
Scheme 1.
Selective activation of γ-C(sp2)–H bonds in the present of β-C(sp2)–H bonds.
Weakly coordinating group directed rhodium-catalyzed unconventional site-selective C–H olefination of indolizines at the 8-position
Xue Feng , Jiaxin Tian , Ying Sun , Huayou Hu , Mingzhu Lu , Yuhe Kan , Danjun Fang , Chao Wang

In recent decades, C–H activation reactions have seen much progress and become a hot topic in organic chemistry [1-13]. In most cases, the selectively activation of C–H bonds is achieved by introducing a directing group. The directing group coordinates with a transition metal to form a kinetically and thermodynamically favorable five-membered cyclic intermediate, which promotes the selective activation of β–C–H bonds of the directing group. The activation of γ–C–H bonds can also be achieved via a six-membered cyclic intermediate in the absence of a β–C–H bond [14-18]. However, examples of selectively activation of γ–C–H bonds in the presence of β–C–H bonds are rare. In 2011, Liu and coworkers first reported an example of the palladium-catalyzed C–H amidation of 2,2-dimethyl-1-(1-tosyl-1H-indol-3-yl)propan-1-one at the 4-position [19]. Subsequently, a series of transition-metal-catalyzed C-4 functionalization reactions of 3-carbonyl indole derivatives have been developed by various groups [20-31]. In 2015, Ma and coworkers reported the rhodium-catalyzed C–H activation of indole selectively at the 7-position over the 2-position, which benefited from the steric hindrance of the directing group [32]. Several similar protocols have since been reported by the same and other groups [33-35]. Although Shi [36] and Yu [37] have reported the palladium-catalyzed site-selective C—H activation of γ-C(sp3)–H bonds in the presence of more accessible β-C(sp3)−H bonds via a six-membered palladacycle, the development of selective γ-C–H bond activation in the presence of β–C–H H bonds still remains in much demand (Scheme 1).
Indolizine is an important heterocycle that has long drawn the attention of synthetic and theoretical chemists owing to its unique properties [38-48]. C–H activation of indolizines was first reported by the Gevorgyan group [49] in 2004 and has since become a hot topic. However, only C–H bonds at the 3-position [50-60] and 1-positions [61-66] of indolizine have been successfully activated. Although C–H activation of indolizine at the 5-position has been achieved via tandem reactions, as reported by Zhang and coworkers [54] and our group [59], the selective activation of C–H bonds in the six-membered ring of indolizine remains challenging. Herein, we report a rhodium-catalyzed selective C–H bond activation at the 8-position of indolizine.
In the preliminary study, the model system selected com-prised N, N-dimethyl-3-phenylindolizine-1-carboxamide (1a, 0.20 mmol) and butyl acrylate (2a, 1.2 equiv.) with [Cp*RhCl2]2 (2 mol%) as catalyst, silver hexafluoroantimonate as additive, copper acetate monohydrate (0.20 mmol) as oxidant, and 1,2-dichloroethane (DCE) as solvent. Pleasingly, desired product 3a was detected in 75% yield by 1H NMR when the reaction was performed at 80 ℃ under air for 8 h (Table 1, entry 1). When [Ru(p-cymene)Cl2]2 was used as catalyst, 3a was detected in 27% yield (entry 2). However, no 3a was detected when Pd(OAc)2 was used as catalyst (entry 3). Additive silver hexafluoroantimonate was shown not to be necessary (entry 4). Several solvents were tested, with 1,1,1-trifluoroethanol (TFE) proven be the best single solvent (entry 7). However, the yield of 3a only reached 78% in TFE. Therefore, we tested mixtures of two different solvents. The mixture of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and 1,2-dichloroethane (DCE) gave the best result, affording a 91% yield of 3a (entry 11). Increasing the amount of copper acetate monohydrate to 2.1 equiv. and running the reaction under an argon atmosphere further improved the yield of 3a to 94%. Finally, the following optimized reaction conditions were established: [Cp*RhCl2]2 (2 mol%), copper acetate monohydrate (2.1 equiv.), mixed solvent HFIP/DCE (0.8 mL; 1:9, v/v), reacted under Argon at 80 ℃. Under these conditions, 3a was isolated in 87% yield (entry 12).
With optimal conditions in hand, a series of alkenes were reacted with 1a as coupling partner (Scheme 2). Acrylates gave the corresponding products in good yields, even when bulky t-butyl acrylate (2c) was used as the alkene source. (Vinylsulfonyl)benzene (2d) was also well tolerated, giving indolizine 3d in 93% yield. Acrylonitrile only gave corresponding indolizine 3e in moderate yield, even when the catalyst loading was increased to 4 mol% and the reaction time was prolonged to 24 h. Other electron deficient alkenes, such as dimethyl vinylphosphonate, N, N-dimethylacrylamide, and N-methyl-N-phenylacrylamide gave indolizines in low yields at 80 ℃. However, increasing the reaction temperature to 110 ℃ and the reaction time to 24 h greatly improved the yields (3f–3h). Notably, styrene also reacted with 1a at 110 ℃ to give indolizine 3i in 59% yield.
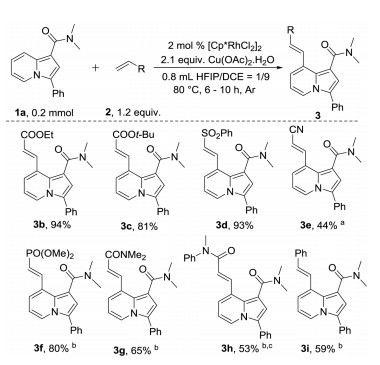
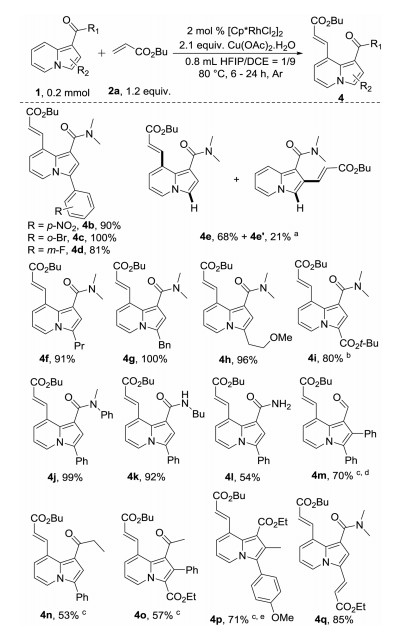
Next, the scope of indolizines was explored (Scheme 3). In-dolizines with different substituents at the phenyl group of the 3-position gave good yields of corresponding products, with even the strongly electron-withdrawing nitro group tolerated (4b). Interestingly, N, N-dimethylindolizine-1-carboxamide (1e) was more reactive than other indolizines, affording corresponding product 4e in 68% yield and 2-olefination by-product 4e′ in 21% yield at 70 ℃. This result indicates that the selectivity of the reaction, although benefiting from steric hindrance, does not depend on steric hindrance of 3-position. Indolizines with different alkyl groups at the 3-position were also well tolerated (4f–4h). The indolizine with an electron withdrawing ester group at the 3-position was less reactive, giving desired product 4i in 80% yield at 90 ℃. Interestingly, olefination only occurred at the 8-position of indolizine, even when a phenyl group was attached to the nitrogen atom of the amide directing group (4j). Monosubstituted and unsubstituted amides were also well tolerated as directing groups, giving the corresponding products in moderate to excellent yields (4k and 4l). Aldehyde and ketone were also tolerated as directing groups giving corresponding products 4m–4n in moderate to good yields at 110 ℃. Ester was an inefficient directing group under standard reaction conditions. However, the corresponding product (4p) was isolated in 71% yield when AgSbF6 was added as an additive. 3,8-Di-olefination of indolizine also was show in Scheme 3 (4q) via combination of this work with our previous work [55].
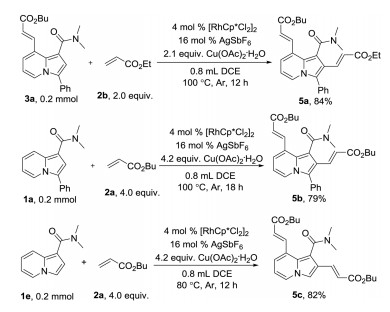
C–H olefination at 2-position was failed under the standard re-action conditions. However, 5a was isolated in 84% yield using 8-olefinated-indolizine 3a as the starting material and AgSbF6 as the additive. Under similar conditions, 2,8-di-olefination of 1a and 1e were successful and give 5b and 5c in 79% and 82% yields (Scheme 4).
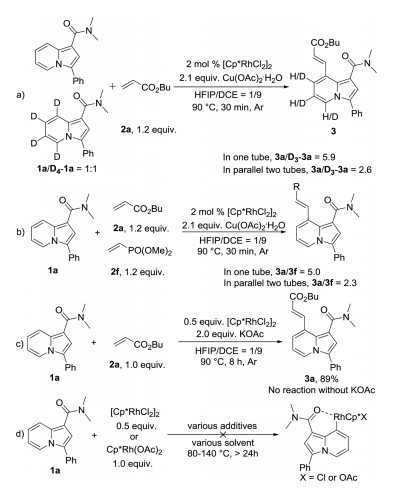
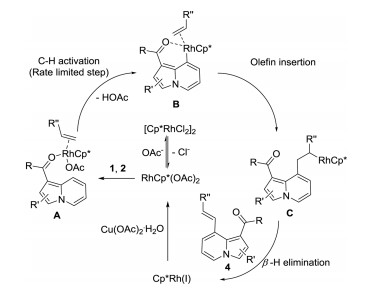
To determine the reaction mechanism, a series of kinetic and control experiments were designed (Scheme 5). First, to determine the rate-limiting step of this reaction, 1a and deuterated 1a (D4-1a) were reacted with 2a in one tube or in two parallel tubes under the optimized conditions at 90 ℃ for 30 min. The obtained ratios of 3a/D3-3a were 5.9 and 2.6 respectively. Second, 1a were reacted with 2a and 2f in one tube or in two parallel tubes under the optimized conditions at 90 ℃ for 30 min. The obtained ratios of 3a/3f were 5.0 and 2.3 respectively. Third, 1a was reacted with 2a using 0.5 equiv. of [RhCp*Cl2]2 under standard reaction conditions. 3a was isolated in 89% yield when 2.0 equiv. of potassium acetate was added and no reaction in the absence of potassium acetate. However, 1a did not react with [RhCp*Cl2]2 or RhCp*(OAc)2 in various solvents with various additives (water, potassium acetate, AgSbF6, copper acetate). These results indicated that the C–H activation was the rate-limiting step, both alkene and acetate anion were involved in the transition state of C–H activation. Furthermore, a Rh(Ⅲ)/Rh(Ⅰ) catalytic cycle was more likely other than the recently reported Rh(Ⅴ)/Rh(Ⅲ) catalytic cycle [67].
Based on the above results and recently reported theoretical calculation results [68], a plausible reaction mechanism was pro-posed (Scheme 6). First, precatalyst [Cp*RhCl2]2 reacts with acetate anion to form activated catalyst Cp*Rh(OAc)2. The activated catalyst then coordinates with the starting material (1) and alkene (2) to form intermediate A. Then A undergoes preferential C–H activation to form intermediate B. Intermediate B then gives intermediate C via olefin insertion. Intermediate C gives the desired product and low-valence RhCp*(Ⅰ) catalyst via β-H elimination. Copper acetate then re-oxidizes RhCp*(Ⅰ) to Cp*Rh(OAc)2 and finish the catalytic cycle.
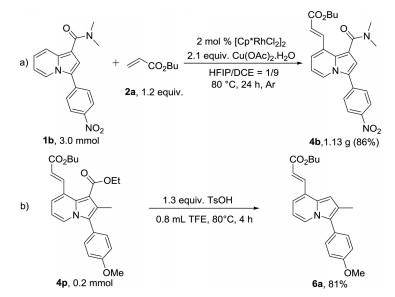
Under the standard reaction conditions, 3.0 mmol of 1b was reacted with 2a to afford 4b in an isolated yield of 86% (1.13 g), which was similar to that of the small-scale reaction. The ester directing group can be removed under acid conditions using trifluoroethanol (TFE) as the solvent and yield 6a in 81%. These results indicate that the reaction was promising in organic synthesis (Scheme 7).
In conclusion, a rhodium-catalyzed weakly coordinating group-directed C–H olefination of indolizines at the 8-position and di-olefination of indolizines at the 2,8-positions were developed. A broad range of functional groups were tolerated in this reaction. Preliminary mechanistic studies showed that the C–H action was the rate-limiting step and alkene was involved in the transition state of C–H activation. The study of specific reaction mechanism of this reaction is ongoing. The versatility of olefinated indolizine moieties should make this protocol highly attractive for use in both materials and medicinal chemistry.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledged Jiangsu Province (No. BK20161307 and "333" Talents Project for H. Hu) and Huaiyin Normal University (No. JSKC18014) for their financial support.
Supplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.039.
F.S. Melkonyan, V. Gevorgyan, Y. Nakao, in: J. Yu (Ed.), Science of Synthesis, Catalytic Transformations via C-H Activation, Georg Thieme Verlag, Stuttgart, 2016.
D.J. Abrams, P.A. Provencher, E.J. Sorensen, Chem. Soc. Rev. 47 (2018) 8925-8967. doi: 10.1039/C8CS00716K
C. Sambiagio, D. Schönbauer, R. Blieck, et al., Chem. Soc. Rev. 47 (2018) 6603-6743. doi: 10.1039/C8CS00201K
P. Gandeepan, T. Mueller, D. Zell, et al., Chem. Rev. 119 (2019) 2192-2452. doi: 10.1021/acs.chemrev.8b00507
S. Rej, N. Chatani, Angew. Chem., Int. Ed. 58 (2019) 8304-8329. doi: 10.1002/anie.201808159
T. Gensch, M.N. Hopkinson, F. Glorius, J. Wencel-Delord, Chem. Soc. Rev. 45 (2016) 2900-2936. doi: 10.1039/C6CS00075D
C.G. Newton, S. Wang, C.C. Oliveira, N. Cramer, Chem. Rev. 117 (2017) 8908-8976. doi: 10.1021/acs.chemrev.6b00692
J. Lv, X. Chen, X. Xue, et al., Nature 575 (2019) 336-340. doi: 10.1038/s41586-019-1640-2
X. Qiu, P. Wang, D. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 1504-1508. doi: 10.1002/anie.201813182
Y. Hu, B. Zhou, H. Chen, C. Wang, Angew. Chem. Int. Ed. 57 (2018) 12071-12075. doi: 10.1002/anie.201806287
X. Qiu, H. Deng, Y. Zhao, Z. Shi, Sci. Adv. 4 (2018) aau6468. doi: 10.1126/sciadv.aau6468
A.J. Borah, Z. Shi, J. Am. Chem. Soc. 140 (2018) 6062-6066. doi: 10.1021/jacs.8b03560
Y. Hu, B. Zhou, C. Wang, Acc. Chem. Res. 51 (2018) 816-827. doi: 10.1021/acs.accounts.8b00028
C. Ghosh, P.J. Nagtilak, M. Kapur, Org. Lett. 21 (2019) 3237-3241. doi: 10.1021/acs.orglett.9b00958
L. Zou, P. Li, B. Wang, L. Wang, Chem. Comm. 55 (2019) 3737-3740. doi: 10.1039/C9CC01014A
Y. Shi, J. Liu, Y. Yang, J. You, Chem. Comm. 55 (2019) 5475-5478. doi: 10.1039/C9CC01733J
G. Wu, W. Ouyang, Q. Chen, Y. Huo, X. Li, Org. Chem. Front. 6 (2019) 284-289. doi: 10.1039/C8QO01105B
Q. Bu, T. Rogge, V. Kotek, L. Ackermann, Angew. Chem. Int. Ed. 57 (2018) 765-768. doi: 10.1002/anie.201711108
B. Xiao, T. Gong, J. Xu, Z. Liu, L. Liu, J. Am. Chem. Soc. 133 (2011) 1466-1474. doi: 10.1021/ja108450m
V. Lanke, K.R. Prabhu, Org. Lett. 15 (2013) 6262-6265. doi: 10.1021/ol4031149
V. Lanke, K.R. Bettadapur, K.R. Prabhu, Org. Lett. 18 (2016) 5496-5499. doi: 10.1021/acs.orglett.6b02698
M.S. Sherikar, R. Kapanaiah, V. Lanke, K.R. Prabhu, Chem. Commun. 54 (2018) 11200-11203. doi: 10.1039/C8CC06264A
X. Chen, G. Zheng, Y. Li, G. Song, X. Li, Org. Lett. 19 (2017) 6184-6187. doi: 10.1021/acs.orglett.7b03099
J. Lv, B. Wang, K. Yuan, Y. Wang, Y. Jia, Org. Lett. 19 (2017) 3664-3667. doi: 10.1021/acs.orglett.7b01681
S. Chen, B. Feng, X. Zheng, et al., Org. Lett. 19 (2017) 2502-2505. doi: 10.1021/acs.orglett.7b00730
N. Thrimurtulu, A. Dey, A. Singh, et al., Adv. Syn. & Cat. 361 (2017) 1441-1446.
K.R. Bettadapur, R. Kapanaiah, V. Lanke, K.R. Prabhu, J. Org. Chem. 83 (2018) 1810-1818. doi: 10.1021/acs.joc.7b02719
T. Okada, A. Sakai, T. Hinoue, et al., J. Org. Chem. 83 (2018) 5639-5649. doi: 10.1021/acs.joc.8b00638
A.J. Boraha, Z. Shi, Chem. Commun. 53 (2017) 3945-3948. doi: 10.1039/C7CC01274H
Y. Yang, P. Gao, Y. Zhao, Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 3966-3971. doi: 10.1002/anie.201612599
V. Lanke, K.R. Prabhu, Chem. Commun. 53 (2017) 5117-5120. doi: 10.1039/C7CC00763A
L. Xu, C. Zhang, Y. He, L. Tan, D. Ma, Angew. Chem. Int. Ed. 55 (2016) 321-325. doi: 10.1002/anie.201508117
Y. Kim, J. Park, S. Chang, Org. Lett. 18 (2016) 1892-1895. doi: 10.1021/acs.orglett.6b00662
Z. Song, A.P. Antonchick, Org. Biomol. Chem. 14 (2016) 4804-4808. doi: 10.1039/C6OB00926C
L. Xu, L. Tan, D. Ma, J. Org. Chem. 81 (2016) 10476-10483. doi: 10.1021/acs.joc.6b01856
J. Xu, Z. Zhang, W. Rao, B. Shi, J. Am. Chem. Soc. 138 (2016) 10750-10753. doi: 10.1021/jacs.6b05978
G. Xia, J. Weng, L. Liu, et al., Nat. Chem. 11 (2019) 571-577. doi: 10.1038/s41557-019-0245-6
E. Kim, Y. Lee, S. Lee, S.B. Park, Acc. Chem. Res. 48 (2015) 538-547. doi: 10.1021/ar500370v
M. Meazza, L.A. Leth, J.D. Erickson, K.A. Jorgensen, Chem. -Eur. J. 23 (2017) 7905-7909. doi: 10.1002/chem.201701820
D. Wu, L. Chen, S. Ma, et al., Org. Lett. 20 (2018) 4103-4106. doi: 10.1021/acs.orglett.8b01663
S. Roy, S.K. Das, B. Chattopadhyay, Angew. Chem. Int. Ed. 57 (2018) 2238-2243. doi: 10.1002/anie.201711209
X. Li, J. Zhao, X. Xie, Y. Liu, Org. Biomol. Chem. 15 (2017) 8119-8133. doi: 10.1039/C7OB02102J
J. Gayton, S.A. Autry, W. Meador, et al., J. Org. Chem. 84 (2019) 687-697. doi: 10.1021/acs.joc.8b02521
S. Asako, T. Kobashi, K. Takai, J. Am. Chem. Soc. 140 (2018) 15425-15429. doi: 10.1021/jacs.8b09297
T. Jin, Z. Tang, J. Hu, et al., Org. Lett. 20 (2018) 413-416. doi: 10.1021/acs.orglett.7b03696
J. Vaitla, A. Bayer, K.H. Hopmann, Angew. Chem. Int. Ed. 57 (2018) 16180-16184. doi: 10.1002/anie.201810451
W. Wang, J. Han, J. Sun, Y. Liu, J. Org. Chem. 82 (2017) 2835-2842. doi: 10.1021/acs.joc.6b02455
Y. Liu, H. Hu, J. Zhou, et al., Org. Biomol. Chem. 15 (2017) 5016-5024. doi: 10.1039/C7OB00980A
C.H. Park, V. Ryabova, I.V. Seregin, A.W. Sromek, V. Ge-vorgyan, Org. Lett. 6 (2004) 1159-1162. doi: 10.1021/ol049866q
I.V. Seregin, V. Ryabova, V. Gevorgyan, J. Am. Chem. Soc. 129 (2007) 7742-7743. doi: 10.1021/ja072718l
J. Xia, X. Wang, S. You, J. Org. Chem. 74 (2009) 456-458. doi: 10.1021/jo802227u
D. Lapointe, K. Fagnou, Org. Lett. 11 (2009) 4160-4163. doi: 10.1021/ol901689q
Y. Yang, K. Cheng, Y. Zhang, Org. Lett. 11 (2009) 5606-5609. doi: 10.1021/ol902315w
Y. Yang, L. Chen, Z. Zhang, Y. Zhang, Org. Lett. 13 (2011) 1342-1345. doi: 10.1021/ol200025k
H. Hu, Y. Liu, H. Zhong, et al., Chem. -Asian J. 7 (2012) 884-888. doi: 10.1002/asia.201101050
B. Zhao, Org. Biomol. Chem. 10 (2012) 7108-7119. doi: 10.1039/c2ob25643f
Y. Ma, J. You, F. Song, Chem. Eur. J. 19 (2013) 1189-1193. doi: 10.1002/chem.201203354
H. Hu, Y. Liu, J. Xu, et al., RSC Adv. 4 (2014) 24389-24393. doi: 10.1039/c4ra03799e
H. Hu, G. Li, W. Hu, et al., Org. Lett. 17 (2015) 1114-1117. doi: 10.1021/ol503681n
K. Nishino, S. Tsukahara, Y. Ogiwara, N. Sakai, Eur. J. Org. Chem. 2019 (2019) 1588-1593. doi: 10.1002/ejoc.201801765
B. Liegault, D. Lapointe, L. Caron, A. Vlassova, K. Fagnou, J. Org. Chem. 74 (2009) 1826-1834. doi: 10.1021/jo8026565
H. Do, O. Daugulis, J. Am. Chem. Soc. 133 (2011) 13577-13586. doi: 10.1021/ja2047717
B. Koszarna, R. Matczak, M. Krzeszewski, et al., Tetrahedron 70 (2014) 225-231. doi: 10.1016/j.tet.2013.11.088
E.J. Choi, E. Kim, Y. Lee, A. Jo, S.B. Park, Angew. Chem. Int. Ed. 53 (2014) 1346-1350. doi: 10.1002/anie.201308826
E. Choi, S. Park, Org. Biomol. Chem. 13 (2015) 5202-5208. doi: 10.1039/C5OB00551E
M. Lu, F. Shi, M. Ji, Y. Kan, H. Hu, J. Org. Chem. 8 (2019) 1555-1560.
J. Kim, K. Shin, S. Jin, D. Kim, S. Chang, J. Am. Chem. Soc. 141 (2019) 4137-4146. doi: 10.1021/jacs.9b00364
R.A. Alharis, C.L. McMullin, D.L. Davies, K. Singh, S.A. Macgregor, J. Am. Chem. Soc. 141 (2019) 8896-8906. doi: 10.1021/jacs.9b02073

Scheme 2 Scope of alkenes. Isolated yields. a Using 4 mol% catalyst for 24 h. b Conducted at 110 ℃ for 24 h. c Using 1.5 equiv. of alkene.

Scheme 3 Scope of indolizines. a Conducted at 70 ℃. b Conducted at 90 ℃. c Conducted at 110 ℃. d Using 4 mol% catalyst. e with 8 mol% AgSbF6.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:




 下载:
下载: