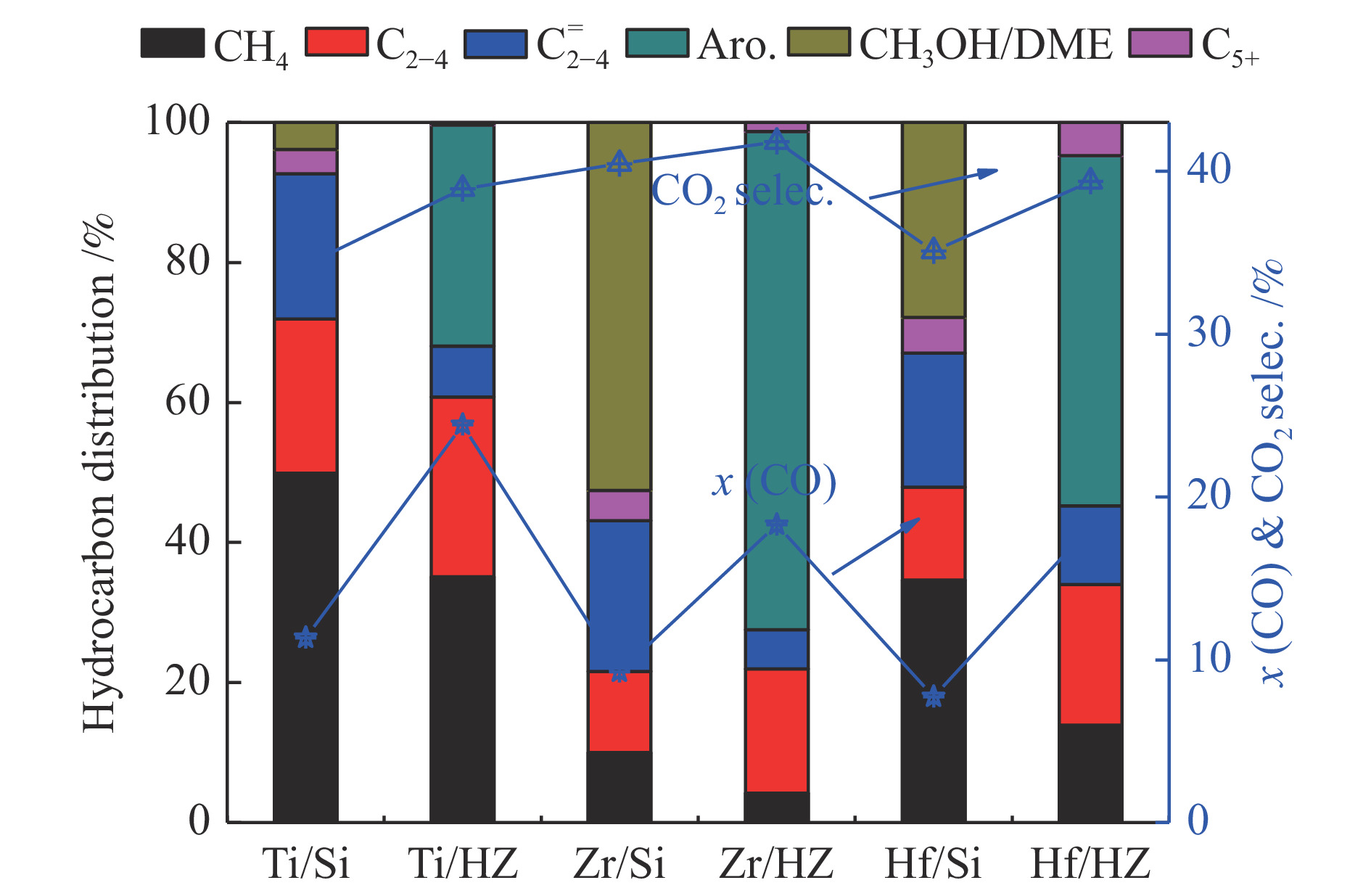
图 1.
金属氧化物催化剂和双能催化剂的催化性能
Figure 1.
Catalytic performance of metal oxides and bifunctional catalysts

芳烃作为大宗石化产品的基础原料,目前,其生产主要依赖于石油催化重整、石脑油蒸气裂解副产物以及少量的煤焦油加氢和催化裂化[1]。为了应对石油资源的减少以及芳烃市场需求量的不断增加,寻求芳烃生产的新方法和新技术变的日益迫切[2]。相较于最先提出的合成气两步法制芳烃(即合成气制甲醇、甲醇制芳烃),合成气一步法制芳烃在热力学利用率和经济效益上具有明显的优势[3, 4]。
目前,根据合成气一步法制芳烃反应过程中中间体产物的不同[4],可以将合成气制芳烃简单的分成费托路线和甲氧基物路线两种途径[5]。费托路线就是利用费托催化剂与ZSM-5分子筛复合,反应过程中产生的烯烃(或烯醇)中间体产物扩散到分子筛中进行芳构化;甲氧基路线则是利用甲醇催化剂与ZSM-5分子筛复合,反应过程中产生甲氧基(或甲醇等含氧中间体产物,即CHxO*)扩散到分子筛中进行芳构化[6, 7]。因此,这种可以直接催化合成气获得芳烃的催化剂也被称作双功能催化剂。在合成气催化反应中,原料气体CO和H2的活化是整个反应能够发生的关键因素,通常认为,金属氧化物或碳化金属等晶体的棱、顶点以及晶体中的缺陷位点是合成气的活性中心[8-10]。因此,研究者们提出,双功能催化剂中金属氧化物的氧空位是影响合成气制芳烃的关键因素之一[11, 12]。TiO2金属氧化物,不仅具有价格低廉、环境友好、化学性质稳定等特点,还具有较高浓度的表面氧空位和内部氧空位,因此,常被用于光催化、烷烃脱氢以及CO的催化氧化[13-15]。在HfO2金属氧化物中,氧空位是HfO2中最为常见的一种空位缺陷,高浓度的氧空位可以有效地提高HfO2的电荷捕获能力。TiO2、ZrO2和HfO2均属于钛副族氧化物,不仅均具有较高浓度的氧空位,而且其金属氧化物的价态也相同(一般为+4),但是目前只有ZrO2与ZSM-5分子筛复合作为双功能催化剂[16, 17],却很少见作为同族的TiO2和HfO2与ZSM-5分子筛复合用于催化合成气反应。
此外,在甲醇路线的双功能催化剂中,一般认为,CO和H2首先在金属氧化物表面生成中间体产物,然后进一步扩散到分子筛中进行反应[18, 19]。由于合成气制甲醇反应机理的复杂性[20, 21],导致甲醇催化剂与分子筛复合的双功能催化剂的反应机理存在较多争议,而争议点主要集中在金属氧化物表面中间体产物的种类[4, 12, 22]。
因此,本研究通过超临界的方法制备了纳米金属氧化物TiO2、ZrO2和HfO2,研究了三种钛副族金属氧化物的结构性质、表面氧空位以及金属氧化物表面中间体产物的种类对双功能催化剂催化CO加氢性能的影响。
五水硝酸锆(Zr(NO3)4·5H2O,REO),氯化钛(TiCl4,AR)和氯化铪(HfCl4,AR)上海麦克林生化科技有限公司。乙醇(CH3CH2OH,≧99.7%,AR),山西同杰化学试剂有限公司。氨水(NH3·H2O,NH3含量25%−28%),洛阳市化学试剂厂。硝酸铵(NH4NO3,AR),台云市粤侨试剂塑料有限公司。ZSM-5分子筛(SiO2/Al2O3=30),南开大学化工试剂厂。以上所用试剂均未进行进一步提纯处理。
金属氧化物的制备:分别取一定量的Zr(NO3)4·5H2O、TiCl4和HfCl4加入到乙醇中,在室温下搅拌溶解。将配制的氨水(体积比NH3·H2O =1∶3)缓慢地滴加到上述溶液中至pH值为8左右,快速搅拌得到溶胶。取一定量的上述溶胶,置于反应釜中,在温度为240 ℃时,采用超临界乙醇的方法分别得到ZrO2、TiO2和HfO2的前驱体,置于烘箱中110 ℃充分干燥。取出ZrO2、TiO2和HfO2的前驱体置于马弗炉中以5 ℃/min的升温速率将温度缓慢地升高到430 ℃,继续在430 ℃下煅烧6 h得到纳米ZrO2、TiO2和HfO2。
H-ZSM-5分子筛的制备:取一定量的ZSM-5分子筛,使用1 mol/L的NH4NO3溶液80 ℃搅拌6 h,抽滤,继续加入NH4NO3溶液,搅拌抽滤,该过程重复三次后取出,烘干。将烘干的ZSM-5分子筛置于马弗炉中,在550 ℃下煅烧6 h,取出备用。后续文中所使用的ZSM-5分子筛,如无特别指出,均为此处所制备的H-ZSM-5分子筛。
双功能催化剂的制备:分别取金属氧化物(即上述纳米ZrO2、TiO2和HfO2)与ZSM-5进行研磨混合,其中,金属氧化物与ZSM-5分子筛的质量比为1∶2,将金属氧化物与ZSM-5分子筛在研钵中充分研磨混合均匀后压制成40−60目的颗粒得到双功能催化剂Zr/HZ、Ti/HZ和Hf/HZ。采用同样的方法,将石英砂(小于100目)分别与金属氧化物置于研钵中充分研磨混合后压制成40−60目的颗粒得到金属氧化物催化剂,依次记作Zr/Si、Ti/Si和Hf/Si。
采用日本理学的Smartlab型X射线衍射仪对所制备的金属氧化物的物相及组分进行测试。衍射光源为Cu Kα射线,电压和电流分别为40 kV和30 mA。测试5°−70°,扫描速率为10(°)/min。
采用美国麦克仪器公司型号为ASAP2460对金属氧化物进行低温N2物理吸附-脱附测试。0.1 g的金属氧化物置于样品管中,在真空环境中加热至200 ℃保持12 h脱出样品中的杂质,然后在77 K条件下进行N2的物理吸附-脱附测试。比表面积、孔体积和孔径尺寸依次采用BET、t-plot和BJH法分析。
采用日本株式会社型号为JEM-2100F的TEM对样品的微观形貌进行表征。取少量金属氧化物粉末置于无水乙醇中,使用超声分散5 min后,取少量待测样品滴至碳支持膜上,干燥至乙醇完全脱出,然后将金属氧化物至于样品台上进行TEM测试。
采用美国麦克仪器公司型号为AutoChemⅡ的全自动多功能吸附仪对样品的H2吸附能力进行测试。取0.1 g金属氧化物置于U型管中,在200 ℃下通入Ar脱出金属氧化物中的杂质,然后将Ar切换成10%H2/Ar,同时将样品继续升温到400 ℃,并保持4 h。将温度降至50 ℃再次切换至Ar气氛进行吹扫1 h。将Ar切换到10%H2/Ar使样品吸附饱和。再次使用Ar吹扫至基线稳定。最后以10 ℃/min的升温速率升温至800 ℃,同时记录脱出气体的信号值。CO的吸附测试与氢气吸附测试方法基本相同,在降到50 ℃ Ar吹扫后将吸附气体切换为10%CO/Ar使样品吸附饱和。
采用美国Thermo Fisher Scientific的Thermo Kalpha对金属氧化物进行XPS测试。取0.5 g金属氧化物在合成气(CO和H2的体积比为CO∶H2=1∶2)的气氛下400 ℃进行反应4 h,并持续通入合成气至室温。密封后,在真空手套箱中取出反应后的样品,在真空手套箱中将粉末压入铟箔中。然后,通过真空盒将压制的样品转移至XPS样品池中。在5 × 10−9 mbar的真空条件下对样品进行测试,记录谱图。所得谱图根据C 1s(284.6 eV)进行校正。
采用德国Bruker公司的VERTEX 70型红外光谱仪进行原位红外测试。取0.05 g样品置于氧化铝坩埚中,然后将坩埚置于原位池中。在200 ℃下通入Ar 30 min脱出杂质气体,继续升温到400 ℃,同时通入合成气(CO和H2的体积比为1∶2)反应3 h。然后通入合成气降温至50 ℃,测试背景。接着,以5 ℃/min的升温速率分别将温度升至100、200、300、360和400 ℃。最后以4 cm−1的分辨率扫描64次进行图谱采集。
将所制备的催化剂置于不锈钢管式固定床中,通入合成气(体积比为H2/CO/N2=64∶32∶4)进行吹扫,背压阀调节系统压力。将反应条件设置为温度400 ℃,压力3 MPa,空速为2000 mL/(h·g)。采用冷却分离的方式进行产物分析。在线气相色谱气体SP-3420A的TCD和FID检测器检测H2、N2、CO、CO2和C1–6的烃类。冷肼分离的液相产物进行油相和水相分离,并使用NexisGC-2030进行检测,色谱柱为HP-INNOWAX。
根据碳原子数计算CO转化率(xCO)、CO2选择性(CO2 selec.)和产物选择性(Ci selec.):
|
|
(1) |
|
|
(2) |
|
|
(3) |
式中,COinlet、COoutlet和CO2outt分别代表进入的CO的摩尔量、尾气中CO的摩尔量和尾气中CO2的摩尔量。
图1为金属氧化物催化剂和双功能催化剂的催化性能。Ti/Si、Zr/Si和Hf/Si的CO转化率依次为11.36%、9.26%和7.72%,双功能催化剂中CO转化率是对应金属氧化物催化剂的两倍左右。其原因可能是由于双功能催化剂中金属氧化物表面产生的中间体产物(烯烃或甲醇)扩散到ZSM-5分子筛中进行反应消耗,中间体产物浓度降低,CO的加氢反应持续向正向移动[18, 19],导致双功能催化剂中CO转化率增加,催化剂反应活性提高。对比金属氧化物和双功能催化剂中CO2的选择性,可以发现两者的CO2选择性具有正相关性。其主要原因CO2是CO在金属氧化物表面的水煤气变换的主要来源[16]。
对比金属氧化物催化剂和双功能催化剂的产物分布,可以看出双功能催化剂中ZSM-5分子筛对催化产物的分布具有显著的影响。相较于Ti/Si、Zr/Si和Hf/Si,双功能催化剂Ti/HZ、Zr/HZ和Hf/HZ中CH4和C2–C4烯烃(即
对比Ti/HZ、Zr/HZ和Hf/HZ中芳烃的选择性和Ti/Si、Zr/Si和Hf/Si中甲醇的选择性,Zr/HZ的芳烃选择性最高为71.15%,Zr/Si的甲醇选择性也最高为52.55%,Ti/HZ中芳烃的选择性最低为31.57%,Ti/Si中甲醇的选择性也仅有3.86%,因此可以推断,金属氧化物中甲醇的选择性与双功能催化剂中芳烃的选择性具有正相关的关系。在Ti/Si中甲醇的选择性较低,但是Ti/HZ的芳烃选择性依然可以达到31.57%,其原因可能是金属氧化物中产生的烯烃或
图2为金属氧化物的结构特征。图2(a)中TiO2在25.38°、38.08°和48°处的特征峰归属于锐钛矿型TiO2(PDF#21-1276)。ZrO2在24.22°、28.25°和34.58°处的特征衍射峰归属于单斜晶相(m-ZrO2)(PDF#37-1484),在30.28°、35.28°、50.36°、59.96°处的特征峰则归属于四方晶相(t-ZrO2)(PDF#50-1089)。HfO2在24.44°、28.60°、31.84°和50.58°处的特征峰归属于单斜晶相(m-HfO2)(PDF#06-0318)。采用K值法计算纳米ZrO2中m-ZrO2和t-ZrO2的占比分别为14.1%和85.9%,因此,ZrO2中以t-ZrO2为主。另外根据谢乐公式D=Kλ/(βcosθ)计算纳米TiO2(2θ取25.38°)、ZrO2(2θ取31.84°)和HfO2(2θ取28.60°)中的晶粒尺寸依次为9.9、4.2和3.9 nm。结合图2(b)和表1可以看出,三种金属氧化物均为介孔结构材料。随着Ti、Zr、Hf原子核的不断增加,纳米TiO2、ZrO2和HfO2的比表面积和孔容均在不断增加,ZrO2孔径尺寸较大为13.52 nm,TiO2的孔径尺寸较小为9.73 nm。
图2(c)–(h)为纳米TiO2、ZrO2和HfO2的TEM照片。由图2(c)、2(e)和2(g)可以看出,三种金属氧化物以近似球形的单颗粒分散,颗粒尺寸较小,仅有5 nm左右。图2(d)的晶面尺寸为0.35 nm,属于典型的锐钛矿型TiO2的(101)晶面[25]。在图2(f)中的晶面间距为0.29和0.28 nm,分别属于t-ZrO2的(011)晶面和m-ZrO2的(111)晶面[26],而在HfO2的中的晶面间距为0.30 nm,属于单斜相HfO2的(111)晶面[27],该结果与XRD结果相互印证。
一般来说,催化剂与反应物之间的吸附作用对催化剂的反应有着重要的影响[28]。化学吸附为多相催化的关键步骤,固体催化剂表面的配位不饱和原子与气体分子形成的化学键,其吸附能明显大于物理吸附。吸附在固体催化剂表面的气体分子活性明显增强,反应能垒降低,反应速率增加。吸附在固体催化剂表面的气体分子一般存在两种状态:分子态化学吸附和分解态化学吸附,分子态化学吸附的气体与催化剂表面的相互作用较弱,在程序升温过程中通常在127 ℃下就可以脱附[29]。结合催化剂的反应温度,选择对脱附温度低于400 ℃的H2-TPD和CO-TPD的脱附峰进行分析对比。结合图3和表2的结果可以看出,ZrO2和HfO2的H2脱附温度均在127 ℃以上,因此可以认为,H2分子在ZrO2和HfO2的表面属于分解态化学吸附,而TiO2在50−220 ℃有一个H2脱附峰,因此可以推测,存在一定量的H2以分子态吸附TiO2中。对比TiO2、ZrO2和HfO2中H2的吸附量,其中,TiO2中H2的吸附量最大(65.96 μmol/g)。
在过渡金属催化剂中,CO的吸附类型不同,其热稳定性存在显著差异,脱附温度低于100 ℃为线式吸附,属于非解离吸附,高温区域的脱附峰,一般认为是CO的解离吸附,这种吸附的CO反应活性较高[30, 31]。对比三种金属氧化物中CO的脱附温度,HfO2中50−139 ℃的CO脱附峰可能属于CO的线式吸附也即非解离吸附。相较于ZrO2、TiO2和HfO2的CO脱附温度较低,CO与TiO2和HfO2氧化金属之间的相互作用强度较低,而ZrO2的CO脱附温度较高,ZrO2对CO的吸附作用较强。ZrO2在高温区域CO脱附量最大,因此,ZrO2对CO的解离吸附能力较强。单位质量金属氧化物脱附的H2和CO的物质量之间的比值记作H/C比。以H/C比来表示金属氧化物表面吸附的H2和CO的能力,由表2可以看出ZrO2的H2/CO的比值最小为0.01,TiO2的比值最大为0.75。

 下载:
导出CSV
下载:
导出CSV
| Sample | SBET /(m2·g−1) | vtotal /(cm3·g−1) | Dave /nm | Cave /nm |
| TiO2 | 74 | 0.18 | 9.73 | 9.9 |
| ZrO2 | 97 | 0.32 | 13.52 | 4.2 |
| HfO2 | 111 | 0.33 | 12.06 | 3.9 |

(a): H2-TPD; (b): CO-TPD
下载:
导出CSV
| Sample | t1 /℃ | t2 /℃ | I1 /(μmol·g−1) | I2 /(μmol·g−1) | H/C (ratio) | |
| TiO2 | H2-TPD | 50−220 | 220−410 | 60.79 | 5.17 | 0.75 |
| CO-TPD | 161−405 | 87.69 | ||||
| ZrO2 | H2-TPD | 201−367 | 2.49 | 0.01 | ||
| CO-TPD | 172−432 | 233.55 | ||||
| HfO2 | H2-TPD | 145−382 | 46.13 | 0.66 | ||
| CO-TPD | 50−139 | 238−416 | 53.81 | 16.56 |
在催化反应中,氧空位不仅可以调节催化剂的电子结构和吸附能,甚至可以作为直接的吸附位点,进而直接影响催化剂的反应效率和产物的选择性[32, 33]。在CO加氢的催化反应中,研究者认为,金属氧化物表面的氧空位是CO吸附解离的主要影响因素[8, 11]。Rahman等[34]和Wang等[35]在研究金属氧化物的氧空位时将氧空位分成单电子氧空位(O–)和双电子氧空位(O2−)以便于观察氧空位电子性质的变化。因此,本研究借鉴该方法将O 1s根据结合能由低到高进行分峰,依次为晶格氧(O)、单电子氧空位(O')、双电子氧空位(O'')和表面羟基/碳酸盐(Oc),如图4所示。结合表3结果,对比纳米TiO2、ZrO2和HfO2三种金属氧化物中氧空位总分数(即I2+I3),由表3可以看出,ZrO2的氧空位分数最高为45.65%,TiO2的氧空位分数最低为35.08%,其原因除了与金属氧化物自身的可还原性质有关,还可能与金属氧化物的结构性质相关,其中TiO2晶体颗粒较大,相对比表面积较小,因此表面氧空位分数较低,而ZrO2不仅晶体颗粒小,比表面积大,同时颗粒中还存在两种晶相(m-ZrO2和t-ZrO2),这可能导致晶相错位而形成结构缺陷,从而提高氧空位浓分数[36]。相较于TiO2和HfO2的双电子氧空位的分数(分别为12.82%和12.10%),ZrO2具有相对较高的双电子氧空位(26.00%)。此外,ZrO2的单电荷氧空位和双电荷氧空位的结合能分别位于532.16和533.18 eV,而TiO2和HfO2的表面氧空位结合能均低于ZrO2。综合上述的对比可以看出,ZrO2表面不仅具有高浓度的表面氧空位,同时表面氧空位平均电荷密度较低,表现出较为明显的缺电子性质。
下载:
导出CSV
| Sample | Binding energy /eV | I /% | ||||||||
| BE1 | BE2 | BE3 | BE4 | I1 | I2 | I3 | I4 | I2+I3 | ||
| TiO2 | 529.28 | 530.80 | 531.68 | 532.68 | 49.52 | 22.26 | 12.82 | 15.39 | 35.08 | |
| ZrO2 | 530.14 | 532.16 | 533.18 | 535.14 | 46.66 | 19.65 | 26.00 | 7.68 | 45.65 | |
| HfO2 | 530.02 | 531.60 | 532.80 | 533.82 | 55.40 | 26.41 | 12.10 | 6.08 | 38.51 | |
图5为金属氧化物的原位红外光谱谱图。由图5(a)–(c)可以看出,随着反应温度的不断升高,3400 cm−1附近归属于羟基(–OH)的峰强度不断降低,其主要原因是随着反应温度的不断升高,金属氧化物表面的部分羟基与CO结合形成甲酸盐类物质或结合水不断挥发导致的游离羟基减少[37]。根据CO和CO2加氢制甲醇的DFT研究结果,可以推断在CO加氢的反应过程中存在三种碳氢氧中间体产物(CHxO*),分别为CHO*、CH2O*和CH3O*[38],而CO2加氢反应时,中间产物则会含有一定量的HCOO*[39]。

在图5(a)中,TiO2在3016 cm−1附近出现的峰为CH3*物种,在2339 cm−1附近的峰为金属氧化物与CO相互作用形成的峰[40]。随着反应温度的不断提高,吸附在TiO2表面的CO不断被脱附出来,因此,2339 cm−1附近的峰不断降低。结合2.3中H2-TPD和CO-TPD的结果可以推断,由于反应温度升高,吸附在TiO2表面的H2和CO减少,因此,导致CH3*在400 ℃时CH3*的量较低。由图5(b)可以看出,在ZrO2中CO加氢反应的中间体产物主要为含氧中间体产物,其中,在2748、2877、2929、2962 cm−1附近的峰依次为HCOO*[18, 41]、CHO*、CH2O*和CH3O*[42, 43]。在低温时(低于200 ℃),ZrO2中CO吸附解离能力较低(ZrO2的CO-TPD)导致中间产物CHO*、CH2O*和CH3O*的吸收峰强度较低。随着反应温度的不断升高,CHO*和CH3O*的峰强度不断增加,CH2O*的相对强度降低。其原因可能是温度升高,CHO*的加氢反应活性增强,CHO*不再逐步加氢,而是直接加氢生成CH3O*。对比ZrO2反应温度在360和400 ℃时CHO*和CH3O*的吸收峰强度可以发现,反应温度在400 ℃时CHO*和CH3O*吸收峰的强度降低,CH3O*的峰向高波数偏移。其原因可能是由于反应温度的升高,中间体CH3O*的加氢能力增强,从而导致中间体更加倾向于生成CH3*。在反应温度高于300 ℃时,HCOO*的峰出现,其原因可能是CO2相对CO更加稳定,加氢反应需要较高的温度,或者反应温度的不断升高,逆水煤气反应加快,CO2含量增加[16]。在图5(c)中,当反应温度升高到300 ℃时,在2890 cm−1附近开始出现吸收峰。随着反应温度升高到360℃时,在3043、2972和2890 cm−1出现的吸收峰依次归属于CH3*、CH3O*和CHO*。
图5(d)为TiO2、ZrO2和HfO2反应为400 ℃的原位红外光谱谱图。对比三种金属氧化物的红外谱图可以看出,在相同的反应温度下,ZrO2表面产生的中间体CH3O*、CH2O*和CHO*含量较高,而TiO2的表面只有CH3*,HfO2的表面虽然产生了CH3O*和CHO*,但是含量较低,且出现了CH3*的峰。另外,对比ZrO2和HfO2中CH3O*、CHO*的峰位置可以看出,相较于ZrO2,HfO2中CH3O*和CHO*的峰位置分别蓝移了10和13 cm−1,说明HfO2对中间体CH3O*和CHO*的吸附作用增强,导致产生的中间体不能及时的扩散出去,而是进一步加氢反应,这可能是HfO2中出现CH3*的一个原因。
在反应过程中,金属氧化物表面吸附解离的H2与反应分子中的不饱和键进行加氢反应[44]。虽然TiO2中氧空位的结合能较低,但是其表面的H/C比较高(H/C比为0.75),导致了反应CO的转化率较高,CO深度加氢形成CH3*。此外,TiO2表面吸附的H2较高,从而有效抑制了TiO2表面的水煤气反应,因此,相较于Zr/Si和Hf/Si,Ti/Si中CO2最低。由于ZrO2表面的氧空位浓度高和氧空位呈现相对缺电子的性质,导致吸附在ZrO2表面的CO的C–O键结合能降低,从而促进了CO的加氢反应。因此,虽然ZrO2金属氧化物的表面H/C比较低(H/C比仅有0.01),但是CO转化率依然可以高达9.26%。此外,由于受到H2吸附解离的影响,CO的深度加氢反应受到限制,CO加氢的中间体产物主要以CHO*为主,也有少部分CHO*加氢生成CH2O*和CH3O*,而不是深度加氢生成CH3*。在HfO2中,虽然金属氧化物表面氧空位浓高达38.51%,但是由于HfO2的氧空位结合能较低(分别为531.68和532.80 eV),导致HfO2中存在少量非解离吸附的CO,同时H/C比高达0.66,因此,在HfO2中不仅有中间体产物CHxO*,还有一定量的CH3*中间体。
通过上述对比讨论可以发现,CO加氢反应不仅和氧空位浓度有关,还和氧空位的电子性质以及金属氧化物吸附解离H2的能力相关。氧空位的缺电子性质导致CO的碳氧键结合能降低,形成高活性CO。与此同时,金属氧化物对H2的吸附解离又直接制约着高活性CO的加氢程度。金属氧化物表面H2吸附解离活性高,CO的吸附解离活性低,则会导致CO深度加氢,形成稳定性较高的CH3*,因此,导致Ti/Si产物中CH4选择性较高(49.92%),甲醇选择性较低(3.86%),从而导致Ti/HZ双功能催化剂产物中烷烃选择性相对较高,芳烃选择性较低。反之,低的H2吸附解离和高的CO吸附解离则会有利于形成碳氢氧产物(CHxO*),而CHxO*则可以经扩散进入ZSM-5分子筛中进行反应生成芳烃[45, 46],因此,Zr/Si催化CO加氢产物中甲醇的选择性较高(52.55%),同时Zr/HZ中芳烃的选择性也相对的较高(71.15%)。
采用超临界法制备的钛副族纳米金属氧化物TiO2、ZrO2、HfO2对CO和H2的吸附解离能力存在着显著的区别,但是均具有催化CO加氢的反应活性。双功能催化剂中的ZSM-5分子筛可以有效的提高CO加氢的反应活性,降低催化产物中CH4和C2–4烯烃的选择性。
金属氧化物表面氧空位浓度和氧空位的电子性质同时影响着CO的吸附解离,缺电子性质的氧空位导致吸附的CO的碳氧键结合能降低,从而有利于CO的加氢反应。
金属氧化物表面氧空位浓度和氧空位电子性质以及表面吸附的H/C比共同决定着金属氧化物表面CO加氢的中间体产物。ZrO2表面产生的CHxO*有利于Zr/HZ获得71.15%的芳烃选择性,而TiO2和HfO2中的CH3*则导致Ti/HZ和Hf/HZ的催化产物CH4选择性较高。
WANG J, JIA Y, KAN Z, LIU S, PING L. Catalytic conversion of methanol to aromatics over nanosized HZSM-5 zeolite modified by ZnSiF6·6H2O[J]. Catal Sci Technol,2017,7(8):1776−1791. doi: 10.1039/C7CY00143F
XU H, LI M, NAWAZ M. Doping of K and Zn elements in FeZr-Ni/ZSM-5: Highly selective catalyst for syngas to aromatics[J]. Catal Commun,2019,23(121):95−99.
ZHANG P, TAN L, YANG G. One-pass selective conversion of syngas to para-xylene[J]. Chem Sci,2019,5(3):213−218.
CHENG K, ZHOU W, KANG J, HE S, SHI S, ZHANG Q, PAN Y, WEN W, WANG Y. Bifunctional catalysts for one-step conversion of syngas into aromatics with excellent selectivity and stability[J]. Chem-US,2017,3(2):334−347.
YANG X, SU X, CHEN D, ZHANG T, HUANG Y. Direct conversion of syngas to aromatics: A review of recent studies[J]. Chin J Catal,2020,41(4):561−573. doi: 10.1016/S1872-2067(19)63346-2
KASIPANDI S, BAE J W. Recent advances in direct synthesis of value-added aromatic chemicals from syngas by cascade reactions over bifunctional catalysts[J]. Adv Mater,2019,31(34):1803390. doi: 10.1002/adma.201803390
BROSIUS R, CLAEYS M. Aromatics from syngas: CO taking control[J]. Chem-US,2017,3(2):202−204.
JIANG F, WANG S, LIU B, LIU J, WANG L, XIAO Y, XU Y, LIU X. Insights into the influence of CeO2 crystal facet on CO2 hydrogenation to methanol over Pd/CeO2 catalysts[J]. ACS Catal,2020,10(19):11493−11509. doi: 10.1021/acscatal.0c03324
FU Y, NI Y, ZHU W, LIU Z. Enhancing syngas-to-aromatics performance of ZnO&H-ZSM-5 composite catalyst via Mn modulation[J]. J Catal,2020,383:97−102.
LI M, NAWAZ M A, SONG G, ZAMAN W Q, LIU D. Influential role of elemental migration in a composite iron-zeolite catalyst for the synthesis of aromatics from syngas[J]. Ind Eng Chem Res,2020,59(19):9043−9054. doi: 10.1021/acs.iecr.0c01282
LIU J, HE Y, YAN L, LI K, ZHANG C, XIANG H, WEN X, LI Y. Nano-sized ZrO2 derived from metal-organic frameworks and their catalytic performance for aromatic synthesis from syngas[J]. Catal Sci Technol,2019,9(10):1−12.
HUANG Z, WANG S, QIN F, HUANG L, YUE Y, HUA W, QIAO M, HE H, SHEN W, XU H. Ceria-zirconia/zeolite bifunctional catalyst for highly selective conversion of syngas into aromatics[J]. J Subst Abuse Treat,2018,13(3):287−288.
GUNTIDA A, WANNAKAO S, PRASERTHDAM P, PANPRANOT J. Acidic nanomaterials (TiO2, ZrO2, and Al2O3) are coke storage components that reduce the deactivation of the Pt-Sn/γ-Al2O3 catalyst in propane dehydrogenation[J]. Catal Sci Technol,2020,10(2010):5100−5112.
DASA A, KUMAR M, BHAGAVATHIACHARI M, NAIR R G. Hierarchical ZnO-TiO2 nanoheterojunction: A strategy driven approach to boost the photocatalytic performance through the synergy of improved surface area and interfacial charge transport[J]. Appl Surf Sci,2020,534(2020):147321.
LEE S, LEE J, NAM K, SHIN W G, SOHN Y. Application of Ni-Oxide@TiO2 core-shell structures to photocatalytic mixed dye degradation, CO oxidation, and supercapacitors[J]. Materials,2016,9(12):1024. doi: 10.3390/ma9121024
ZHOU W, ZHOU C, YIN H, SHI J, ZHANG G, ZHENG X, MIN X, ZHANG Z, CHENG K. Direct conversion of syngas into aromatics over a bifunctional catalyst: inhibiting net CO2 release[J]. Chem Commun,2020,56(39):5239−5242.
ZHOU W, SHI S, WANG Y, ZHANG L, WANG Y, ZHANG G, MIN X, CHENG K, ZHANG Q, KANG J, WANG Y. Selective conversion of syngas to aromatics over a Mo-ZrO2/H-ZSM-5 bifunctional catalyst[J]. J Subst Abuse Treat,2019,13(3):287−288.
WANG Y, ZHAN W, CHEN Z, CHEN J, LI X, LI Y. Advanced 3D hollow-out ZnZrO@C combined with hierarchical zeolite for highly active and selective co hydrogenation to aromatics[J]. Acs Catal,2020,10(13):7177−7187. doi: 10.1021/acscatal.0c01418
WANG X, CAO R, CHEN K, SI C, BAN H, ZHANG P, MENG F, JIA L, MI J, LI Z. Synthesis gas conversion to lower olefins over ZnCr‐SAPO‐34 catalysts: Role of ZnOZnCr2O4 interface[J]. ChemCatChem,2020,12(17):4387−4395. doi: 10.1002/cctc.202000473
CHENG Y, MA Z, NING Z, WEI W, SUN Y. Methanol synthesis from CO2-rich syngas over a ZrO2 doped CuZnO catalyst[J]. Catal Today,2006,115(1):222−227.
KIEFFER R, FUJIWARA M, UDRON L, SOUMA Y. Hydrogenation of CO and CO2 toward methanol, alcohols and hydrocarbons on promoted copper-rare earth oxides catalysts[J]. Catal Today,1997,36(1):15−24. doi: 10.1016/S0920-5861(96)00191-5
SU J, WANG D, WANG Y, ZHOU H, LIU C, LIU S, WANG C, YANG W, XIE Z, HE M. Direct conversion of syngas to light olefins over Zr-In2O3 and SAPO-34 bifunctional catalysts: Design of oxide component and construction of reaction network[J]. ChemCatChem,2018,10(7):1536−1541. doi: 10.1002/cctc.201702054
COMAS-VIVES A, VALLA M, COPÉRET C, SAUTET P. Cooperativity between Al Sites promotes hydrogen transfer and carbon-carbon bond formation upon dimethyl ether activation on alumina[J]. Acs Central Sci,2015,1(6):313−319. doi: 10.1021/acscentsci.5b00226
WANG G, WU W, ZAN W, BAI X, WANG W, QI X, KIKHTYANIN O V. Preparation of Zn-modified nano-ZSM-5 zeolite and its catalytic performance in aromatization of 1-hexene[J]. Trans Nonferr Metal Soc,2015,25(5):1580−1586. doi: 10.1016/S1003-6326(15)63761-X
TOMOKI A, MITSUTAKA O, KOJI T, SUSUMU T, MASATAKE H. Analytical TEM observation of Au and Ir deposited on rutile TiO2[J]. J Electr Micro,2003,52(2):119−124.
ZHAO L, HONG J, JIE T, WENJUN M, YIN Y, HAITAO Z. Synergistic effect of oxygen vacancies and ni species on tuning selectivity of Ni/ZrO catalyst for hydrogenation of maleic anhydride into succinic anhydride and γ-butyrolacetone[J]. Nanomaterials-Basel,2019,9(3):406. doi: 10.3390/nano9030406
RAMADOSS A, SANG J K. Synthesis and characterization of HfO2 nanoparticles by sonochemical approach[J]. J Alloy Compd,2012,544(2012):115−119.
QIU H. Interaction of adsorbates with clean and metal-covered oxide surfaces: Vibrational spectroscopic studies[D]. Bochum: Ruhr-University Bochum of Germany, 2009.
徐飞. 基于程序升温脱附谱的TiO2(110)和ZnO(0001)表面光化学研究[D]. 北京: 中国科学技术大学, 2020.XU-fei. Studies of Photochemistry on R-TiO2 (110) and ZnO (0001) surface using temperature programmed desorption[D]. Beijing: University of Science and Technology of China, 2020.
ZHANG C, ZHAO G, LIU K, YONG Y, XIANG H, LI Y. Adsorption and reaction of CO and hydrogen on iron-based Fischer-Tropsch synthesis catalysts[J]. J Mol Catal A: Chem,2010,328(1/2):35−43. doi: 10.1016/j.molcata.2010.05.020
XU J, BARTHOLOMEW C H. Temperature-programmed hydrogenation (TPH) and in situ Mssbauer spectroscopy studies of carbonaceous species on silica-supported iron Fischer-Tropsch catalysts[J]. J Phys Chem B,2005,109(6):2392−2403. doi: 10.1021/jp048808j
LIU B, LI C, ZHANG G, YAO X, CHUANG S, LI Z. Oxygen vacancy promoting dimethyl carbonate synthesis from CO2 and methanol over Zr-doped CeO2 nanorods[J]. Acs Catal,2018,8(11):10446−10456. doi: 10.1021/acscatal.8b00415
ZHU J, MU S. Defect engineering in the carbon-based electrocatalysts: Insight into the intrinsic carbon defects[J]. Adv Funct Mater,2020,30(25):2001097. doi: 10.1002/adfm.202001097
RAHMAN M A, ROUT S, THOMAS J P, MCGILLIVRAY D, LEUNG K T. Defect-rich dopant-free ZrO2 nanostructures with superior dilute ferromagnetic semiconductor properties[J]. J Am Chem Soc,2016,138(36):11896−11906. doi: 10.1021/jacs.6b06949
WANG J, XIA Y, DONG Y, CHEN R, XIANG L, KOMARNENI S. Defect-rich ZnO nanosheets of high surface area as an efficient visible-light photocatalyst[J]. Appl Catal B: Environ,2016,192(5):8−16.
SETVÍN M, WAGNER M, SCHMID M, PARKINSON G S, DIEBOLD U. Surface point defects on bulk oxides: atomically-resolved scanning probe microscopy[J]. Chem Soc Rev,2017,46(7):1772−1784. doi: 10.1039/C7CS00076F
WANG S, FANG Y, HUANG Z, XU H, SHEN W. The effects of the crystalline phase of zirconia on C-O activation and C-C coupling in converting syngas into aromatics[J]. Catalysts,2020,10(2):262. doi: 10.3390/catal10020262
WU P, YANG B. Theoretical insights into the promotion effect of subsurface boron for the selective hydrogenation of CO to methanol over Pd catalysts[J]. Phys Chem Chem Phys,2016,35(12):20833−21996.
YIN K, SHEN Y. Theoretical insights into CO2 hydrogenation to HCOOH over FexZr1−xO2 solid solution catalyst[J]. Appl Surf Sci,2020,528:146926.
PAN Q, PENG J, SHENG W, WANG S. In situ FTIR spectroscopic study of the CO2 methanation mechanism on Ni/Ce0.5Zr0. 5O2[J]. Catal Sci Technol,2014,4(12):412−415.
LIU X, ZHOU W, YANG Y, CHENG K, KANG J, ZHANG L, ZHANG G, MIN X, ZHANG Q, WANG Y. Design of efficient bifunctional catalysts for direct conversion of syngas into lower olefins via methanol/dimethyl ether intermediates[J]. Chem Sci,2018,9:4708−4718.
LIU X, WANG M, ZHOU C, ZHOU W, CHENG K, KANG J, ZHANG Q, DENG W, WANG Y. Selective transformation of carbon dioxide into lower olefins with a bifunctional catalyst composed of ZnGa2O4 and SAPO-34[J]. Chem Commun,2017,54(2):140−143.
WANG J, LI G, LI Z, TANG C, FENG Z, AN H, LIU H, LIU T, LI C. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol[J]. Sci Adv,2017,3(13):34−36.
TOSONI S, CHEN H Y T, PACCHIONI G. A DFT study of Ni clusters deposition on titania and zirconia (101) surfaces[J]. Surf Sci,2016,646(34):230−238.
ARSLAN M T, QURESHI B A, GILANI S Z A, CAI D, MA Y, USMAN M, CHEN X, WANG Y, WEI F. Single-step conversion of H2-deficient syngas into high yield of tetramethylbenzene[J]. Acs Catal,2019,9(3):2203−2212. doi: 10.1021/acscatal.8b04548
YANG X, SUN T, MA J, SU X, WANG R, ZHANG Y, DUAN H, HUANG Y, ZHANG T. The influence of intimacy on the iterative reactions during OX-ZEO process for aromatic production[J]. J Energy Chem,2019,35:60−65.

图 1 金属氧化物催化剂和双能催化剂的催化性能
Figure 1 Catalytic performance of metal oxides and bifunctional catalysts

图 2 金属氧化物的结构特征(a)XRD谱图,(b)N2吸附-脱附等温曲线,(c)和(d)为TiO2的TEM照片,(e)和(f)为ZrO2的TEM照片,(g)和(h)为HfO2的TEM照片
Figure 2 Structural characteristics of metal oxides, (a) XRD pattern, (b) N2-adsorption and desorption isotherm, (c) and (d) TEM pictures of TiO2, (e) and (f) TEM pictures of ZrO2, (g) and (h) TEM pictures of HfO2

图 3 金属氧化物的吸附性能
Figure 3 Desorption performance of metal oxides
(a): H2-TPD; (b): CO-TPD

图 5 金属氧化物的原位红外光谱谱图
Figure 5 In-situ infrared spectra of metal oxides in syngas (a) TiO2, (b) ZrO2 and (c) HfO2 at different temperatures, and (d) in-situ infrared spectra of metal oxides at 400 ℃
表 1 金属氧化物的结构性质
Table 1. Textural properties of metal oxides
| Sample | SBET /(m2·g−1) | vtotal /(cm3·g−1) | Dave /nm | Cave /nm |
| TiO2 | 74 | 0.18 | 9.73 | 9.9 |
| ZrO2 | 97 | 0.32 | 13.52 | 4.2 |
| HfO2 | 111 | 0.33 | 12.06 | 3.9 |
 下载: 导出CSV
下载: 导出CSV
表 2 金属氧化物的H2、CO脱附温度及相应的脱附量
Table 2. H2, CO desorption temperatures and corresponding desorption amounts of metal oxides
| Sample | t1 /℃ | t2 /℃ | I1 /(μmol·g−1) | I2 /(μmol·g−1) | H/C (ratio) | |
| TiO2 | H2-TPD | 50−220 | 220−410 | 60.79 | 5.17 | 0.75 |
| CO-TPD | 161−405 | 87.69 | ||||
| ZrO2 | H2-TPD | 201−367 | 2.49 | 0.01 | ||
| CO-TPD | 172−432 | 233.55 | ||||
| HfO2 | H2-TPD | 145−382 | 46.13 | 0.66 | ||
| CO-TPD | 50−139 | 238−416 | 53.81 | 16.56 |
下载: 导出CSV
表 3 金属氧化物中O 1s的结合能及相对氧物种的分数
Table 3. Binding energy and the corresponding surface atomic concentration of O 1s in metal oxides
| Sample | Binding energy /eV | I /% | ||||||||
| BE1 | BE2 | BE3 | BE4 | I1 | I2 | I3 | I4 | I2+I3 | ||
| TiO2 | 529.28 | 530.80 | 531.68 | 532.68 | 49.52 | 22.26 | 12.82 | 15.39 | 35.08 | |
| ZrO2 | 530.14 | 532.16 | 533.18 | 535.14 | 46.66 | 19.65 | 26.00 | 7.68 | 45.65 | |
| HfO2 | 530.02 | 531.60 | 532.80 | 533.82 | 55.40 | 26.41 | 12.10 | 6.08 | 38.51 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: