Figure 1.
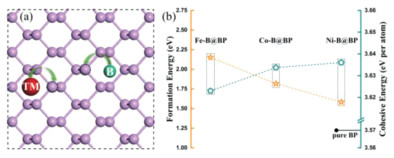
(a) Schematic diagram of designed TM-B co-doped BP catalysts with paraconfiguration. (b) The calculated formation energies and cohesive energies of designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts.
Efficient electrochemical reduction of CO to C2 products on the transition metal and boron co-doped black phosphorene
Lingyi Kong , Zhe Chen , Qinghai Cai , Lichang Yin , Jingxiang Zhao

Electrochemical conversion of carbon dioxide (CO2) has been regarded as an attractive solution to reduce carbon emission called for by the Paris Agreement, as well as a renewable and sustainable carbon cycle route [1-3], in which green renewable electricity with low cost can be used to produce valuable carbon-based fuels and chemicals using high-efficiency electrocatalysts [4-6]. In this field, the technologies of converting CO2 to CO have already reached the level of industrial application [7], because many synthetic electrocatalysts can achieve this process with high-efficiency and low energy consumption, such as various Ni single-atom catalysts [8-11]. However, it is difficult to obtain C2 products (e.g., C2H4, C2H6 and C2H5OH) with higher economic value and wider industrial applications, due to the limitations of C-C coupling process during the CO2 electroreduction [12, 13].
Therefore, the electrocatalytic reduction of CO (COER) to produce C2 chemicals has become an effective alternative to the direct CO2 conversion, because CO has been an increasingly low-cost feedstock through CO2 electroreduction, as well as the key reaction intermediate for the C-C coupling process through two possible mechanisms, including the direct CO dimerization and "carbene" mechanism, in which a second CO molecule couples with some carbon-containing intermediates, such as CHO*, CH* and CH2* [13-16]. Remarkably, many theoretical and experimental works have recently explored the high feasibility and attractive prospects of CO reduction (including the improved reaction selectivity and yield compared with direct CO2 reduction), to obtain valuable C2 products with high energy density and value [14-19]. However, in order to drive the direct electrochemical conversion of CO molecules, it is highly desirable to develop efficient electrocatalyst to overcome the huge kinetic barrier of CO coupling.
Currently, the exploration and fabrication of efficient electrocatalysts for COER are mainly focus on transition metals (TM) and boron (B) atoms due to their coexistence of empty and occupied orbitals, following the "acceptance-donation" mechanism to activate and convert CO molecules [20-24]. However, the CO coupling process is difficult to be achieved on the single isolated TM or B catalytically active site [18, 25-27]. Especially, during the CO2 electroreduction, the products obtained by effective single-atom catalysts are mainly concentrated in C1 products [8-10, 28], which can be ascribed to the lack of more active sites to promote CO coupling. Therefore, it is very promising to construct high-efficiency electrocatalysts with dual active sites to boost the C-C coupling process [29-31].
With this in mind, we proposed three low-cost electrocatalysts with dual active sites for CO coupling and reduction via co-doping TM and B atoms into the two-dimensional black phosphorus (BP), namely TM-B@BP, in which the low-cost Fe, Co and Ni atoms were taken as examples. Notably, the 2D BP has been widely chosen as a suitable substrate for single atom deposition due to its consecutive gully structure, and excellent optical/electrical properties, as well as the facile preparation by mechanical exfoliation or liquid phase exfoliation [32-34]. In particular, the single B doped BP has been theoretically reported to exhibit wide applications in catalysis field, which could be synthesized in the future under suitable conditions due to its low formation energy [18, 35-37]. Based on the constructed TM-B@BP catalysts, we theoretically investigated the synergistic effect of the dual active sites composed of TM and B atom for the CO coupling, as well as corresponding catalytic activity and product distribution for COER. Our DFT results demonstrate that moderate kinetic energy barriers and significant exothermicity during the CO coupling process can be observed on the three designed dual-site catalysts, implying the high feasibility for C-C coupling process. Furthermore, the formed CO*-dimer intermediates can be easily hydrogenated to various C2 products, such as C2H5OH, C2H4 and C2H6, with very low limiting potentials of −0.41, −0.20 and −0.28 V, respectively, on the designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts, suggesting their superior catalytic performance for COER to C2 products. Besides, the three proposed electrocatalysts can greatly suppress the production of the competing C1 products and hydrogen, indicating their high selectivity towards C2 products. Thus, our findings propose that the TM-B@BP (TM = Fe, Co and Ni) can be utilized as efficient electrocatalysts for the capture and conversion of CO molecules into high-value C2 chemicals, which opens a new door for effective and sustainable carbon-cycle utilization.
The Vienna Ab Initio Simulation Package (VASP) code was used to perform all spin-polarized DFT calculations [38]. Perdew-Burke-Ernzerhof (PBE) functional was employed to describe the exchange correlation interactions within the generalized gradient approximation. The electron-ion interactions were represented by the projector augmented wave (PAW) method [39, 40]. The kinetic energy cutoff of the plane wave was set to be 500 eV. The convergence criterion for forces on each atom and electronic structure iteration were set to be 0.03 eV/Å and 10−5 eV, respectively. Van der Waals interaction was described by the empirical correction in Grimme's method (DFT+D3) [41]. A 3 × 4 supercell of BP monolayer consisting of 48 P atoms was constructed for the co-doping of TM (Fe, Co and Ni) atom and B atom. The vacuum space in the z direction was set to be 15 Å, which is large enough to minimize the interaction between periodic images. The k-point in the Brillouin zone was sampled with a 3×3×1 Monkhorst-Pack. The climbing image nudged elastic band (CI-NEB) method was used to search for transition state with only one imaginary frequency [42]. Ab initio molecular dynamics simulations (AIMD) were performed in the canonical ensemble (NVT) with Nose-Hoover thermostat at 300 K for a time period of 5 ps [43, 44]. The charge population and transfer were calculated by using the Bader Charge analysis [45]. Other computational details including adsorption energy, cohesive energy, formation energy, and free energy calculations as well as the influence of strong correlation interaction and solvation effect were summarized in Supporting information.
The structural stability and experimental feasibility of a given catalyst are the prerequisite for its long-term applications. To this end, based on the 2D structure of BP with gully feature [18], we substituted two separate P atoms with one TM atom and one B atom by considering three configurations, including para-, meta- and ortho-doping configuration (Fig. S1 in Supporting information). After fully structural relaxation without any constraint, we found that the para-doping configuration is more energetically favorable than other two structures by about 0.20~0.50 eV (Table S1 in Supporting information), suggesting that the former is more stable. Notably, a negative correlation between the computed total energy and the TM-B distance can be obtained. For example, the Fe-B distance in the para-configuration with the lowest total energy is 3.66 Å, which is larger than those of meta- (3.37 Å) and ortho-doping one (1.85 Å), and similar phenomenon can also be observed for Co-B@BP and Ni-B@BP. Thus, in the following discussion, the para-doping configuration will be mainly focused (Fig. 1a).
After confirming the structures of the three designed TM-B@BP catalysts, we further examined their corresponding formation energies and cohesive energies to evaluate their experimental feasibility and structural stability. As shown in Fig. 1b, the formation energies of Fe-B@BP, Co-B@BP and Ni-B@BP catalyst were calculated to be 2.15, 1.82 and 1.58 eV, respectively, which are significantly lower than those of experimentally synthesized Fe@N4 and Co@N4 catalysts (2.26 eV and 2.27 eV, respectively) [46], implying their high synthesis feasibility under experimental conditions. Furthermore, their cohesive energies are calculated to be 3.62, 3.63 and 3.64 eV per atom, respectively, which are slightly higher than that of pure 2D BP (3.57 eV per atom), indicating the high structural stabilities of the three catalysts. In addition, the AIMD simulations were also performed to evaluate their thermodynamic stabilities. As shown in Fig. S2 (Supporting information), the atomic structures of the three TM-B@BP are well preserved after 5 ps AIMD simulations, further verifying their outstanding structural stabilities.
Since the electronic properties of one designed catalyst play an important role in its catalytic performance, we further examined the total density of states (TDOS) of Fe-B@BP, Co-B@BP and Ni-B@BP with corresponding local density of states (LDOS) of TM and B dopants as presented in Fig. S3 (Supporting information). As obviously shown here, Fe dopant shows higher occupied and unoccupied states near the Fermi level, indicating that it can adsorb the target CO molecule more stably, based on the "acceptance-donation" mechanism, followed by Co and Ni dopant. Furthermore, regarding the B dopants in these three designed catalysts, basically the same LDOS (B-LDOS) can be observed, with obvious unoccupied orbital above the Fermi level, implying their similar capture ability for CO molecule. In general, combined with excellent stabilities and electronic properties, three designed catalysts are expected to be highly possible for subsequent CO adsorption, coupling and reduction.
According to previous studies [17-19], the coupling of two CO molecules into a CO*-dimer is a key elementary step to affect and determine the reduction of CO to C2 products due to its possible huge energy barrier for the C-C bond formation. Therefore, to evaluate the possibility of CO coupling to CO-dimer species under reaction conditions, we explored this process on the three TM-B@BP catalysts from both the viewpoints of thermodynamics and kinetics. As shown in Fig. 2, we found that the first CO molecule is more stably adsorbed on the TM sites, with the adsorption energies of −1.81, −1.59 and −1.18 eV on Fe, Co and Ni site, respectively. Subsequently, the second CO molecule can be captured at the B sites, with the correspondingly released energies of −0.89, −0.93 and −0.93 eV. Our DFT results clearly reveal that the designed TM-B@BP catalysts show a strong capture capability towards two CO molecules. Remarkably, the calculated adsorption strengths of CO molecules on the TM and B active sites are fully consistent with our speculations based on the DOS analysis. Besides, detailed LDOS and COHP (crystal orbital Hamilton population) after two CO molecules adsorption and further analysis can be found in Fig. S4 (Supporting information).

Furthermore, we examined the C-C bond formation via the coupling of the two separately adsorbed CO molecules. According to the CI-NEB method, moderate kinetic energy barriers of 0.91, 0.77 and 0.52 eV should be overcome for the CO coupling to generate C-C bond (i.e., CO*-dimer) on the designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts, respectively. Interestingly, these kinetic energy barriers of CO coupling on the TM-B@BP catalysts (0.91, 0.77 and 0.52 eV) are comparable (even lower) to other potential catalysts for CO coupling, such as B/C2N (0.73 eV) [17], B-N@BP (0.60) [18], Cu-B@g-C3N4 (0.99 eV) [30], boron nitride nanoribbon (1.30 eV) [47] and Cu (211) surface (1.62 eV) [48]. Thus, the direct dimerization of two adsorbed CO* species on TM-B@BP catalysts can easily proceed in kinetics. In addition, this step of 2CO(g) → CO*-dimer is highly exothermic by 1.99, 1.96 and 1.70 eV, respectively, on the designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts, respectively (Fig. 2). After considering the contribution of zero-point energy and entropy, the free energy changes for the formation of CO*-dimer species are computed to be −0.86, −0.83 and −0.56 eV, respectively, on the three catalysts. Especially, the newly formed C-C bond lengths of CO*-dimer on Fe-B@BP, Co-B@BP and Ni-B@BP are 1.56, 1.63 and 1.66 Å, respectively, which is slightly longer than that of ethane (1.54 Å), further testifying the formation of stable C-C bond via CO coupling. Overall, the low kinetic barrier, high exothermicity, and short C-C bond length indicate that the CO coupling is favorable on these three designed catalysts both kinetically and thermodynamically, which normally help to trigger the generation of multi-carbon products.
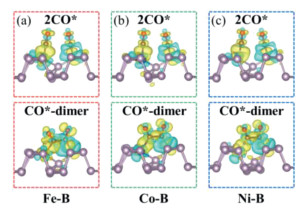
To gain deep insight into the CO coupling, we further computed the difference charge density of the two separated adsorbed CO* and CO*-dimer (Fig. 3). Our results demonstrate that obvious charge accumulation (yellow color) occurs between the TM dopant and the adsorbed CO molecule, while both the charge accumulation and charge depletion appear between the B dopant and another adsorbed CO molecule, especially the obvious surrounding charge depleted regions (cyan color). Such coexisting charge accumulation and depletion regions will greatly promote the C-C coupling due to the attraction of positive and negative charges between each other. In addition, significant charge transfer (about 0.83, 0.72 and 0.68|e|, respectively) can be observed between the CO*-dimer and the designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts, implying the high chemical reactivity of the three TM-B@BP catalysts towards the C-C bond formation. Therefore, the co-doping of TM (Fe, Co and Ni) and B can lead to the CO coupling to form stable C-C bond on three TM-B@BP catalysts, which will play an important role on yielding the multi-carbon products through the continuous hydrogenation processes.
Since the CO coupling can be easily achieved in terms of thermodynamics and kinetics on the three designed electrocatalysts, we further examined the subsequent hydrogenation step of CO*-dimer to generate C2 products via multiple proton-coupled electron transfer steps (H+ + e−). Fig. 4 summarizes the most energetically favorable reaction pathway of CO reduction (with the lowest positive free energy change (ΔG) between any two elementary steps) towards C2 products on these TM-B@BP catalysts, and the corresponding atomic structures of the involved reaction intermediates are presented in Figs. S5-S7 (Supporting information).

As discussed above, two CO molecules are firstly coupled to form CO*-dimer species with the ΔG values of −0.86, −0.83 and −0.56 eV on Fe-B@BP, Co-B@BP and Ni-B@BP, respectively. Subsequently, on the three designed TM-B@BP catalysts, various hydrogenation reaction pathways will proceed, thus generating different C2 products. Specifically, on the Fe-B@BP catalyst (Fig. 4a and Fig. S5), the first few steps of reducing CO*-dimer to C2 products are as follows: *CO-dimer → *COHCO → *COHCOH → *CHOHCOH → *CHCOH → *CH2COH, with the ΔG values of −0.16, +0.05, −0.03, −0.49 and −0.45 eV, respectively. Subsequently the formed *CH2COH species will be hydrogenated to *CH3COH or *CH2CHOH. As shown in Fig. 4a, along the *CH3COH path, the final product is ethanol (CH3CH2OH), while ethylene (CH2CH2) will be achieved along *CH2CHOH pathway. Especially, the hydrogenation of *CH2COH species to *CH3COH or *CH2CHOH is identified as the potential-determining step (PDS) during the whole CO electroreduction due to its largest positive free energy change (0.39 or 0.41 eV) among all elementary reactions. Therefore, due to the small difference in the free energy change for the two steps, these two valuable products can be achieved through CO electroreduction on the designed Fe-B@BP catalyst at a low limiting potential of −0.41 V. Notably, the obtained CH3CH2OH and CH2CH2 products can be spontaneously separated due to their different phases at room temperature.
As for the Co-B@BP catalyst (Fig. 4b and Fig. S6), via the continuous hydrogenation processes, CO*-dimer is finally reduced to CH2CH2 product, following the reaction paths as: CO*-dimer → *COHCO → *COHCOH → *CHOHCOH → *CHCOH → *CH2COH → *CH2CHOH → *CH2CH → *CH2CH2, in which the first hydrogenation of CO*-dimer to produce *COCOH species is the potential-determining step, with the uphill free energy change of 0.20 eV, corresponding to the limiting potential of −0.20 V. In addition, on the Ni-B@BP catalyst, as presented in Fig. 4c and Fig. S7, the CO*-dimer species is gradually hydrogenated to achieve *CH3CH2OH species, in which the formation of *COHCO species exhibits the largest free energy change of 0.28 eV. Subsequently, there are two competing reactions for the further hydrogenation of obtained *CH3CH2OH species: (1) it will release from the catalyst surface to produce ethanol, or (2) be further hydrogenated to generate CH3CH3 + *OH. According to their free energy changes (0.16 eV vs. −0.06 eV), we predicted that CH3CH3 is the dominant product for COER on the Ni-B@BP catalyst with a very low limiting potential of −0.28 V. Finally, the remaining *OH species can be easily reduced to H2O molecule with the free energy change of −0.81 eV.
Overall, on the three electrocatalysts (Fe-B@BP, Co-B@BP and Ni-B@BP), extremely low energy inputs (0.41, 0.20 and 0.28 eV, respectively) are required to boost the conversion of CO molecules into multiple valuable C2 products (ethanol, ethylene and ethane, respectively), which are lower than (or comparable to) those of previously reported Cu (100) surface (0.31 eV) [49], defective Mo2TiC2O2 (0.32 eV) [50], Cu-B@g-C3N4 (0.45 eV) [30], and copper/borophene interface (0.61 eV) [51], thus suggesting their high catalytic activities of COER to generate high-value C2 products.
In addition to their high catalytic activities of the three electrocatalysts for achieving C2 products during the COER, another important issue is their selectivity towards C2 products in the practical applications. In this regard, two competing reactions should be considered, including the COER to C1 products and the hydrogen evolution reaction (HER) at the active sites [18]. For the former, the free energy change for the hydrogenation of the single CO at different active sites to form *CHO species was examined. As shown in Table S2 (Supporting information), we found that the ΔG values for *CO → *CHO are at least 0.55 eV at both TM and B active sites, which are always larger than the maximum ΔG value for COER to C2 products (0.20~0.41 eV), suggesting their higher selectivity for C2 products. On the other hand, for the HER, we calculated the adsorption energy of proton (H*) to evaluate the competitive adsorption with CO molecules (Table S2). Our results show that the H* species is unstably adsorbed at the TM sites with the adsorption energies ranging from −0.06 eV to +0.24 eV due to the repulsion between the positively charged TM and H+. On the contrary, the B site show a strong interaction with H* species with the adsorption energy of −0.67 eV. These adsorption energies of hydrogen are less negative than those of two CO molecules on TM and B sites (at least −0.89 eV), suggesting that the active sites are more energetically favorable to be covered by CO molecules, greatly suppressing the competitive HER and thus suggesting high selectivity for CO electroreduction towards C2 products.
In summary, by performing comprehensive DFT computations, we designed a new kind of electrocatalyst for the CO reduction to generate valuable C2 chemicals via co-doping the 2D BP with the TM (TM = Fe, Co and Ni) and B atoms. Our results demonstrate that two CO molecules can be effectively coupled into the key CO*-dimer species both thermodynamically and kinetically, due to the synergistic effect between the TM and B active sites. Furthermore, based on the free energy computations, we found that all three TM-B@BP catalysts exhibit the desired high catalytic activities for COER with the rather low limiting potentials (−0.41, −0.20 and −0.28 V), and ethanol, ethylene and ethane were identified as the main products. In addition, due to the lower energy inputs during COER to C2 products and the stronger interactions between designed catalysts and CO molecules, the competing C1 products in COER and H2 product in HER can be effectively suppressed, endowing our proposed TM-B@BP electrocatalysts high selectivity for C2 products. Therefore, our simulations show that the three as-designed TM-B@BP catalysts can be utilized as a new type of low-cost electrocatalysts with high activity and selectivity for CO conversion to high-value multi-carbon products, providing useful guidance for sustainable carbon fixation and energy storage in future.
The authors declare no competing financial interest.
This work is supported by the National Natural Science Foundation of China (NSFC, Nos. 51972312 and U20A20242) and the Natural Science Foundation of Liaoning Province of China (No. 2020-MS-003). The authors acknowledge the computation support from TianHe-1(A) at the National Supercomputer Center in Tianjin and Tianhe-2 at the National Supercomputer Center in Guangzhou.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.09.010.
M.G. Kibria, J.P. Edwards, C.M. Gabardo, et al., Adv. Mater. 31 (2019) 1807166. doi: 10.1002/adma.201807166
L. Zhang, Z.J. Zhao, T. Wang, et al., Chem. Soc. Rev. 47 (2018) 5423-5443. doi: 10.1039/C8CS00016F
O.S. Bushuyev, P. De Luna, C.T. Dinh, et al., Joule 2 (2018) 825-832. doi: 10.1016/j.joule.2017.09.003
Q. Wang, Y. Lei, D. Wang, et al., Energy Environ. Sci. 12 (2019) 1730-1750. doi: 10.1039/C8EE03781G
Y. Wang, Y. Liu, W. Liu, et al., Energy Environ. Sci. 13 (2020) 4609-4624. doi: 10.1039/D0EE02833A
W. Zhang, Y. Hu, L. Ma, et al., Adv. Sci. 5 (2018) 1700275. doi: 10.1002/advs.201700275
X. Tan, C. Yu, Y. Ren, et al., Energy Environ. Sci. 14 (2021) 765-780. doi: 10.1039/D0EE02981E
T. Zheng, K. Jiang, N. Ta, et al., Joule 3 (2019) 265-278. doi: 10.1016/j.joule.2018.10.015
Z. Chen, X. Zhang, W. Liu, et al., Energy Environ. Sci. 14 (2021) 2349-2356. doi: 10.1039/D0EE04052E
T. Möller, W. Ju, A. Bagger, et al., Energy Environ. Sci. 12 (2019) 640-647. doi: 10.1039/C8EE02662A
X. Zhang, Y. Wang, M. Gu, et al., Nat. Energy 5 (2020) 684-692. doi: 10.1038/s41560-020-0667-9
Y. Zheng, A. Vasileff, X. Zhou, et al., J. Am. Chem. Soc. 141 (2019) 7646-7659. doi: 10.1021/jacs.9b02124
Q. Fan, M. Zhang, M. Jia, et al., Mater. Today Energy 10 (2018) 280-301. doi: 10.1016/j.mtener.2018.10.003
M. Jouny, G.S. Hutchings, F. Jiao, Nat. Catal. 2 (2019) 1062-1070. doi: 10.1038/s41929-019-0388-2
W. Luc, X. Fu, J. Shi, et al., Nat. Catal. 2 (2019) 423-430. doi: 10.1038/s41929-019-0269-8
L. Wang, S. Nitopi, A.B. Wong, et al., Nat. Catal. 2 (2019) 702-708. doi: 10.1038/s41929-019-0301-z
Z. Chen, J. Zhao, J. Zhao, et al., Nanoscale 11 (2019) 20777-20784. doi: 10.1039/C9NR07559C
Z. Chen, X. Liu, J. Zhao, et al., J. Mater. Chem. A 8 (2020) 11986-11995. doi: 10.1039/D0TA03991H
L. Chen, C. Tang, K. Davey, et al., Chem. Sci. 12 (2021) 8079-8087. doi: 10.1039/D1SC01694F
C. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci. 509 (2020) 145392. doi: 10.1016/j.apsusc.2020.145392
W. Zhang, Y. Hu, L. Ma, et al., Nano Energy 53 (2018) 808-816. doi: 10.1016/j.nanoen.2018.09.053
W. Zhang, C. Xu, Y. Hu, et al., Nano Energy 73 (2020) 104796. doi: 10.1016/j.nanoen.2020.104796
W. Zhang, S. Yang, M. Jiang, et al., Nano Lett 21 (2021) 2650-2657. doi: 10.1021/acs.nanolett.1c00390
J. Zhao, Z. Chen, J. Zhao, et al., Appl. Surf. Sci. 498 (2019) 143868. doi: 10.1016/j.apsusc.2019.143868
H. Yang, C. He, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3202-3206. doi: 10.1016/j.cclet.2021.03.038
Q. Li, Y.C. Wang, J. Zeng, et al., Rare Met. 40 (2021) 3442-3453. doi: 10.1007/s12598-021-01772-7
V. -H. Nguyen, B.S. Nguyen, Z. Jin, et al., Chem. Eng. J. 402 (2020) 126184. doi: 10.1016/j.cej.2020.126184
H. Shang, T. Wang, J. Pei, et al., Angew. Chem. In. Ed. 59 (2020) 22465-22469. doi: 10.1002/anie.202010903
L. Li, H. Guo, G. Yao, et al., J. Mater. Chem. A 8 (2020) 22327-22334. doi: 10.1039/D0TA05821A
T. He, K. Reuter, A. Du, J. Mater. Chem. A 8 (2020) 599-606. doi: 10.1039/C9TA12090D
Y. Zhao, S. Zhou, J. Zhao, iScience 23 (2020) 101051. doi: 10.1016/j.isci.2020.101051
M.Z. Rahman, C.W. Kwong, K. Davey, et al., Energy Environ. Sci. 9 (2016) 709-728. doi: 10.1039/C5EE03732H
M. Liu, S. Feng, Y. Hou, et al., Mater. Today 36 (2020) 91-101. doi: 10.1016/j.mattod.2019.12.027
X. Mu, J. Wang, M. Sun, Mater. Today Phys. 8 (2019) 92-111. doi: 10.1016/j.mtphys.2019.02.003
C. Liu, Q. Li, C. Wu, et al., J. Am. Chem. Soc. 141 (2019) 2884-2888. doi: 10.1021/jacs.8b13165
Y. Cheng, Y. Song, Y. Zhang, Phys. Chem. Chem. Phys. 21 (2019) 24449-24457. doi: 10.1039/C9CP04647J
L. Shi, Q. Li, C. Ling, et al., J. Mater. Chem. A 7 (2019) 4865-4871. doi: 10.1039/C8TA11025E
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169. doi: 10.1103/PhysRevB.54.11169
P.E. Blöchl, Phys. Rev. B 50 (1994) 17953. doi: 10.1103/PhysRevB.50.17953
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865. doi: 10.1103/PhysRevLett.77.3865
L. Goerigk, S. Grimme, Phys. Chem. Chem. Phys. 13 (2011) 6670-6688. doi: 10.1039/c0cp02984j
G. Henkelman, B.P. Uberuaga, H. Jónsson, J. Chem. Phys. 113 (2000) 9901-9904. doi: 10.1063/1.1329672
G. Bussi, D. Donadio, M. Parrinello, J. Chem. Phys. 126 (2007) 014101. doi: 10.1063/1.2408420
S. Nosé, J. Chem. Phys. 81 (1984) 511-519. doi: 10.1063/1.447334
G. Henkelman, A. Arnaldsson, H. Jónsson, Comp. Mater. Sci. 36 (2006) 354-360. doi: 10.1016/j.commatsci.2005.04.010
C. Choi, S. Back, N. -Y. Kim, et al., ACS Catal. 8 (2018) 7517-7525. doi: 10.1021/acscatal.8b00905
S. Tang, X. Zhou, S. Zhang, et al., ACS Appl. Mater. Interfaces 11 (2018) 906-915.
J.H. Montoya, A.A. Peterson, J.K. Nørskov, ChemCatChem 5 (2013) 737-742. doi: 10.1002/cctc.201200564
R. Chen, H.Y. Su, D. Liu, et al., Angew. Chem. 132 (2020) 160-166. doi: 10.1002/ange.201910662
X. Qian, L. Li, Y. Li, et al., Phys. Chem. Chem. Phys. 23 (2021) 12431-12438. doi: 10.1039/D1CP01291F
T. He, C. Tang, A.R.P. Santiago, et al., J. Mater. Chem. A 9 (2021) 13192-13199. doi: 10.1039/D1TA02355A

Figure 1 (a) Schematic diagram of designed TM-B co-doped BP catalysts with paraconfiguration. (b) The calculated formation energies and cohesive energies of designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts.

Figure 2 (a) The calculated relative energy changes of CO adsorption and coupling process on the designed Fe-B@BP, Co-B@BP and Ni-B@BP catalysts, in which * represents catalytic active site. (b) The corresponding atomic structures of 2CO*, transition state (TS) and CO*-dimer.

Figure 3 The difference charge density plots for 2CO* and CO*-dimer on the designed (a) Fe-B@BP, (b) Co-B@BP and (c) Ni-B@BP catalyst. The isosurface value is set to be 0.003 e/Å3, the charge accumulated and depleted regions are shown in yellow and cyan, respectively.

Figure 4 Free energy diagrams of CO reduction into C2 products at 0 V potentials on the designed (a) Fe-B@BP, (b) Co-B@BP and (c) Ni-B@BP catalyst, in which the dash lines represent the proton-coupled electron transfer processes and the solid lines indicate the pure adsorption or desorption processes.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: