinum prodrug. CholesPt(IV)-Liposome possessed a higher cytotoxicity against the cancer cells in comparison with cisplatin. In addition, CholesPt(IV)-Liposome induced a substantial decrease in the size of patient-derived xenograft tumors of hepatocellular carcinoma in mice. Furthermore, CholesPt(IV)-Liposome had lower toxicity to normal organs, revealing it's good biocompatibility. In view of its appreciable competence in improving platinum efficiency and reducing side effects, the proposed formulation should be emphasized as a worthy strategy in development of potent platinum delivery systems for cancer treatment.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the GDNRC [Guangdong Nature Resource Center] (2020) (037), National Natural Science Foundation of China (Nos. 81773642, 52073139), the Natural Science Foundation of Guangdong Province (No. 2019A1515011619), Guangdong Provincial Science and Technology Department (No. 2016A030311015), the Key R & D Plan of Chenzhou (No. ZDYF202008), and the Discipline Leader Startup Fund of Huazhong University of Science and Technoloy Union Shenzhen Hospital (No. YN2021002). BABL/c nude mice are used in accordance with the guidelines of ICE for Clinical Research and Animal Trials of the First Affiliated Hospital of Sun Yat-sen University Approval Letter for Research Protocol (No. 2018240).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.10.077.
I.S. Song, N. Savaraj, Z.H. Siddik, et al., Mol. Cancer Ther. 3 (2004) 1543–1549.
Figure 1
Schematic illustration and characterizations of cholesPt(IV)-functionalized liposome. Liposomes integrated by cholesPt(IV) were prepared by sequential film hydration and extrusion approach. (a) Anchor of cholesPt(IV) into the lipid bilayer of liposome to yield CholesPt(IV)-Liposome. (b) Integration percentage of cholesPt(IV) as a function of varying feed ratios of cholesPt(IV)/lipids. (c) Representative DLS result of cholesPt(IV) integrated liposome at the identified feed ratio of cholesPt(IV)/lipid of 0.4:1. (d) Colloidal stability of CholesPt(IV)-Liposome (at the identified feed ratio of cholesPt(IV)/lipid ratio of 0.4:1). The size derivation of the formulated liposome was monitored by DLS measurement.
Figure 2In vitro toxicity of diverse platinum formulations of cisPt(IV), cisplatin and CholesPt(IV)-Liposome in A2780 and A2780 DDP cells. Cell viability of A2780 (a) and A2780 DDP (b) under treatment of cisPt(IV), cisplatin and cholesPt-NPs for 72 h. The IC50 of cisPt(IV), cisplatin and CholesPt(IV)-Liposome on A2780 and A2780 DDP cells at 72 h were shown in (c). (d) The drug resistance fold of each platinum drug formulation was calculated by the ratio of IC50 at A2780 DDP to IC50 at A2780.
Figure 3
Impact of drug resistance on cellular uptake of diverse platinum derivatives. Quantification of intracellular platinum in A2780 (a) and A2780 DDP (b) after 2 and 6 h drug incubation with cisplatin, cisPt(IV) and CholesPt(IV)-Liposome. The uptake-reducing index was summarized in (c). The uptake-reducing was defined as the ratio of platinum uptake at A2780 to platinum uptake at A2780 DDP. (d) Cellular uptake activity of CholesPt(IV)-Liposome at 37 and 4 ℃.
Figure 4In vivo antitumor effects of CholesPt(IV)-Liposome. (a) Tumor growth curves of HCC PDX-bearing mice during the treatment process. (b) Body weight curves of mice. Weight (c) and image (d) of the tumors harvested from mice on day 10.
***P < 0.001.
Figure 5
Therapeutic efficacy and biocompatibility evaluation of CholesPt(IV)- Liposome on HCC PDX models. (a) H&E staining, immunohistochemistry of TUNEL (brown), and γ -H2A.X (green) analyses of tumor sections. Scale bar: 50 μm. (b) Biochemical analysis of serum after the treatments on 10 d.
Figure 6
Antitumor mechanism investigation of CholesPt(IV)-Liposome. Western blot analysis (a) and quantification (b) of antitumor-associated proteins in HepG2 cells after various treatments. Western blot analysis (c) and quantification (d) of DNA damage related proteins. *P < 0.05, **P < 0.01, ***P < 0.001.


 DownLoad:
DownLoad:




 下载:
下载:







 下载:
下载:
