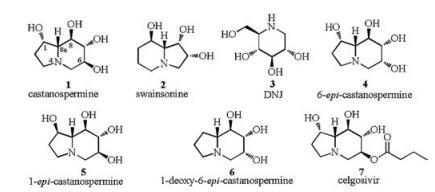
图 1
Castanospermine and its related compounds.
Figure 1.
Castanospermine and its related compounds.

Polyhydroxylated indolizidines are an important class of naturally occurring alkaloids with castanospermine (1), swainsonine (2), 6-epi-castanospermine (4) etc. as representatives. Most of these alkaloids are known as potent glycosidase inhibitors [1], and exhibit diverse biological activity especially as potential therapeutic agents in the treatment of cancer [2], diabetes [3], obesity [4], and HIV [5].
As one of the most-studied polyhydroxylated indolizidines, castanospermine (1) (Fig. 1) was first isolated in 1981 from Australian legume Castanospermum australe [6] and then from the dried pod of Alexa leiopetala [7] as a powerful inhibitor of α-and β-glucosidases [8]. In order to clarify the structure activity relationship (SAR) and to search for pharmaceutical lead compounds, a number of natural or unnatural analogues of 1 have been synthesized and studied for their biological activities. Amongst these analogues, the naturally occurring 6-epi-castanospermine (4) was proved to be a potent inhibitor of amyloglucosidase [9], the unnaturally occurring 1-epi-castanospermine (5) was found to be a potential anti-AIDS lead compound [10] and 1-deoxy-6-epi-castanospermine (6) exhibited competitive inhibition of lysosomal α-mannosidase [11]. Of course, the most successful castanospermine analogue developed till now must be 6-O-butanoyl-castanospermine (7, celgosivir), which is currently undergoing phase Ⅱ clinical trials for treatment of hepatitis C [12]. All of the above analogues, including compound 5, the synthetic C-1 diastereomer of castanospermine, have been extensively studied for more efficient synthetic methodologies in order to establish detailed structure-activity relationship (SAR) [10, 13]. Herein we reported the stereoselective synthesis of 1-epi-castanospermine (5) from readily available starting material 2, 3, 4, 6-tetra-O-benzyl-1-deoxynojirimycin (11).
Since 1-epi-castanospermine (5) could be regarded as a bicyclic analogue of 1-deoxynojirimycin (DNJ, 3) [14], a retrosynthetic analysis can easily break the target 1-epi-castanospermine (5) to the readily available D-glucose-derived 2, 3, 4, 6-tetra-O-benzyl-1-deoxynojirimycin (11). Compound 11 could be converted to alcohol 10 according to literature methods, and then oxidized to aldehyde 9. Subsequent allyl addition and reductive amination would provide the target product 5 (Scheme 1).
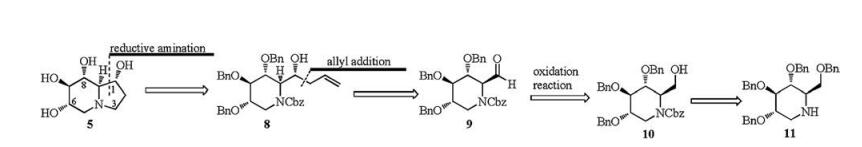
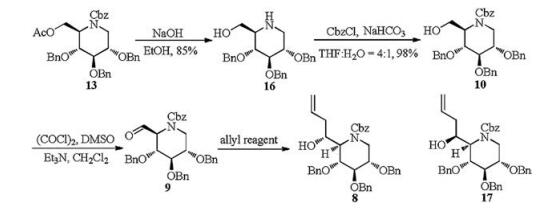
2, 3, 4, 6-Tetra-O-benzyl-1-deoxynojirimycin (11) was prepared from commercially available 2, 3, 4, 6-tetra-O-benzyl-D-glucopyranose in three steps over 72% total yield [15]. Treatment of compound 11 with different protecting reagents, such as AcCl, (Boc)2O, BnBr and CbzCl gave the corresponding N-Ac, N-Boc, N-Bn and N-Cbz products respectively, and then put under selective debenzylation and in situ acetylation by the AcOH/Ac2O/2%H2SO4 condition [15, 16] (Scheme 2). Compounds with N-Ac, or N-Boc, or N-Bn recovered only their corresponding starting materials. However, compound 12 with N-Cbz protecting group proceeded smoothly and provided the target selective debenzylation and in situ acetylation product 13 in 81% yield. Hydrolysis of compound 13 under basic conditions (NaOMe or K2CO3) gave the oxazolidinone 15 instead of the expected alcohol 10, which may be obtained by the intramolecular nucleophilic attack of the oxygen anion to the benzyloxycarbonyl of intermediate 14
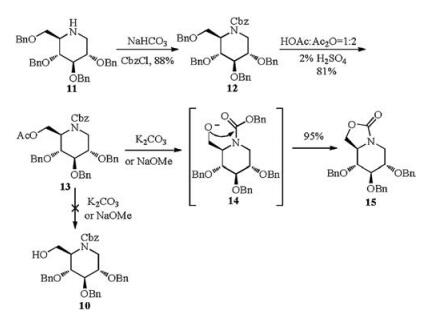
A two-step procedure was then tried for synthesis of alcohol 10 (Scheme 3). 6-O-Ac and N-Cbz groups were first removed under strong basic condition in one pot to furnish compound 16, and then treated with CbzCl in weak basic condition to give the desired product 10 in high yield (98%). Swern oxidation of compound 10 gave aldehyde 9, which was unstable and directly used in the next step without purification. A series of allylation conditions were attempted to finish the synthesis of compound 8 and its epimer 17. Addition of allylmagnesium bromide to aldehyde 9 afforded isomer 8 exclusively (Table 1) in 78% yield for 2 steps (entry 1). Similarly, reaction of allylzinc bromide (entry 2) [17] with aldehyde 9 under Barbier conditions [17, 18] gave also the single isomer 8, but in better yield (88% for 2 steps). The configuration of the newlyformed chiral center may be rationalized by a Felkin-Anh transition state, which is known to be the preferred pathway in the addition of organometallic reagents to carbonyl compounds (Fig. 2). However, other conditions including selective chelationcontrolled [19], Sakurai (entries 3 and 4) [20, 21] and Keck reaction (entry 5) [22] did not give any expected product.
 下载:
导出CSV
下载:
导出CSV

|
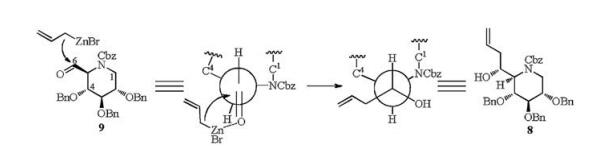
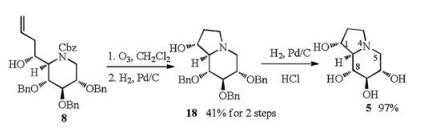
Alcohol 8 was then converted to aldehyde by ozonolysis (Scheme 4), which was directly used in the intramolecular reductive amination [23] procedure to establish the bicyclic compound 18 in 41% yield over 2 steps. Subsequent hydrogenolysis of 18 afforded 1-epi-castanospermine (5) in good yield (97%). The C-1 configuration of 5 was determined as R by the NOESY experiment of 5, which showed strong interaction between H-1 and H-8 (Supporting information). The 1H NMR and 13C NMR spectra of the synthetic 1-epi-castanospermine (5) were identical to those reported in literature with optical rotation ([α]D20 +6.0 (c 0.5 in MeOH)) in consistent with literature [13d] ([α]D20 +6.2 (c 0.15 in MeOH)).
In summary, we have synthesized 1-epi-castanospermine from the common precursor 2, 3, 4, 6-tetra-O-benzyl-1-deoxynojirimycin (11) in 9 steps and 21% overall yield, with selective debenzylation, Barbier reaction and reductive amination as the key steps. The synthetic method is efficient and general for the synthesis of analogues of castanospermine, which will be valuable for in-depth study of the structure-activity relationship of this class of compounds.
All reagents were used as received from commercial sources or prepared according to the literature. Analytical TLC were performed with 0.20 mm silica gel 60F plates and visualized by ultraviolet light or by treatment with a spray of Pancaldi reagent (NH4)6MoO4, Ce(SO4)2, H2SO4, H2O. Column chromatographic purification of products was carried out on silica gel (200-300 mesh). Melting points were determined using an electrothermal melting point apparatus and were uncorrected. Acidic ion exchange chromatography was performed on a Dowex 50WX2-400, H+ form. Infrared spectra were recorded on an FT-IR spectrometer. 1H NMR spectra were measured in CDCl3 (with TMS as an internal standard) or D2O on a magnetic resonance spectrometer (1H at 300, 400, or 500 MHz, 13C at 75, 100, or 125 MHz). High resolution mass spectra (HRMS) were recorded on an LTQ/FT linear ion trap mass spectrometer. Polarimetry was carried out using an Optical Activity AA-10R polarimeter and the measurements were made at the sodium D-line with a 0.5 dm path length cell. Concentrations (c) are given in gram per 100 mL.
The detailed information of synthesis of compounds 11-13, 15 and 16, the spectra of all compounds are deposited in Supporting information.
To a stirred solution of 16 (7.4 g, 16.5 mmol) in 50 mL THF/H2O (4:1, v/v) was added NaHCO3 (2.8 g, 33.3 mmol), followed by dropwise addition of CbzCl (3.6 mL, 24.8 mmol) under 0 ℃. The reaction mixture was stirred at room temperature for 1 h until TLC showed completion of the reaction. The mixture was extracted with EtOAc (3 × 40 mL). The organic phases were combined, dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (EtOAc/petroleum ether = 1:4) to afford compound 10 (9.2 g, 98% yield) as a white solid.
Data for 10: [α]D20 +4.0 (c 1.0 in CH2Cl2); m.p.101-103 ℃; IR (KBr, cm-1): 3446, 2878, 1698, 1089, 1071, 735, 697; 1H NMR (400 MHz, CDCl3): δ 7.46-7.39 (m, 20H), 5.24 (s, 2H), 4.87-4.75 (m, 5H), 4.61 (d, 1H, J = 11.8 Hz), 4.12 (s, 1H), 3.99 (s, 2H), 3.88-3.87 (m, 3H), 3.76-3.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 156.44, 138.28, 138.16, 136.50, 128.65, 128.59, 128.54, 128.51, 128.18, 128.04, 128.01, 127.92, 127.90, 127.85, 127.79, 81.92, 77.55, 75.35, 73.44, 73.32, 71.25, 67.61, 61.36, 58.87, 42.57; HRMS-ESI (m/z): calcd. for C35H37O6NNa+: 590.2513 [M+Na]+, found: 590.2504.
To a stirred solution of oxalyl chloride (0.24 mL, 2.52 mmol) in dry CH2Cl2 (15 mL) was added DMSO (0.24 mL, 3.36 mmol) in CH2Cl2 (2 mL) dropwise via syringe at -78 ℃ under an atmosphere of argon. After 20 min, a solution of 10 (950 mg, 1.68 mmol) in CH2Cl2 (3 mL) was added dropwise via syringe. After 20 min, the reaction mixture was quenched with Et3N (0.70 mL, 5.04 mmol), then the mixture was stirred under -78 ℃ for another 15 min. Upon warming to room temperature, the reaction was stirred for 45 min. The mixture was extracted with CH2Cl2 (3 × 8 mL). The organic phases were dried over MgSO4 and concentrated in vacuo. The resulting crude product aldehyde 9 was used directly in the next step without purification. To a stirred solution of aldehyde 9 in THF (5 mL) was added activated zinc (550 mg, 8.46 mmol) and allyl bromide (0.6 mL, 6.72 mmol), then the mixture was added saturated aq. NH4Cl dropwise and the reaction were initiated. The reaction mixture was stirred for 1 h. The mixture was filtrated and extracted with EtOAc (3 × 8 mL). The organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (EtOAc/petroleum ether = 1:10) to afford compound 8 (900 mg, 88% yield for 2 steps) as a light yellow syrup.
Data for 8: [α]D20 +6.0 (c 1.7 in CH2Cl2); IR (KBr, cm-1): 3447, 3031, 2927, 1697, 1454, 1070, 735, 697; 1H NMR (500 MHz, CDCl3): δ 7.32-7.18 (m, 20H), 5.80 (s, 1H), 5.10 (s, 4H), 4.67-4.46 (m, 6H), 4.25 (s, 1.4H), 4.03-3.97 (m, 2.6H), 3.78 (d, 1H, J = 3.6 Hz), 3.64 (s, 1H), 3.37 (s, 1H), 2.72 (s, 0.5H), 2.30-2.12 (m, 2.5H); 13C NMR (125 MHz, CDCl3): δ 156.74, 138.33, 138.29, 137.74, 136.59, 136.37, 134.88, 129.00, 128.76, 128.61, 128.51, 128.39, 128.21, 128.17, 128.04, 128.01, 127.97, 127.89, 127.72, 127.57, 118.27, 79.42, 75.96, 74.38, 73.42, 72.85, 71.93, 71.46, 70.87, 70.17, 69.64, 67.53, 59.07, 58.73, 41.44, 39.71, 39.11, 38.24; HRMS-ESI (m/z): calcd. for C38H41O6NNa+: 630.2826 [M+Na]+, found: 630.2820.
The solution of compound 8 (668 mg, 1.1 mmol) in methanol (70 mL) was cooled to -60 ℃ and purged with oxygen, then submitted to the ozonization procedure until TLC showed completion of the reaction. The reaction mixture was purged with argon and quenched by Me2S (1 mL). The resulting mixture was stirred for 2 h at room temperature, and then concentrated in vacuo. The resulting aldehyde was dissolved in methanol (10 mL), followed by 10% Pd/C (30 mg) under an argon atmosphere. The suspension was stirred at room temperature under hydrogen atmosphere overnight. TLC showed completion of the reaction. The reaction mixture was purged with nitrogen, catalyst was removed by filtration, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (EtOAc/petroleum ether = 1:3) to afford compound 18 (207 mg, 41% yield for 2 steps) as a yellow syrup.
Data for 18: 
To a stirred solution of 18 (50 mg, 0.11 mmol) in MeOH (5 mL) was added 10% Pd/C (10 mg) and concentrated HCl (1 mL) under an atmosphere of argon. The resulting suspension was stirred under an atmosphere of H2 for 24 h. Hydrogen was then replaced with nitrogen, and the catalyst was removed from the reaction mixture by filtration, and then washed with MeOH (3 × 3 mL). The filtrate was concentrated in vacuo, then the residue was dissolved in MeOH (1 mL), neutralized with aqueous ammonium solution, and concentrated in vacuo. The above procedure was repeated for three times to ensure complete neutralization. The residue was purified by an acidic ion exchange column (Dowex 50W × 2-400, H+ form), eluting with distilled water (50 mL) and then 1 mol/L NH4OH (50 mL), affording 5 (20 mg, 97%) as a light yellow solid.
Data for 5: [α]D20 +6.0 (c 0.5 in MeOH); mp 77-79 ℃; 1H NMR (400 MHz, D2O): δ 4.22 (s, 1H), 3.60 (dd, 1H, J = 14.2, 9.4 Hz), 3.39-3.29 (m, 2H), 3.15 (dd, 1H, J = 10.9, 5.0 Hz), 2.94 (t, 1H, J = 8.7 Hz), 2.60 (q, 1H, J = 9.1 Hz), 2.33-2.23 (m, 2H), 2.15 (t, 1H, J = 7.2 Hz), 1.68 (t, 1H, J = 10.7 Hz); 13C NMR (100 MHz, D2O): δ 78.50, 73.56, 73.07, 72.70, 69.65, 54.57, 50.79, 32.30; HRMS-ESI (m/z): calcd. for C8H16O4N+ 190.1074 [M+H]+, found 190.1071. All the data are paralleled with literatures [13d, 24].
Financial support from the National Natural Science Foundation of China (No. 21272240), National Science and Technology Major Projects for "Major New Drugs Innovation and Development" (No. 2013ZX09508104) and National Engineering Research Center for Carbohydrate Synthesis of Jiangxi Normal University are gratefully acknowledged.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2017.05.013.
(a) V. H. Lillelund, H. H. Jensen, X. Liang, M. Bols, Recent developments of transition-state analogue glycosidase inhibitors of non-natural product origin, Chem. Rev. 102(2002) 515-554;
(b) G. W. J. Watson, N. Asano, R. J. Molyneux, R. J. Nash, Polyhydroxylated alkaloids-natural occurrence and therapeutic applications, Phytochemistry 56(2001) 265-295.
Pili R., Chang J., Partis R.A.. The α-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and Inhibits tumor growth[J]. Cancer Res., 1995, 55: 2920-2926.
(a) H. Nojima, I. Kimura, F. J. Chen, et al. , Antihyperglycemic effects of Ncontaining sugars from xanthocercis zambesiaca, morus bombycis, aglaonema treubii, and castanospermum australe in streptozotocin-diabetic mice, J. Nat. Prod. 61(1998) 397-400;
(b) J. A. Balfour, D. McTavish, Acarbose, Drugs 46(1993) 1025-1054.
Truscheit E., Frommer W., Junge B.. Chemistry and biochemistry of microbial α-glucosidase iInhibitors[J]. Angew. Chem. Int. Ed., 1981, 20: 744-761. doi: 10.1002/(ISSN)1521-3773
Fleet G.W.J., Karpas A., Dwek R.A.. Inhibition of HIV replication by aminosugar derivatives[J]. FEBS Lett., 1988, 237: 128-132. doi: 10.1016/0014-5793(88)80185-6
Hohenschutz L.D., Bell E.A., Jewess P.J.. Castanospermine A 1, 6, 7, 8-tetrahydroxyoctahydroindolizine alkaloid, from seeds of castanospermum australe[J]. Phytochemistry, 1981, 20: 811-814. doi: 10.1016/0031-9422(81)85181-3
Nash R.J., Fellows L.E., Dring J.V.. Castanospermine in alexa species[J]. Phytochemistry, 1988, 27: 1403-1404. doi: 10.1016/0031-9422(88)80203-6
(a) B. C. Campbell, R. J. Molyneux, K. C. Jones, Differential inhibition by castanospermine of various insect disaccharidases, J. Chem. Ecol. 13(1987) 1759-1770;
(b) Y. T. Pan, H. Hori, R. Saul, et al. , Castanospermine inhibits the processing of the oligosaccharide portion of the influenza viral hemagglutinin, Biochemistry 22(1983) 3975-3984;
(c) R. Saul, J. P. Chambers, R. J. Molyneux, A. D. Elbein, Castanospermine, a tetrahydroxylated alkaloid that inhibits β-glucosidase and β-glucocerebrosidase, Arch. Biochem. Biophys. 221(1983) 593-597;
(d) T. Szumilo, G. P. Kaushal, A. D. Elbein, Purification and properties of glucosidase I from mung bean seedlings, Arch. Biochem. Biophys. 247(1986) 261-271.
(a) G. W. J. Fleet, N. G. Ramsden, R. J. Molyneux, G. S. Jacob, Synthesis of 6-epicastanospermine and 1, 6-diepicastanospermine from l-gulonolactone and synthesis of l-6-epicastanospermine and l-1, 6-diepicastanospermine from dgulonolactone, Tetrahedron Lett. 29(1988) 3603-3606;
(b) G. W. J. Fleet, N. G. Ramsden, R. J. Nash, et al. , Synthesis of the enantiomers of 6-epicastanospermine and 1, 6-diepicastanospermine from d-and lgulonolactone, Carbohydr. Res. 205(1990) 269-282.
Bernotas R.C., Ganem B.. Total syntheses of (+)-castanospermine and (+)-deoxynojirimycin[J]. Tetrahedron Lett., 1984, 25: 165-168. doi: 10.1016/S0040-4039(00)99830-7
Winchester B.G., Cenci di Bello I., Richardson A.C.. The structural basis of the inhibition of human glycosidases by castanospermine analogues[J]. Biochem. J., 1990, 269: 227-231. doi: 10.1042/bj2690227
Whitby K., Taylor D., Patel D., Ahmed P., Tyms A.S.. Action of celgosivir (6-Obutanoyl castanospermine) against the pestivirus BVDV:implications for the treatment of hepatitis C[J]. Antiviral Chem. Chemother., 2004, 15: 141-151. doi: 10.1177/095632020401500304
(a) J. Mulzer, H. Dehmlow, J. Buschmann, P. Luger, Stereocontrolled total synthesis of the unnatural enantiomers of castanospermine and 1-epicastanospermine, J. Org. Chem. 57(1992) 3194-3202;
(b) H. Ina, C. Kibayashi, Total syntheses of (+)-castanospermine and (+)-1-epicastanospermine and their 1-O-αcyl derivatives from a common chiral building block, J. Org. Chem. 58(1993) 52-61;
(c) S. E. Denmark, B. Herbert, Synthesis of (—)-7-epiaustraline and (—)-1-epicastanospermine, J. Org. Chem. 65(2000) 2887-2896;
(d) L. Cronin, P. V. Murphy, Novel synthesis of castanospermine and 1-epicastanospermine, Org. Lett. 7(2005) 2691-2693;
(e) T. J. Wu, P. Q. Huang, A concise approach to (+)-1-epi-castanospermine, Tetrahedron Lett. 49(2008) 383-386;
(f) G. Liu, T. J. Wu, Y. P. Ruan, P. Q. Huang, A flexible approach to azasugars: asymmetric total syntheses of (+)-castanospermine, (+)-7-deoxy-6-epicastanospermine, and (+)-1-epi-castanospermine, Chem. Eur. J. 16(2010)5755-5768;
(g) N. B. Kalamkar, V. G. Puranik, D. D. Dhavale, Synthesis of C1-and C8 aepimers of (+)-castanospermine from d-glucose derived γ, δ-epoxyazide: intramolecular 5-endo epoxide opening approach, Tetrahedron 67(2011) 2773-2778.
Wrodnigg T.M.. From lianas to glycobiology tools:twenty-five years of 2, 5-dideoxy-2, 5-imino-d-mannitol[J]. Monatsh. Chem., 2002, 133: 393-426. doi: 10.1007/s007060200018
Wennekes T., van den Berg R.J.B.H.N., Donker W.. Development of adamantan-1-yl-methoxy-functionalized 1-deoxynojirimycin derivatives as selective inhibitors of glucosylceramide metabolism in man[J]. J. Org. Chem., 2007, 72: 1088-1097. doi: 10.1021/jo061280p
(a) R. J. Tennant-Eyles, B. G. Davis, A. J. Fairbanks, Peptide templated glycosylation reactions, Tetrahedron: Asymmetry 11(2000) 231-243;
(b) N. Miquel, G. Doisneau, J. M. Beau, Reductive samariation of anomeric 2-pyridyl sulfones with catalytic nickel: an unexpected improvement in the synthesis of 1, 2-trans-diequatorial C-glycosyl compounds, Angew. Chem. 112(2000) 4277-4280;
(c) G. Yang, X. Ding, F. Kong, Selective 6-O-debenzylation of mono-and disaccharide derivatives using ZnCl2-Ac2O-HOAc, Tetrahedron Lett. 38(1997) 6725-6728.
Petrier C., Luche J.L.. Allylzinc reagent additions in aqueous media[J]. J. Org. Chem., 1985, 50: 910-912. doi: 10.1021/jo00206a047
Mattes H., Benezra C.. Reformatsky-type reactions in aqueous media. Use ofbronometryl-acrylic acid for the synthesis of α-methylene-γ-butyrolactones[J]. Tetrahedron Lett, 1985, 26: 5697-5698. doi: 10.1016/S0040-4039(01)80923-0
Hamana H., Ikota N., Ganem B.. Chelate selectivity in chelation-controlled allylations:a new synthesis of castanospermine and other bioactive indolizidine alkaloids[J]. J. Org. Chem., 1987, 52: 5492-5494. doi: 10.1021/jo00233a045
(a) M. D. Groaning, A. I. Meyers, Remote steric effects in the Sakurai reaction, Tetrahedron Lett. 40(1999) 8071-8074;
(b) P. H. Lee, K. Lee, S. Y. Sung, S. Chang, The catalytic sakurai reaction, J. Org. Chem. 66(2001) 8646-8649.
Bates R.W., Khanizeman R.I.N., Hirao H., Tay Y.S., Sae-Lao P.. A total synthesis of (+)-negamycin through isoxazolidine allylation[J]. Org. Biomol. Chem., 2014, 12: 4879-4884. doi: 10.1039/c4ob00537f
Kalita P.K., Phukan P.. Facile chemoselective carbonyl allylation of chalcones with allyltributylstannane catalyzed by CuI[J]. Tetrahedron Lett., 2013, 54: 4442-4445. doi: 10.1016/j.tetlet.2013.06.037
Li Y.X., Shimada Y., Sato K.. Synthesis and glycosidase inhibition of australine and its fluorinated derivatives[J]. Org. Lett., 2015, 17: 716-719. doi: 10.1021/ol503728e
Izquierdo I., Tamayo J.A., Rodríguez M., Franco F., Lo Re D.. Synthesis of (+)-1-epi-castanospermine from l-sorbose[J]. Tetrahedron, 2008, 64: 7910-7913. doi: 10.1016/j.tet.2008.06.021

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载:





 下载:
下载: