
Scheme 1.
Halogenation reactions of diazo compounds.
Facile synthesis of (β-chlorodifluoroethyl)phosphonates via chlorination reaction of difluoroalkyl diazo derivatives with HCl
Jiang Liu , Romana Pajkert , Li Wang , Haibo Mei , Gerd-Volker Röschenthaler , Jianlin Han

Diazo compounds belong to the most versatile building blocks and have been extensively applied in organic synthesis for the preparation of complex compounds as well as biological molecules [1-10]. Many protocols for diverse transformations of diazo derivatives have been reported, among which, halogenation of diazo compounds via carbene or carbene-free pathways has been developed as an alternative strategy for the synthesis of organo halides [11-14]. In the past decades, several halogen sources, including N-fluorobenzenesulfonimide (NFSI), PhSeF, N-chlorosuccinimide (NCS), PhICl2, N-bromosuccinimide (NBS), oxalyl dibromide, N-iodosuccinimide (NIS), and others have been used in the respective transformations to successfully furnish the corresponding halo products (Scheme 1a) [11-21]. On the other hand, fluoroalkyl-substituted diazo compounds have been demonstrated to be valuable tools for the rapid construction of varieties of fluoroalkyl substituted organic molecules [22-26]. Although several types of reactions of 2, 2, 2-trifluorodiazoethane (CF3CHN2) have been well developed [27-36], there are only a few halogenation reactions reported until now. It was reported that CF3CHN2 could react with molecular iodine in ether at room temperature to give gem-iodo compound as the product (Scheme 1b) [37]. Also, the reactions between CF3CHN2 and NOCl/NO2Cl were developed, which were conducted at −10 ℃ affording the chlorinated products in poor chemical yields (Scheme 1b) [38], [39]. A similar fluoroalkyl substituted diazo, difluorodiazoethane (CF2HCHN2), has been well reported for the easy introduction of CF2H moiety to the molecules in recent years [40-54]. However, to the best of our knowledge, there is no example of halogenation reaction of difluorodiazoethane or its analogs until now. Thus, the development of new protocols of difluorodiazoethane is still being actively pursued in the context of synthetic fluorine chemistry.
Today, difluoromethylphosphonate (DFMP)-containing compounds have been widely used as versatile high-tech tools in biomedical research, as they have been established as molecular probes in biochemical and biophysical assays to examine enzymes and substrate-enzyme interactions [55], [56]. Thus, the development of efficient methodologies and related reagents for the rapid assembling of these compounds becomes very urgent and a great challenge. Very recently, we designed and synthesized an unstable difluoroalkyl-substituted diazo analog, (β-diazo-α, α-difluoroethyl)phosphonate, which has been applied in the synthesis of DFMP-containing sulfonic esters, carboxylic acids, and pyrazoles [57-59].
Inspired by the elegant works on difluoroalkyl-substituted diazo derivatives [40-54] and our continuous interest to develop efficient syntheses of DFMP-containing compounds [57-59], we questioned that if halogenation of our unstable (β-diazo-α, α-difluoroethyl)phosphonates is possible with easily available hydrochloric acid as a halogen source. A significant difficulty for the halogenation reaction originates from the fact that the unstability of the diazo compounds [58] and the competition between the reaction of halo anions and other nucleophiles and anions existing in the reaction system. Herein, we would like to report an efficient halogenation reaction of (β-diazo-α, α-difluoroethyl)phosphonates with hydrochloric acid as a chlorine source affording (β-chlorodifluoroethyl)phosphonate as the product in high chemical yields (Scheme 1c). The reaction is performed under mild conditions without the need of a protecting inert atmosphere, which provides a new strategy for the synthesis of α, α-difluorophosphonate derivatives. It should be mentioned that nucleophilic chloride is involved, which is different from the previous reports with electrophilic halogen reagents used [37-39]. Furthermore, this reaction represents the first example of halogenation of difluoroalkyl-substituted diazo compounds.
According to our previous reports on the reactions of (β-diazo-α, α-difluoroethyl) phosphonates [57-59], we hypothesize that the in situ generated diazo intermediate can react with hydrochloric acid via the sequence of protonation and chlorination. Thus, we started to investigate the chlorination reaction with (2-amino-1, 1-difluoro-2-phenylethyl)phosphonate (1a) as the model substrate and hydrochloric acid as the chlorine source. Concerned with the incomplete conversion of amine 1a and unstability of the in situ generated compound 2a, we used 1.2 equiv. excess of amine 1a for optimization. The reaction was carried out by using 1.2 equiv. of tert-butyl nitrite for the diazotization of amino phosphonates at 60 ℃ in CCl4, and then chlorination at room temperature. It was found that a lot of gas bubbles (N2) rushed out simultaneously after addition of hydrochloric acid, and the corresponding (β-chlorodifluoroethyl)phosphonate 3a was obtained in 45% yield after 3 h (entry 1, Table 1). Inspired by this positive result, we continued to optimize the conditions by increasing the loading amount of amine 1a from 1.2 equiv. to 2.0 equiv. Then the reaction proceeded smoothly to afford the desired chlorinated product with a dramatically increased yield (96%, entry 2). Further increasing the amount of amine 1a to 3.0 equiv. did not provide an obvious improvement and a similar yield was found (97%, entry 3). Finally, the reaction conditions were screened by variation of reaction temperature and time (entries 4–7). The yield decreased to 87% when the reaction temperature was increased to 60 ℃ (entry 5), which may be due to the decomposition of the difluorodiazonium intermediate at high temperature. Stopping the reaction at 1 h or prolonging the reaction time to 6 h did not give any improvement (entries 6 and 7).
After obtaining the optimal conditions, we then turned our attention to investigate the substrate scope for this chlorination reaction. A series of (β-amino-α, α-difluoroethyl)phosphonates 1 were used as substrates for the reaction with hydrochloric acid (Scheme 2). It was pleased that this reaction shows satisfactory functional group compatibility, and several types of substituents on phenyl group, including methyl, methoxyl, halo, and cyano, were well tolerated affording the corresponding (β-chlorodifluoroethyl)phosphonates 3 in 76%–97% yield. Generally, amines with electron-donating substituents on the phenyl moiety, such as 1b-1f, could react well with hydrochloric acid and successfully furnish the desired product (3b–3f) in excellent yields (84%–99%). In contrast, amines with electron-withdrawing substituents 1l-1n provided lower yields (76%–88%). Interestingly, steric hindrance almost had no effect on the reaction outcome. The substrates featuring ortho-, meta- and para-substituted phenyl could all be smoothly converted into the desired product in the same level of yields. These positive results suggest the potential application of this halogenation method in the late-stage modification of complex molecules and preparation of DFMP-containing compounds.

To further explore the halogen source and demonstrate the synthetic utility of this halogenation reaction, the reaction in a one-step manner and using of other halogen sources were tried for this transformation (Scheme 3). First, the reaction with all the reagents, including amine 1a, t-BuONO, AcOH, hydrochloric acid and acetonitrile/carbon tetrachloride mixed at the same time, instead of one-pot manner, was performed at 60 ℃. The reaction also proceeded smoothly affording the desired product 3a in 69% yield (Scheme 3a). Then, we used an easy handling combination of sodium chloride and sulfonic acid as a chlorine source, instead of hydrochloric acid, to react with the in situ generated diazo derivative 2a under the standard reaction conditions. This combination can also work well in this reaction, affording the desired product 3a in 67% yield (Scheme 3b). Besides chlorination, we also want to check the possibility of iodonation reaction of (β-diazo-α, α-difluoroethyl)phosphonate 2a (Scheme 3c). Unfortunately, the desired iodination reaction was almost not observed, and the reaction generated a complex mixture and no main adducts can be isolated. A similar result was obtained when sodium bromide was used as a halogen source, and almost no desired product 5a was found (Scheme 3d). It should be mentioned that bromination with the use of aqueous HBr has also been examined. Although the desired bromination product was detected, the reaction mixture was too complex and the clean product could not be isolated and obtained. This reaction is also suitable for large-scale synthesis, and a good yield (82%) was obtained when the reaction scale of HCl was increased from 0.1 mol to 1.0 mmol (Scheme 3e).

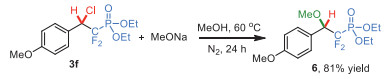
On the other hand, the further chemistry of the obtained (β-chlorodifluoroethyl)phosphonate products 3 was also investigated. The product 3f was subjected to the substitution reaction with sodium methoxide in methanol at 60 ℃ under nitrogen (Scheme 4). The reaction proceeded smoothly and afforded the corresponding (β-methoxyl difluoroethyl)phosphonate 6 in 81% yield at after 24 h.
Then, the following control experiments were performed to gain insight into the mechanism of the reaction. The 19F NMR was used for the investigation of the reaction process (for the 19F NMR spectra, Fig. S1 in Supporting information). First, new peaks (−102 ppm) occurs after addition of tert-butyl nitrite into the amine 1a solution, which indicates the generation of diazo intermediate 2a. After addition of sodium chloride, the peak of diazo 2a does not disappear. However, when sulfonic acid is added, the peak of diazo 2a disappears rapidly along with the conversion of diazo 2a to chlorination product 3a, which means protonation is the key step for the formation of the desired product.
In the light of previous reports on diazo compound [11-14, 57-59] and the above results, a plausible mechanism was proposed for this halogenation reaction and is shown in Scheme 5. Treatment of amine 1a by tert-butyl nitrite generates the diazo intermediate 2a, which then undergoes the protonation reaction affording the intermediate A. Subsequently, nucleophilic reaction of the intermediate A by chlorine anion happens to complete the halogenation reaction affording the desired halo product 3a with the release of nitrogen.

In conclusion, we have developed an efficient chlorination reaction of in situ generated (β-diazo-α, α-difluoroethyl)phosphonates with hydrochloric acids as a chlorine source. A wide range of (β-amino-α, α-difluoroethyl)phosphonates are competent substrates in this direct halogenation affording a rich library of (β-chlorodifluoroethyl)phosphonate products with up to 99% yields. This transformation uses easily available hydrochloric acid as a halogen source, instead of previously reported electrophilic chlorine reagent. Furthermore, this reaction represents the first example of the halogenation of difluoroalkyl-substituted diazo compounds and also provides an efficient pathway for the synthesis of high-value DFMP-containing compounds.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge the financial supports from the National Natural Science Foundation of China (No. 21761132021) and German Research Foundation (No. RO362/74–1). Qin Lan project from Jiangsu Province for JLH is also acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.10.066.
Y. Xiang, C. Wang, Q. Ding, Y. Peng, Adv. Synth. Catal. 361(2019) 919-944. doi: 10.1002/adsc.201800960
S.F. Zhu, Q.L. Zhou, Natl. Sci. Rev. 1(2014) 580-603. doi: 10.1093/nsr/nwu019
Ł. W. Ciszewski, K. Rybicka-Jasinska, D. Gryko, Org. Biomol. Chem. 17(2019) 432-448. doi: 10.1039/C8OB02703J
F. Wei, C. Song, Y. Ma, et al., Sci. Bull. 60(2015) 1479-1492. doi: 10.1007/s11434-015-0874-0
S. Wu, H.X. Song, C.P. Zhang, Chem. Asian J. 15(2020) 1660-1677. doi: 10.1002/asia.202000305
Z. Yang, M.L. Stivanin, I.D. Jurber, R.M. Koenigs, Chem. Soc. Rev. 49(2020) 6833-6847. doi: 10.1039/D0CS00224K
M. Marinozzi, F. Pertusati, M. Serpi, Chem. Rev. 116(2016) 13991-14055. doi: 10.1021/acs.chemrev.6b00373
S. Jana, Y. Guo, R.M. Koenigs, Chem. Eur. J. 27(2021) 1270-1281. doi: 10.1002/chem.202002556
R.K. Mykhailiuk, R.M. Koenigs, Chem. Eur. J. 26(2020) 89-101. doi: 10.1002/chem.201903335
K.A. Mix, M.R. Aronoff, R.T. Raines, ACS Chem. Biol. 11(2016) 3233-3244. doi: 10.1021/acschembio.6b00810
H.D. Khanal, R.S. Thombal, S.M.B. Maezono, Y.R. Lee, Adv. Synth. Catal. 360(2018) 3185-3212. doi: 10.1002/adsc.201800340
S.V. Kohlhepp, T. Gulder, Chem. Soc. Rev. 45(2016) 6270-6288. doi: 10.1039/C6CS00361C
V. Burianova, D. Dar'in, M. Krasavin, Tetrahedron Lett. 61(2020) 152255. doi: 10.1016/j.tetlet.2020.152255
R. Zhao, L. Shi, Angew. Chem. Int. Ed. 59(2020) 12282-12292. doi: 10.1002/anie.202003081
C. Ma, D. Xing, W. Hu, Org. Lett. 18(2016) 3134-3137. doi: 10.1021/acs.orglett.6b01346
T.B. Poynder, A.I.C. Orué, T.L. Sharp-Bucknall, et al., Chem. Commun. 57(2021) 4970-4973. doi: 10.1039/D1CC01567B
X. He, C. Yang, Y. Wu, et al., Org. Biomol. Chem. 18(2020) 4178-4182. doi: 10.1039/D0OB00683A
Y. Zhou, Y. Zhang, J. Wang, Org. Biomol. Chem. 14(2016) 10444-10453. doi: 10.1039/C6OB01858K
A. Kazantsev, D. Zhukovsky, A. Bubyrev, D. Dar'in, M. Krasavin, Tetrahedron Lett. 69(2021) 152969. doi: 10.1016/j.tetlet.2021.152969
H. Wang, X. Sun, M. Hu, et al., Adv. Synth. Catal. 362(2020) 3347-3351. doi: 10.1002/adsc.202000009
F. He, C. Pei, R.M. Koenigs, Chem. Commun. 56(2020) 599-602. doi: 10.1039/C9CC08888A
L. Mertens, R.M. Koenigs, Org. Biomol. Chem. 14(2016) 10547-10556. doi: 10.1039/C6OB01618A
L. Mertens, K.J. Hock, R.M. Koenigs, Chem. Eur. J. 22(2016) 9542-9545. doi: 10.1002/chem.201601707
J. Li, D. Zhang, J. Chen, C. Ma, W. Hu, ACS Catal. 10(2020) 4559-4565. doi: 10.1021/acscatal.0c00972
J. Li, C. Ma, D. Xing, W. Hu, Org. Lett. 21(2019) 2101-2105. doi: 10.1021/acs.orglett.9b00382
Z. Chen, Y. Zheng, J.A. Ma, Angew. Chem. Int. Ed. 56(2017) 4569-4574. doi: 10.1002/anie.201700955
P.K. Mykhailiuk, Chem. Rev. 120(2020) 12718-12755. doi: 10.1021/acs.chemrev.0c00406
X. Wang, X. Wang, J. Wang, Tetrahedron 75(2019) 949-964. doi: 10.1016/j.tet.2019.01.016
H. Gilman, R. G Jones, J. Am. Chem. Soc. 65(1943) 1458-1460. doi: 10.1021/ja01248a005
B. Morandi, E.M. Carreira, Angew. Chem. Int. Ed. 49(2010) 938-941. doi: 10.1002/anie.200905573
R. Guo, N. Lv, F.G. Zhang, J.A. Ma, Org. Lett. 20(2018) 6994-6997. doi: 10.1021/acs.orglett.8b02816
A.V. Arkhipov, V.A. Arkhipov, J. Cossy, V.O. Kovtunenko, P.K. Mykhailiuk, Org. Lett. 18(2016) 3406-3409. doi: 10.1021/acs.orglett.6b01565
U.P.N. Tran, R. Hommelsheim, Z. Yang, et al., Chem. Eur. J. 26(2020) 1254-1257. doi: 10.1002/chem.201904680
H. Luo, G. Wu, Y. Zhang, J. Wang, Angew. Chem. Int. Ed. 54(2015) 14503-14507. doi: 10.1002/anie.201507219
Z. Zhang, Q. Zhou, W. Yu, et al., Chin. J. Chem. 35(2017) 387-391. doi: 10.1002/cjoc.201600888
X. Wang, Y. Xu, Y. Deng, et al., Chem. Eur. J. 20(2014) 961-965. doi: 10.1002/chem.201304143
H. Gilman, R.G. Jones, J. Am. Chem. Soc. 65(1943) 1458-1460. doi: 10.1021/ja01248a005
L.W. Kissinger, W.E. McQuistion, M. Schwartz, L. Goodman, Tetrahedron 19(1963) 131-135. doi: 10.1016/S0040-4020(63)80049-6
L.W. Kissinger, W.E. McQuistion, M. Schwartz, Tetrahedron 19(1963) 137-141.
P.K. Mykhailiuk, R.M. Koenigs, Chem. Eur. J. 25(2019) 6053-6063. doi: 10.1002/chem.201804953
S. Emamian, J. Fluorine Chem. 199(2017) 77-91. doi: 10.1016/j.jfluchem.2017.05.004
P.K. Mykhailiuk, Angew. Chem. Int. Ed. 54(2015) 6558-6561. doi: 10.1002/anie.201501529
K.J. Hock, L. Mertens, F.K. Metze, C. Schmittmann, R.M. Koenigs, Green Chem. 19(2017) 905-909. doi: 10.1039/C6GC03187K
J. Britton, T.F. Jamison, Angew. Chem. Int. Ed. 56(2017) 8823-8827. doi: 10.1002/anie.201704529
J. Li, X.L. Yu, J. Cossy, et al., Eur. J. Org. Chem. 2017(2017) 266-2702017. doi: 10.1002/ejoc.201601462
J. Britton, T.F. Jamison, Eur. J. Org. Chem. 2017(2017) 6566-65742017. doi: 10.1002/ejoc.201700992
K.J. Hock, L. Mertens, R.M. Koenigs, Chem. Commun. 52(2016) 13783-13786. doi: 10.1039/C6CC07745E
X.W. Zhang, W.L. Hu, S. Chen, X.G. Hu, Org. Lett. 20(2018) 860-863. doi: 10.1021/acs.orglett.7b04028
Y. Gao, S.Q. Peng, D.Y. Liu, P.X. Rui, X.G. Hu, Eur. J. Org. Chem. 2019(2019) 1715-17212019. doi: 10.1002/ejoc.201801334
Y. Duan, J.H. Lin, J.C. Xiao, Y.C. Gu, Chem. Commun. 53(2017) 3870-3873. doi: 10.1039/C7CC01636K
X. Peng, F.G. Zhang, J.A. Ma, Adv. Synth. Catal. 362(2020) 4432-4437. doi: 10.1002/adsc.202000776
Z.Q. Zhang, M.M. Zheng, X.S. Xue, et al., Angew. Chem. Int. Ed. 58(2019) 18191-18196. doi: 10.1002/anie.201911701
J.L. Zeng, Z. Chen, F.G. Zhang, J.A. Ma, Org. Lett. 20(2018) 4562-4565. doi: 10.1021/acs.orglett.8b01854
S.J. Zhai, D. Cahard, F.G. Zhang, J.A. Ma, Chin. Chem. Lett. 33(2022) 863-866. doi: 10.1016/j.cclet.2021.08.007
M. Shevchuk, Q. Wang, R. Pajkert, et al., Adv. Synth. Catal. 363(2021) 2912-2968. doi: 10.1002/adsc.202001464
V.D. Romanenko, V.P. Kukhar, Chem. Rev. 106(2006) 3868-3935. doi: 10.1021/cr051000q
H. Mei, J. Liu, R. Pajkert, et al., Org. Chem. Front. 8(2021) 767-772. doi: 10.1039/D0QO01394C
H. Mei, L. Wang, R. Pajkert, et al., Org. Lett. 23(2021) 1130-1134. doi: 10.1021/acs.orglett.1c00150
J. Liu, J. Xu, R. Pajkert, et al., Acta Chim. Sin. 79(2021) 747-750. doi: 10.6023/A21030096

Scheme 2 Substrate scope of various amines. Reaction conditions: amine 1 (0.2 mmol), t-BuONO (0.24 mmol), AcOH (0.1 mmol) were dissolved in CCl4 (2 mL) and stirred at 60 ℃ for 15 min, then 12 mol/L HCl (0.1 mmol) dissolved in MeCN (2 mL) was added and the mixture was stirred at room temperature for 3 h. Isolated yields based on HCl.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



 下载:
下载: