Table 1.
Overview of N-terminal modification method components and key features.
Citation:
Hongfei Jiang, Wujun Chen, Jie Wang, Renshuai Zhang. Selective N-terminal modification of peptides and proteins: Recent progresses and applications[J]. Chinese Chemical Letters,
2022, 33(1): 80-88.
doi:
10.1016/j.cclet.2021.06.011

Selective N-terminal modification of peptides and proteins: Recent progresses and applications
English
Selective N-terminal modification of peptides and proteins: Recent progresses and applications
Abstract:
Numerous strategies for linking desired chemical probes with target peptides and proteins have been developed and applied in the field of biological chemistry. Approaches for site-specific modification of native amino acid residues in test tubes and biological contexts represent novel biological tools for understanding the role of peptides and proteins. Selective N-terminal modification strategies have been broadly studied especially in the last 10 years, as N-terminal positions are typically solvent exposed and provide chemically distinct sites for many peptide and protein targets, making N terminus distinct from other functional groups. A growing number of chemical and enzymatic techniques have been developed to modify N-terminal amino acids, and those techniques have the potential in the fields of medicine, basic research and applied materials science. This review focuses on appraising modification methodologies with the potential for biological applications from the past 10 years.
-
1. Introduction
Selective peptide and protein modification techniques are gaining great significance in the fields of medical diagnostics [1], biopharmaceutical conjugates (e.g., PEGylation, lipidation, and antibody-drug conjugates) [2], bioimaging [3], and material sciences [4]. Selective modification strategies targeting the side chain of lysine [5-7] and cysteine [8-11] are widely studied, thus the preparation of high-quality and well-defined bioconjugates mainly rely on those strategies up to now. Tyrosine modification has been achieved by using chemical reagents targeting the aromatic hydroxyl group or phenyl ring on the side chain and metal catalysts are widely used to increase their selectivity [12-15]. Histidine and tryptophan can be selectively modified via visible-light-promoted C-H alkylation method and biomimetic electron transfer process separately [16, 17]. Lin and Taylor's studies for selective methionine modification are very promising, and they used oxaziridine-based reagents to produce protein conjugates that can serve as a platform for the development of visible-light-mediated bioorthogonal protein functionalization processes and enable precise addition of payloads to proteins for identification of hyperreactive methionine residues in whole proteomes [18, 19]. Those in-chain residue modification techniques enable a marked increase in their functional diversity and structure analysis and approaches for termini modification are still required to expand the protein functionalization toolbox.
N terminus and C terminus modification techniques also aroused widespread interests of researchers. Terminus modifications of proteins have the potential to be more generally applicable, since the majority of protein termini are accessible and have chemical environments distinct from remainder of the protein. A significant advantage of terminal amino acid residues is they are structurally unique in peptides and proteins and single-chain peptides and proteins possess only one N- and C-terminal residue, moreover, they usually have no vital impact on their biological activities, and the widely used protein tags for protein purification and structure characterization are located in protein N or C terminus [20]. Up to now, C terminus modification techniques are very few, visible-light-mediated single-electron transfer (SET) method reported by the MacMillan group to perform decarboxylative alkylation at C-terminal residues for selective C terminus modification is very appealing [21]. By contrast, there are quite a few choices for selective N terminus modification [22]. The α-amine group of N terminus stands out as a uniquely reactive site, and they are typically solvent exposed and provide chemically distinct sites for many protein targets. A large number of site-specific modification techniques targeting protein α-amine position with applications in chemical biology. This review focuses on appraising modification methodologies from about the past 10 years as well as novel biological applications of the strategies. Table 1 gives an overview of selective N-terminal modification method components and key features of them.
Table 1
2. Selective N-terminal modification under catalyst-free conditions
When considering N-terminal modification, the first point is how to make a distinction between N-terminall α-amine and in-chain lysine ε-amine. It is gratifying that the N-terminal α-amine has pKa ~6-8, while the in-chain lysine ε-amine has pKa ~10, which makes the different reactivity of them [23]. Consequently, by careful control of solvent pH, the N-terminal α-amine will be more nucleophilic than lysine ε-amine. In addition, N-terminal α-amine and in-chain lysine ε-amine are structurally different, therefore, N-terminal α-amine can be selectively modified by unique side chain participation strategies.
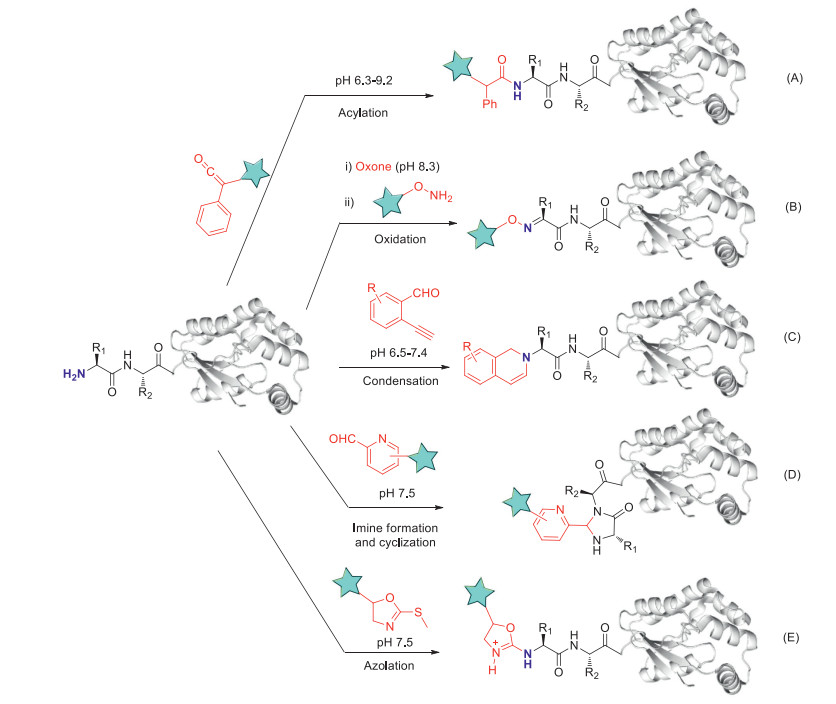
N-Hydroxysuccinimide (NHS) esters directly react with N-terminal amine under acidic conditions (pH 4.5-6.5) to form amide bonds, however, the conversion and selectivity are not satisfactory. Ketenes are discovered to be much more efficient than NHS esters for selective N-terminal modification in the aspects of N-terminal selectivity (500 equiv. ketenes, 12 h, 37 ℃, pH 6.3-9.2, Fig. 1A), and ketenes can react with N-terminal α-amine under mild condition through acylation of the amino group [24]. Proteins including insulin, lysozyme, RNaseA, and a therapeutic protein BCArg are selectively N-terminally modified at room temperature using ketenes. Whereas, the ketene strategy only has moderate conversions (< 50% for 17 N-terminal amino acid residues), ketenes can be further modified to improve their universal N-terminal amino acid residues compatibility.
Figure 1
 Figure 1. Selective modification of protein N termini under catalyst-free conditions. Methods include acylation using ketenes (A); condensation with 2-ethynylbenzaldehydes (2EBAs) (B); oxidation with (i) oxone followed by (ii) oxime exchange by functionalized hydroxylamines (C); imidazolidinone formation with 2-pyridinecarboxaldehyde (2PCA) derivatives (D); azolation with azolines (E).
Figure 1. Selective modification of protein N termini under catalyst-free conditions. Methods include acylation using ketenes (A); condensation with 2-ethynylbenzaldehydes (2EBAs) (B); oxidation with (i) oxone followed by (ii) oxime exchange by functionalized hydroxylamines (C); imidazolidinone formation with 2-pyridinecarboxaldehyde (2PCA) derivatives (D); azolation with azolines (E).Oxidation of peptides and proteins is a complex biological process involving the attack of reactive oxygen species on amino acid residues. The use of oxidizing agents for modification of peptides and proteins still remains limited due to the formation of complicated oxidized products via uncontrolled oxidation. Through screening of common bench-top oxidizing reagents, Wong and co-workers discovered an efficient method using oxone (2KHSO5·KHSO4·K2SO4) as the convenient and chemoselective reagent for selective N-terminal α-amine modification through an oxime formation-exchange reaction (2 equiv. oxones, 1 h, 25 ℃, pH 8.3, 40%-99% conversion, Fig. 1B). The technique uses oxone for selective oxidation of N-terminal amine of peptides to oximes followed by transoximation with O-substituted hydroxylamines [25]. However, oxones also oxidize thioether group of methionine, thiol group of cysteine and indole of tryptophan, thus this strategy has great limitations for protein N-terminal modification. In addition, the Wong group has reported N-terminal modification of peptides and proteins by using 2-ethynylbenzaldehydes (2EBAs) for the production of well defined bioconjugates with moderate to good conversions [26]. A therapeutic recombinant Bacillus caldovelox arginase mutant (BCArg mutant), ribonuclease A and lysozyme are N-terminally modified using alkyne- and fluorescein-linked 2EBA (20 equiv. 2EBAs, 16 h, 37 ℃, pH 6.5-7.4, 10%-84% conversion, Fig. 1C).
Francis and co-workers reported a simple, one-step method suitable for modifying single N-terminal α-amine on a broad scope of structurally and chemically varied proteins by use of 2-pyridinecarboxyaldehydes (400 equiv. 2PCAs, 16 h, 37 ℃, pH 7.5, 33%-99% conversion, Fig. 1D) [27]. This method has become a widely used technique in the field of chemical biology for selective N-terminal amine ligation. Notably, a small amount (< 10%) of a second addition is seen in the case of the N-terminal glycine peptide. Several modified 2PCAs have been reported and applied for selective N-terminus modification [28-31].
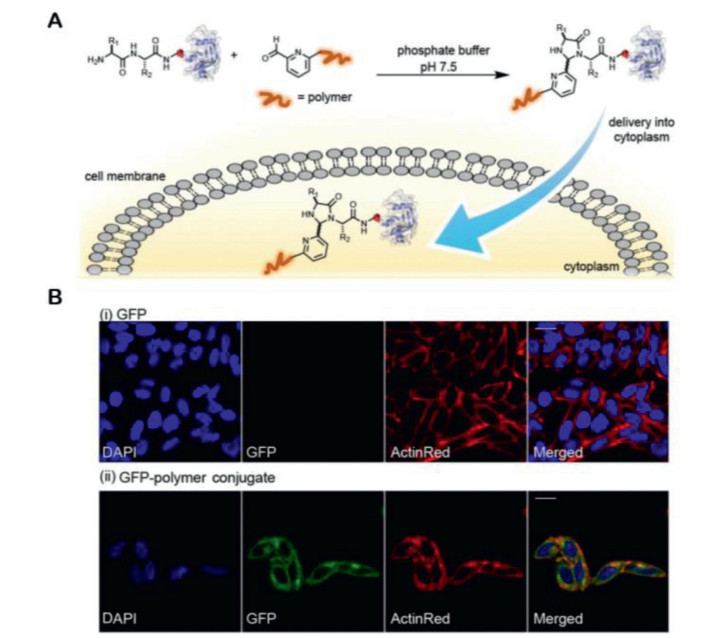
Francis and co-workers applied the 2PCA strategy to intracellular protein delivery. 2PCA linked to amphiphilic polymer can react with N-terminal amine under physical condition for site-selectively attaching amphiphilic polymer to the N-terminal positions of proteins [32]. This ligation is simple under mild, aqueous conditions with no required genetic engineering of the proteins. Polymer-protein conjugates can be delivered into the cytosol, likely through a membrane fusion mechanism (Fig. 2A). The internalization of the GFP-polymer conjugates is investigated using the HeLa cell line to test the protein-polymer intracellular uptake efficiency. Confocal microscopy analysis demonstrates that the GFP molecules conjugated to amphiphilic polymer are efficiently delivered into the cytosolic compartment, whereas GFP alone is not detected (Fig. 2B).
Figure 2
 Figure 2. 2PCA-polymer strategy for protein delivery. (A) Schematic illustration of protein-polymer conjugate preparation using 2PCA for N-terminal modification. (B) Confocal microscopy images are shown for HeLa cells after exposure to GFP or GFP-polymer conjugates, nuclear and cytoplasmic stains are performed on fixed cells using DAPI and ActinRed, respectively. Scale bars represent 20 µm. Reproduced with permission [32]. Copyright 2019, American Chemical Society.
Figure 2. 2PCA-polymer strategy for protein delivery. (A) Schematic illustration of protein-polymer conjugate preparation using 2PCA for N-terminal modification. (B) Confocal microscopy images are shown for HeLa cells after exposure to GFP or GFP-polymer conjugates, nuclear and cytoplasmic stains are performed on fixed cells using DAPI and ActinRed, respectively. Scale bars represent 20 µm. Reproduced with permission [32]. Copyright 2019, American Chemical Society.Mass spectrometry sensitive probes are of great significance to their biological applications [33]. The azolation strategy for site- and chemo-selective labeling of N-terminus of proteins (50 equiv. azolines, 3 h, 37 ℃, pH 7.5°, 50%-99% conversion, Fig. 1E) is able to enhance the mass detection sensitivity of the bioconjugates. Azoles as mass sensitivity probes can selectively modify N-terminal amine under physiological conditions [34]. The conversions of azolation strategy are at moderate to excellent levels (50%-99%). Current strategies using chemical tag to enhance protein detection by multiple orders would lead to the unambiguous analysis of the resulting bioconjugates [35], whereas, azolation presented a single-step, versatile strategy for the selective modification of protein N-termini with mass boosters.
3. Chemical catalysts driven N-terminal modification
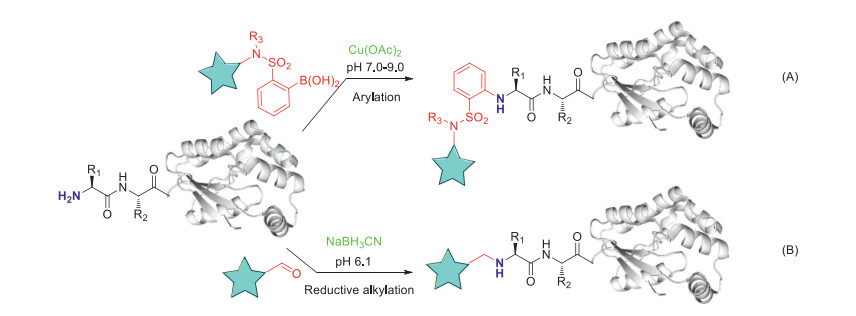
Catalyst-triggered, especially metal-catalyzed or metal-directed, reactions are widely applied in chemoselective peptides and proteins modification [36-38]. Cross-coupling methods, typically mediated by transition metal complexes, are becoming important tools for N-C bond formation and they have become an interesting approach for selective N-terminal amine modification of peptides and proteins. Plenty of chemo-selective approaches targeting cysteine [12, 39] or tyrosine [40, 41] with a variety of transition metal catalysts have been developed. Whereas, cross-coupling at amine groups in native peptides and proteins are limited. Ball and co-workers described a copper-mediated amine arylation method with the utilization of boronic acid reagents bearing certain o-electron withdrawing groups for selective N-terminal α-amine modification of polypeptides under mild conditions in primarily aqueous solution (20 equiv. boronic acid reagents, 0.5 equiv. Cu(OAc)2, 18 h, 37 ℃, pH 7.0, < 5%-97% conversion, Fig. 3A). The method shows complete selectivity for N-terminus in the presence of lysine side chains [42], however, 20%-30% organic solvents (acetonitrile, tetrafluoroethylene or DMSO) are required to facilitate the dissolution of boronic acid reagents, which is adverse for its protein labeling applications. Hung-Chieh Chou and co-workers applied reductive alkylation reaction in selective N-terminal modification (2 equiv. aldehydes, 5 equiv. NaBH3CN, 24 h, 37 ℃, pH 6.1-6.2, 30%-95% conversion, Fig. 3B). The scope of the aldehydes are broadly expanded to various aldehyde derivatives including 2PCAs, benzaldehyde derivatives, alkylaldehyde derivatives, even glucose and maltose [43]. NaBH3CN mediated reductive alkylation technique for peptides N-terminal modification has been discovered to produce 1%-18% di-modified peptides (both on N-terminus) as by-product, whereas the N-terminal selectivity is encouraging (> 99%).
Figure 3
 Figure 3. Selective modification of protein N termini using chemical catalysts. Methods include copper-mediated peptide arylation with boronic acids (A), alkylation by reductive amination with aldehydes (B).
Figure 3. Selective modification of protein N termini using chemical catalysts. Methods include copper-mediated peptide arylation with boronic acids (A), alkylation by reductive amination with aldehydes (B).4. Enzymes catalyzed N-terminal modification
Enzymatic protein labeling techniques are powerful tools for site-specific peptides and proteins modification. While chemical reaction directed peptides and proteins modification methods often yield heterogeneously modified products, enzymatic protein labeling techniques are highly efficient and produce single products under mild reaction conditions. Significant progresses have been made during the last few years in the field of enzymatic protein N-terminal labeling with broad applications [44-48].
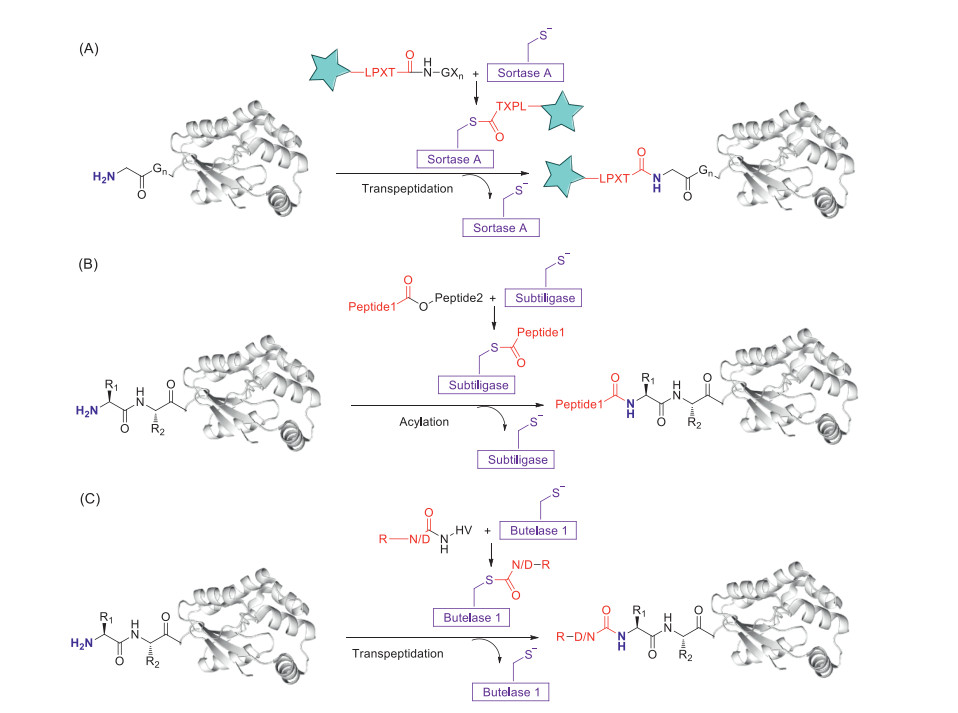
Sortase A (SrtA), a transpeptidase from Staphylococcus aureus, can recognize the LPXTG (known as a sortag) sequence and cleave the amide bond between threonine and glycine residues, forming a new threonine-glycine peptide bond with N-terminal amine of an oligoglycine-terminating peptide [49-51]. Sortase A has been broadly applied in the N-terminal modification of proteins. Several Sortase A variants are commercially available. While most of sortase classes showed negligible activity in vitro, Sortase A can be used in the non-natural environment. Sortase A variants R159G and D165Q/D186G/K196V have increased resistance (2.2-fold) and catalytic efficiency (6.3-fold) in 45% (v/v) dimethylsulfoxide (0.01 equiv. Sortase A, 5 equiv. substrate, 14 h, 25 ℃, co-solvents, Fig. 4A). Moreover, D165Q/D186G/K196V also show increased activity for the conjugation of hydrophobic peptides in normal organic co-solvents (ethanol, methanol, ACN, DMF, and DMSO) [52]. Low catalytic efficiency represents major disadvantage of Sortase A, hence, comparably high enzyme concentrations are required. Ca2+-independent Sortase A variants are prepared with obviously enhanced (up to140-fold) catalytic activity [51]. Immobilized sortases are developed and utilized to further improve the catalytic efficiency, and they enable large scale reactions and are recyclable. Francis and co-workers reported a proline variant of the evolved sortase A named SrtA 7M which is labeled with lithocholic acid (LA) at N-terminal, LA exhibits strong binding to β-cyclodextrin (β-CD) for its further immobilization on resin. The SrtA 7M-resin conjugate is scalable and retained full enzymatic activity even after multiple rounds of recycling [53].
Figure 4
 Figure 4. Enzyme-mediated N-terminal modification. (A) Sortase A (SrtA)-catalyzed transpeptidation of an LPXTG peptide derivative for attachment to an H2N-(G)n-protein. (B) Attachment of a glycolate ester substrate mediated by subtiligase. (C) N-terminal acetylation with an Asn/Asp-thiodepsipeptide using butelase 1.
Figure 4. Enzyme-mediated N-terminal modification. (A) Sortase A (SrtA)-catalyzed transpeptidation of an LPXTG peptide derivative for attachment to an H2N-(G)n-protein. (B) Attachment of a glycolate ester substrate mediated by subtiligase. (C) N-terminal acetylation with an Asn/Asp-thiodepsipeptide using butelase 1.N-Myristoyltransferase (NMT) is widely spread in eukaryotes that catalyzes the co- and post-translational, irreversible attachment of myristic acid to protein N-terminal amine. However, recent work demonstrates that NMT is also a lysine myristoyltransferase [54] indicating that NMT strategy is not a highly N-terminal selective modification technique. Worthwhile, subtiligase mediated strategy is an appealing method for N-terminal amine modification because ligation occurs with complete chemoselectivity for the peptides and proteins N terminus over lysine ε-amines without the requirement for a particular amino acid sequence tag [55-57]. The efficiency of ligation depends mainly on the accessibility and ability of subtiligase substrate N-terminal sequence to be modified (Fig. 4B). Introduction of extended N-terminal sequences, for example AFA sequence, has been employed as a useful strategy for achieving high modification conversions and short reaction time [58].
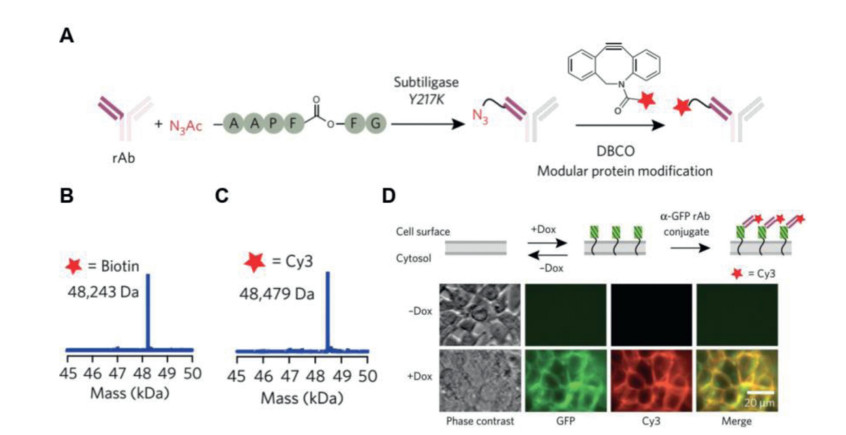
Wells and co-workers have identified a family of 72 mutant subtiligases with N-terminal modification activity and characterized the ligation efficiency for more than 25, 000 enzyme-substrate pairs of peptides. The subtiligase specificity mutant Y217K is applied for high-yield protein bioconjugation to recombinant antibodies (rAbs). A bioconjugation protocol by using subtiligase to incorporate a bio-orthogonal azide group at rAb N-terminus has been developed (Fig. 5A). This azide can be modified through click chemistry with dibenzyocyclooctyne (DBCO) derivatives (biotin-DBCO, Cy3-DBCO etc.). The target modified rAbs, DBCO-biotin-rAb and DBCO-Cy3-rAb have been detected by ESI mass (Figs. 5B and C). The strategy also has utility in biological context, a HEK-293T cell line is modified for doxycycline (Dox)-inducible expression of cell-surface GFP in combination with Cy3-anti-GFP antibody (α-GFP). The Dox-induced cells are observed Cy3-α-GFP binding and colocalization of the Cy3 and GFP signals. Whereas, no binding of Cy3-α-GFP is observed in un-induced cells (Fig. 5D). These results indicate that subtiligase-catalyzed N-terminal modification strategy can be applied in incorporating probes into proteins without affecting their biological functions [57].
Figure 5
 Figure 5. Subtiligase-catalyzed strategy for rAb bioconjugation. (A) Azide-bearing peptide ester reacted with DBCOs, providing a convenient route for modular protein labeling. (B) ESI mass spectra of DBCO-biotin-rAb. (C) ESI mass spectra of DBCO-Cy3-rAb. (D) Cy3-α-GFP rAb staining of a HEK293T cell line modified for Dox-inducible expression of cell surface GFP. Reproduced with permission [57]. Copyright 2017, Springer Nature.
Figure 5. Subtiligase-catalyzed strategy for rAb bioconjugation. (A) Azide-bearing peptide ester reacted with DBCOs, providing a convenient route for modular protein labeling. (B) ESI mass spectra of DBCO-biotin-rAb. (C) ESI mass spectra of DBCO-Cy3-rAb. (D) Cy3-α-GFP rAb staining of a HEK293T cell line modified for Dox-inducible expression of cell surface GFP. Reproduced with permission [57]. Copyright 2017, Springer Nature.Butelase 1 belongs to the C13 subfamily of asparaginyl endopeptidase (AEP) and has been applied as a peptide and protein transpeptidation catalyst, it acts as a ligase under near-neutral conditions to catalyze amide bond formation. Butelase 1 catalyzes the ligation of N-terminal amine and C-terminal Asx (Asp and Asn)-His-Val-motif forming an Asx-Xaa-peptide bond either intra- or inter-molecularly (Fig. 4C) [59, 60]. Butelase 1 has broad tolerance for nearly any N-terminal amino acids Xaa, it is a highly efficient Asx-Xaa-ligase with a catalytic efficiency of up to 1.34 × 106 L mol-1 s-1 [61] and have utility in live cell labeling [62]. James P. Tam and co-workers demonstrate an improved asparaginyl-ligase-catalyzed transpeptidation strategy, metal-complexation based strategy, to increase AEP (including butelase 1) catalyzed N-terminal modification conversions [63]. A major obstacle of butelase 1 is its availability, currently, butelase 1 can only be obtained from C. ternatea via a laborious extraction and theyield is 5 mg/kg of fresh plant. While the recombinant expression of butelase 1 has not been successful [46].
5. Selective N-terminal modification on specific amino acid residues
The unique labeling strategies for specific amino acid residues including serine, threonine, tryptophan, cysteine, proline and glycine at protein N-terminal positions have been reported [22]. While very few studies focus on N-terminal serine, threonine and tryptophan modification recently, quite a few novel strategies for selective N-terminal cysteine, proline and glycine modification have been reported.
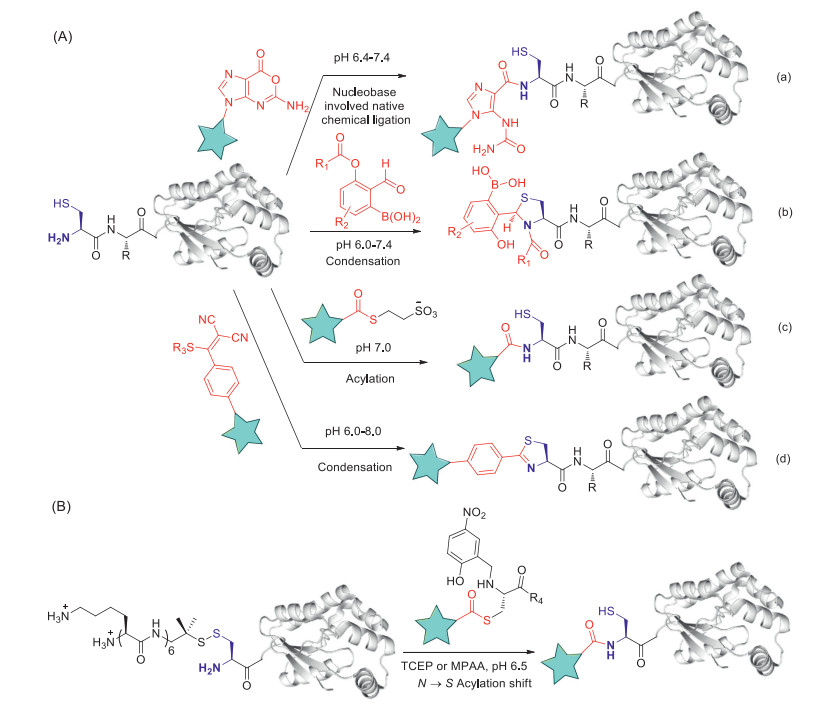
Biocompatible strategies for selective N-terminal cysteine modification are sought after by researchers. Native chemical ligation (NCL) [64-66], hydrazide-based native chemical ligation [67-69] as well as the 2-cyanobenzothiazole (CBT) [70, 71] condensation reactions represent the major strategies for N-terminal cysteine modification previously. Unfortunately, these reactions are less ideal for biological applications due to slow kinetics and/or suboptimal N-terminal cysteine selectivity. Pack and co-workers report nucleobase-involved native chemical ligation (NbCL) that allows a site-specific oligonucleotide-peptide conjugation via a new S-N acyl transfer reaction between an oxanine nucleobase and N-terminal cysteine (Fig. 6A-a). NbCL (2 h) strategy is much faster than NCL strategy (48 h), however, NbCL strategy is performed at 310 K, which should greatly limit its applications especially on proteins [72]. Alternatively, 2-formylphenylboronic acid (2FPBA) mediated N-terminal cysteine modification strategies with the production of a thiazolidino boronate (TzB) complex are reported by the Gao group [73] and the Gois group [74] independently in 2016. However, the TzB formation is dynamic, with the conjugate dissociating over an hour. Recently, the Gao group further optimized this strategy through a TzB-mediated acylation reaction of N-terminal cysteine that gives rise to stable conjugates while retaining the fast kinetics and high selectivity (Fig. 6A-b) [75]. NHS-esters are known to have applications for peptide and protein amine group labeling, however, selectivity of NHS-esters is the principle problem. Cole and co-workers have discovered that commercially available NHS ester can be efficiently transesterified with mercaptoethanesulfonate (MESNA). This newly modified NHS-ester can then be used to specifically label N-terminal amine of recombinant proteins possessing free N-cysteine residues (Fig. 6A-c) [76]. 2-((Alkylthio)(aryl)methylene)malononitrile (TAMM) has been reported to react specifically and rapidly with the N-terminal cysteine under biocompatible conditions through a unique mechanism involving thiol-vinyl sulfide exchange, cyclization, and elimination of dicyanomethanide to form 2-aryl-4, 5-dihydrothiazole (ADT) as a stable unit (Fig. 6A-d). TAMM is also applied to cyclize peptides and proteins containing both an N-terminal and an internal cysteine residue to generate phage-based ADT-cyclic peptide libraries without reducing phage infectivity [77]. Poor solubility of peptide segments is an obvious bottleneck for the chemical synthesis of proteins using NCL. Aucagne and co-workers developed a (Lys)6 tag mediated methodology based on the introduction of an oligolysine tag through a disulfide linkage with the N-terminal cysteine residue to overcome solubility challenges (Fig. 6B). (Lys)6 tag can be cleaved within seconds under NCL conditions to generate in situ the reactive free cysteine for the later ligation reaction [78].
Figure 6
 Figure 6. Modification of specific amino acids: N-terminal cysteine (NCys). (A) Methods include (from top) nucleobase-involved native chemical ligation (NbCL) with oxanine nucleobases; TzB (thiazolidine boronate)-mediated conjugation; NHS-ester transformation with mercaptoethanesulfonate (MESNA) modified NHS esters; 2-aryl-4, 5-dihydrothiazole (ADT) formation with 2-((alkylthio)(aryl)methylene) malononitriles (TAMMs). (B) NCL-based ligation using the (Lys)6 tag methodology.
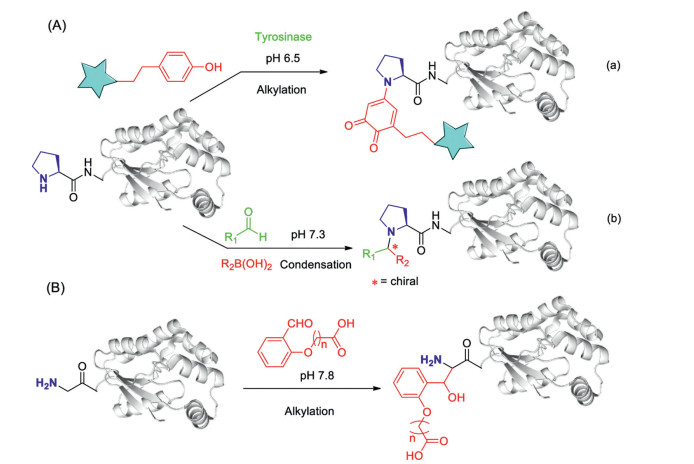
Figure 6. Modification of specific amino acids: N-terminal cysteine (NCys). (A) Methods include (from top) nucleobase-involved native chemical ligation (NbCL) with oxanine nucleobases; TzB (thiazolidine boronate)-mediated conjugation; NHS-ester transformation with mercaptoethanesulfonate (MESNA) modified NHS esters; 2-aryl-4, 5-dihydrothiazole (ADT) formation with 2-((alkylthio)(aryl)methylene) malononitriles (TAMMs). (B) NCL-based ligation using the (Lys)6 tag methodology.Different from other N-terminal residues, the N-terminal proline has a secondary amine group, this feature provides chances for selective N-terminal proline amine modification. Tyrosinase enzyme isolated from Agaricus bisporus (abTYR) can oxidize phenols or catechols to highly reactive o-quinone intermediates that then couple to N-terminal proline residues in high yield under the air condition (Fig. 7A-a) [79]. K3Fe(CN)6 can replace abTYR as a tool for coupling of ortho-aminophenols and N-terminal proline [80], however, free thiols of proteinc ysteines also react rapidly with ortho-aminophenols and should therefore be protected before the modification. In addition, a secondary amine selective Petasis (SASP) bioconjugation method involving a Petasis three-component coupling reaction between proline amine, aldehyde and organoboron reagent has been developed (Fig. 7A-b). The key advantage of the SASP method includes its high chemoselective and stereoselective (> 99% de) nature, moreover, it affords labeled proteins in one pot with broad substrate scope [81]. In addition, aldehydes have been discovered to have application in the site-specific labeling of natural or easy-to-engineer N-terminus glycine in proteins with remarkable efficiency and selectivity (Fig. 7B). The strategy generates a latent nucleophile from N-terminus imine which can react with aldehydes to deliver aminoalcohols under physiological conditions. The mild reaction conditions do not alter the structure and function of the insulin [82].
Figure 7
 Figure 7. Modification of specific amino acids: (A) N-terminal proline and (B) N-terminal glycine. (A) Methods include (from top) tyrosinase mediated site-selective oxidative coupling reactions for the modification of N-terminal proline using phenols; secondary amine selective Petasis (SASP) reaction for selective bioconjugation at N-terminal proline with aldehydes, (B) selective N-terminal glycine modification using aldehydes through the formation of aminoalcohol.
Figure 7. Modification of specific amino acids: (A) N-terminal proline and (B) N-terminal glycine. (A) Methods include (from top) tyrosinase mediated site-selective oxidative coupling reactions for the modification of N-terminal proline using phenols; secondary amine selective Petasis (SASP) reaction for selective bioconjugation at N-terminal proline with aldehydes, (B) selective N-terminal glycine modification using aldehydes through the formation of aminoalcohol.6. Labeling efficiency (conversion) screening on N-terminal amino acid residues
N-Terminal α-amine pKa (6-8) varies from amino acid residues, indicating the different basicity as well as the reactivity. This characteristic results in the N-terminal modification conversion to differ from N-terminal amino acid residues. Table 2 summarized the conversion of partial N-terminal modification strategies in this review. Among the 20 amino acid residues, the N-terminal Gln- and Glu- tend to have relatively higher conversions (average conversion of Gln- and Glu- are 95% and 83% separately) and N-terminal α-amino group selectivity, while N-terminal Thr- and Trp- are more difficult to be modified (average conversion of Thr- and Trp- are 54.3% and 54.5% separately).
Table 2
 Table 2. Summary of partial N-terminal modification strategies' conversions of 20 N-terminal amino acid residues.
Table 2. Summary of partial N-terminal modification strategies' conversions of 20 N-terminal amino acid residues. DownLoad:
CSV
DownLoad:
CSV

7. Conclusion and outlook
Site-specific modification of N-terminus has been attracting significant attention. An increasing number of site-specific modification approaches targeting the α-amine of the N terminus for applications in chemical biology have been developed. However, there is still room for improvement in the existing N-terminal modification strategies. As compared to other bioconjugation strategies, the specific impact of N-terminal modification strategies is their ability to functionalize a wide range of proteins at a single location, which remains challenging in other instances. Alternatively, a single time modification of protein N-terminal location leads to protein less modified especially in protein fluorescence labeling studies, more powerful strategies similar to N-terminal PEGylation and polymerization can represent new aspects of N-terminal modification. Novel N-terminus modification strategies among which the bioconjugation reaction is fast, selective, operates at low-micromolar concentrations and is complementary to existing bioconjugation strategies should be considered for deeper investigation. Another consideration is the accessibility of the reagents used for the N-termini modification. Reagents that are commercially available or can be prepared in simple synthetic steps from commercially available building blocks will have a great vogue. Continued efforts are poised to develop more powerful techniques for N-terminal modification to shed light on critical information of protein profiling and modulating as well as their biological applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was supported by Shandong Provincial Natural Science Foundation, China (No. ZR2020QC081, H. Jiang), and Youth Innovation Team Talent Introduction Program of Shandong Province (No. 20190164, R. Zhang and H. Jiang).
-
-
[1]
N. Krall, F.P. da Cruz, O. Boutureira, et al., Nat. Chem. 8(2016) 103-113. doi: 10.1038/nchem.2393
-
[2]
S.B. van Witteloostuijn, S.L. Pedersen, K.J. Jensen, et al., ChemMedChem 11(2016) 2474-2495. doi: 10.1002/cmdc.201600374
-
[3]
K. Lang, J.W. Chin, Chem. Rev. 114(2014) 4764-4806. doi: 10.1021/cr400355w
-
[4]
L. Witus, M.B. Francis, Acc. Chem. Res. 44(2011) 774-783. doi: 10.1021/ar2001292
-
[5]
S.R. Adusumalli, D.G. Rawale, K. Thakur, et al., Angew. Chem. Int. Ed. 59(2020) 10332-10336. doi: 10.1002/anie.202000062
-
[6]
M.J. Matos, B.L. Oliveira, N. Martinez-Saez, et al., J. Am. Chem. Soc. 140(2018) 4004-4017. doi: 10.1021/jacs.7b12874
-
[7]
D. Hymel, F. Liu, Org. Lett. 22(2020) 3067-3071. doi: 10.1021/acs.orglett.0c00816
-
[8]
A.M. Embaby, S. Schoffelen, C. Kofoed, et al., Angew. Chem. Int. Ed. 57(2018) 8022-8026. doi: 10.1002/anie.201712589
-
[9]
M.J. Matos, C.D. Navo, T. Hakala, et al., Angew. Chem. Int. Ed. 58(2019) 6640-6644. doi: 10.1002/anie.201901405
-
[10]
N. Shindo, H. Fuchida, M. Sato, et al., Nat. Chem. Biol. 15(2019) 250-258. doi: 10.1038/s41589-018-0204-3
-
[11]
M. Zhang, G. Liang, Sci. China Chem. 61(2018) 1088-1098. doi: 10.1007/s11426-018-9277-6
-
[12]
J. Ohata, M.K. Miller, C.M. Mountain, et al., Angew. Chem. Int. Ed. 57(2018) 2827-2830. doi: 10.1002/anie.201711868
-
[13]
D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, et al., J. Am. Chem. Soc. 140(2018) 17120-17126. doi: 10.1021/jacs.8b09372
-
[14]
H.S. Hahm, E.K. Toroitich, A.L. Borne, et al., Nat. Chem. Biol. 16(2020) 150-159. doi: 10.1038/s41589-019-0404-5
-
[15]
S. Sato, K. Nakane, H. Nakamura, Org. Biomol. Chem. 18(2020) 3664-3668. doi: 10.1039/D0OB00650E
-
[16]
X. Chen, F. Ye, X. Luo, et al., J. Am. Chem. Soc. 141(2019) 18230-18237. doi: 10.1021/jacs.9b09127
-
[17]
S.J. Tower, W.J. Hetcher, T.E. Myers, et al., J. Am. Chem. Soc. 142(2020) 9112-9118. doi: 10.1021/jacs.0c03039
-
[18]
M.T. Taylor, J.E. Nelson, M.G. Suero, et al., Nature 562(2018) 563-568. doi: 10.1038/s41586-018-0608-y
-
[19]
S. Lin, X.Y. Yang, S. Jia, et al., Science 355(2017) 597-602. doi: 10.1126/science.aal3316
-
[20]
C.L. Young, Z.T. Britton, A.S. Robinson, Biotechnol. J. 7(2012) 620-634. doi: 10.1002/biot.201100155
-
[21]
S. Bloom, C. Liu, D.K. Kolmel, et al., Nat. Chem. 10(2018) 205-211. doi: 10.1038/nchem.2888
-
[22]
C.B. Rosen, M.B. Francis, Nat. Chem. Biol. 13(2017) 697-705. doi: 10.1038/nchembio.2416
-
[23]
T. Sereda, C.T. Mant, A.M. Quinn, et al., J. Chromatogr. 646(1993) 17-30. doi: 10.1016/S0021-9673(99)87003-4
-
[24]
A.O. Chan, C.M. Ho, H.C. Chong, et al., J. Am. Chem. Soc. 134(2012) 2589-2598. doi: 10.1021/ja208009r
-
[25]
K.K. Kung, K.F. Wong, K.C. Leung, et al., Chem. Commun. 49(2013) 6888-6890. doi: 10.1039/c3cc42261e
-
[26]
J.R. Deng, N.C. Lai, K.K. Kung, et al., Commun. Chem. 3(2020) 67. doi: 10.1038/s42004-020-0309-y
-
[27]
J.I. MacDonald, H.K. Munch, T. Moore, et al., Nat. Chem. Biol. 11(2015) 326-331. doi: 10.1038/nchembio.1792
-
[28]
N. Inoue, A. Onoda, T. Hayashi, Bioconjug. Chem. 30(2019) 2427-2434. doi: 10.1021/acs.bioconjchem.9b00515
-
[29]
B. Koo, N.S. Dolan, K. Wucherer, et al., Biomacromolecules 20(2019) 3933-3939. doi: 10.1021/acs.biomac.9b01002
-
[30]
S.S. Liew, C. Zhang, J. Zhang, et al., Chem. Commun. 56(2020) 11473-11476. doi: 10.1039/D0CC04728G
-
[31]
A. Onoda, N. Inoue, E. Sumiyoshi, et al., ChemBioChem 21(2020) 1274-1278. doi: 10.1002/cbic.201900692
-
[32]
R. Sangsuwan, P. Tachachartvanich, M.B. Francis, J. Am. Chem. Soc. 141(2019) 2376-2383. doi: 10.1021/jacs.8b10947
-
[33]
A. Bagag, J.M. Jault, N. Sidahmed-Adrar, et al., PLoS One 8(2013) e79033. doi: 10.1371/journal.pone.0079033
-
[34]
K.C. Tang, M. Raj, Angew. Chem. Int. Ed. 60(2021) 1797-1805. doi: 10.1002/anie.202007608
-
[35]
M.C. Martos-Maldonado, C.T. Hjuler, K.K. Sorensen, et al., Nat. Commun. 9(2018) 3307. doi: 10.1038/s41467-018-05695-3
-
[36]
O. Boutureira, G.J. Bernardes, Chem. Rev. 115(2015) 2174-2195. doi: 10.1021/cr500399p
-
[37]
D.K. Kolmel, E.T. Kool, Chem. Rev. 117(2017) 10358-10376. doi: 10.1021/acs.chemrev.7b00090
-
[38]
O. Koniev, A. Wagner, Chem. Soc. Rev. 44(2015) 5495-5551. doi: 10.1039/C5CS00048C
-
[39]
K.K. Kung, H.M. Ko, J.F. Cui, et al., Chem. Commun. 50(2014) 11899-11902. doi: 10.1039/C4CC04467C
-
[40]
M.S. Messina, J.M. Stauber, M.A. Waddington, et al., J. Am. Chem. Soc. 140(2018) 7065-7069. doi: 10.1021/jacs.8b04115
-
[41]
K.L. Seim, A.C. Obermeyer, M.B. Francis, J. Am. Chem. Soc. 133(2011) 16970-16976. doi: 10.1021/ja206324q
-
[42]
M.K. Miller, H. Wang, K. Hanaya, et al., Chem. Sci. 11(2020) 10501-10505. doi: 10.1039/D0SC02933E
-
[43]
D. Chen, M.M. Disotuar, X. Xiong, et al., Chem. Sci. 8(2017) 2717-2722. doi: 10.1039/C6SC04744K
-
[44]
E.M. Milczek, Chem. Rev. 118(2018) 119-141. doi: 10.1021/acs.chemrev.6b00832
-
[45]
Y. Zhang, K.Y. Park, K.F. Suazo, et al., Chem. Soc. Rev. 47(2018) 9106-9136. doi: 10.1039/C8CS00537K
-
[46]
S. Lin, C. He, Chin. Chem. Lett. 29(2018) 1017-1021. doi: 10.1016/j.cclet.2018.05.006
-
[47]
M. Schmidt, A. Toplak, P.J. Quaedflieg, et al., Curr. Opin. Chem. Biol. 38(2017) 1-7.
-
[48]
S.H. Henager, N. Chu, Z. Chen, et al., Nat. Methods 13(2016) 925-927. doi: 10.1038/nmeth.4004
-
[49]
Q. Wu, H.L. Ploegh, M.C. Truttmann, ACS Chem. Biol. 12(2017) 664-6673. doi: 10.1021/acschembio.6b00998
-
[50]
Z. Zou, M. Noth, F. Jakob, et al., Bioconjug. Chem. 31(2020) 2476-2481. doi: 10.1021/acs.bioconjchem.0c00486
-
[51]
X. Dai, A. Böker, U. Glebe, RSC Adv. 9(2019) 4700-4721. doi: 10.1039/C8RA06705H
-
[52]
Z. Zou, H. Alibiglou, D.M. Mate, et al., Chem. Commun. 54(2018) 11467-11470. doi: 10.1039/C8CC06017G
-
[53]
C.B. Rosen, R.L. Kwant, J.I. MacDonald, et al., Angew. Chem. Int. Ed. 55(2016) 8585-8589. doi: 10.1002/anie.201602353
-
[54]
T. Kosciuk, H. Lin, ACS Chem. Biol. 15(2020) 1747-1758. doi: 10.1021/acschembio.0c00314
-
[55]
A.M. Weeks, J.A. Wells, Chem. Rev. 120(2020) 3127-3160. doi: 10.1021/acs.chemrev.9b00372
-
[56]
A.M. Weeks, J.A. Wells, Curr. Protoc. Chem. Biol. 12(2020) e79.
-
[57]
A.M. Weeks, J.A. Wells, Nat. Chem. Biol. 14(2018) 50-57. doi: 10.1038/nchembio.2521
-
[58]
X. Tan, R. Yang, C.F. Liu, Org. Lett. 20(2018) 6691-6694. doi: 10.1021/acs.orglett.8b02747
-
[59]
X. Hemu, X. Zhang, J.P. Tam, Org. Lett. 21(2019) 2029-2032. doi: 10.1021/acs.orglett.9b00151
-
[60]
J.P. Tam, N.Y. Chan, H.T. Liew, et al., Sci. China Chem. 63(2020) 296-307. doi: 10.1007/s11426-019-9648-3
-
[61]
G.K. Nguyen, A. Kam, S. Loo, et al., J. Am. Chem. Soc. 137(2015) 15398-15401. doi: 10.1021/jacs.5b11014
-
[62]
X. Bi, J. Yin, G.K. Nguyen, et al., Angew. Chem. Int. Ed. 56(2017) 7822-7825. doi: 10.1002/anie.201703317
-
[63]
F.B. Rehm, T.J. Tyler, K. Yap, et al., Angew. Chem. Int. Ed. 60(2021) 4004-4008. doi: 10.1002/anie.202013584
-
[64]
A.C. Conibear, E.E. Watson, R.J. Payne, et al., Chem. Soc. Rev. 47(2018) 9046-9068. doi: 10.1039/C8CS00573G
-
[65]
R.J. Giesler, P.W. Erickson, M.S. Kay, Curr. Opin. Chem. Biol. 58(2020) 37-44. doi: 10.1016/j.cbpa.2020.04.003
-
[66]
Y. Tan, H. Wu, T. Wei, et al., J. Am. Chem. Soc. 142(2020) 20288-20298. doi: 10.1021/jacs.0c09664
-
[67]
Y. Wu, Y.L. Li, W. Cong, et al., Chin. Chem. Lett. 31(2020) 107-110. doi: 10.1016/j.cclet.2019.05.010
-
[68]
Y. Zheng, F.M. Wu, S.L. Ling, et al., Chin. Chem. Lett. 31(2020) 1267-1270. doi: 10.1016/j.cclet.2019.09.038
-
[69]
C. Zuo, B.C. Zhang, M. Wu, et al., Chin. Chem. Lett. 31(2020) 693-696. doi: 10.1016/j.cclet.2019.08.039
-
[70]
N.A. Patil, J.A. Karas, B.J. Turner, et al., Bioconjug. Chem. 30(2019) 793-799. doi: 10.1021/acs.bioconjchem.8b00908
-
[71]
W. Wang, J. Gao, J. Org. Chem. 85(2020) 1756-1763. doi: 10.1021/acs.joc.9b02959
-
[72]
E.K. Jang, Y. Koike, Y. Ide, et al., Chem. Commun. 56(2020) 5508-5511. doi: 10.1039/C9CC08808C
-
[73]
A. Bandyopadhyay, S. Cambray, J. Gao, Chem. Sci. 7(2016) 4589-4593. doi: 10.1039/C6SC00172F
-
[74]
H. Faustino, M. Silva, L.F. Veiros, et al., Chem. Sci. 7(2016) 5052-5058. doi: 10.1039/C6SC01520D
-
[75]
K. Li, W. Wang, J. Gao, Angew. Chem. Int. Ed. 59(2020) 14246-14250. doi: 10.1002/anie.202000837
-
[76]
D.R. Dempsey, H. Jiang, J.H. Kalin, et al., J. Am. Chem. Soc. 140(2018) 9374-9378. doi: 10.1021/jacs.8b05098
-
[77]
X. Zheng, Z. Li, W. Gao, et al., J. Am. Chem. Soc. 142(2020) 5097-5103. doi: 10.1021/jacs.9b11875
-
[78]
S.A. Abboud, E. h. Cisse, M. Doudeau, et al., Chem. Sci. 12(2021) 3194-3201. doi: 10.1039/D0SC06001A
-
[79]
J.C. Maza, D.L. Bader, L. Xiao, et al., J. Am. Chem. Soc. 141(2019) 3885-3892. doi: 10.1021/jacs.8b10845
-
[80]
A.C. Obermeyer, J.B. Jarman, M.B. Francis, J. Am. Chem. Soc. 136(2014) 9572-9579. doi: 10.1021/ja500728c
-
[81]
Y.E. Sim, O. Nwajiobi, S. Mahesh, et al., Chem. Sci. 11(2020) 53-61. doi: 10.1039/C9SC04697F
-
[82]
L. Purushottam, S.R. Adusumalli, U. Singh, et al., Nat. Commun. 10(2019) 2539. doi: 10.1038/s41467-019-10503-7
-
[1]
-
Figure 1 Selective modification of protein N termini under catalyst-free conditions. Methods include acylation using ketenes (A); condensation with 2-ethynylbenzaldehydes (2EBAs) (B); oxidation with (i) oxone followed by (ii) oxime exchange by functionalized hydroxylamines (C); imidazolidinone formation with 2-pyridinecarboxaldehyde (2PCA) derivatives (D); azolation with azolines (E).
Figure 2 2PCA-polymer strategy for protein delivery. (A) Schematic illustration of protein-polymer conjugate preparation using 2PCA for N-terminal modification. (B) Confocal microscopy images are shown for HeLa cells after exposure to GFP or GFP-polymer conjugates, nuclear and cytoplasmic stains are performed on fixed cells using DAPI and ActinRed, respectively. Scale bars represent 20 µm. Reproduced with permission [32]. Copyright 2019, American Chemical Society.
Figure 3 Selective modification of protein N termini using chemical catalysts. Methods include copper-mediated peptide arylation with boronic acids (A), alkylation by reductive amination with aldehydes (B).
Figure 4 Enzyme-mediated N-terminal modification. (A) Sortase A (SrtA)-catalyzed transpeptidation of an LPXTG peptide derivative for attachment to an H2N-(G)n-protein. (B) Attachment of a glycolate ester substrate mediated by subtiligase. (C) N-terminal acetylation with an Asn/Asp-thiodepsipeptide using butelase 1.
Figure 5 Subtiligase-catalyzed strategy for rAb bioconjugation. (A) Azide-bearing peptide ester reacted with DBCOs, providing a convenient route for modular protein labeling. (B) ESI mass spectra of DBCO-biotin-rAb. (C) ESI mass spectra of DBCO-Cy3-rAb. (D) Cy3-α-GFP rAb staining of a HEK293T cell line modified for Dox-inducible expression of cell surface GFP. Reproduced with permission [57]. Copyright 2017, Springer Nature.
Figure 6 Modification of specific amino acids: N-terminal cysteine (NCys). (A) Methods include (from top) nucleobase-involved native chemical ligation (NbCL) with oxanine nucleobases; TzB (thiazolidine boronate)-mediated conjugation; NHS-ester transformation with mercaptoethanesulfonate (MESNA) modified NHS esters; 2-aryl-4, 5-dihydrothiazole (ADT) formation with 2-((alkylthio)(aryl)methylene) malononitriles (TAMMs). (B) NCL-based ligation using the (Lys)6 tag methodology.
Figure 7 Modification of specific amino acids: (A) N-terminal proline and (B) N-terminal glycine. (A) Methods include (from top) tyrosinase mediated site-selective oxidative coupling reactions for the modification of N-terminal proline using phenols; secondary amine selective Petasis (SASP) reaction for selective bioconjugation at N-terminal proline with aldehydes, (B) selective N-terminal glycine modification using aldehydes through the formation of aminoalcohol.
Table 2. Summary of partial N-terminal modification strategies' conversions of 20 N-terminal amino acid residues.
 下载: 导出CSV
下载: 导出CSV
-









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 77
- 文章访问数: 1423
- HTML全文浏览量: 488
 下载:
下载:
