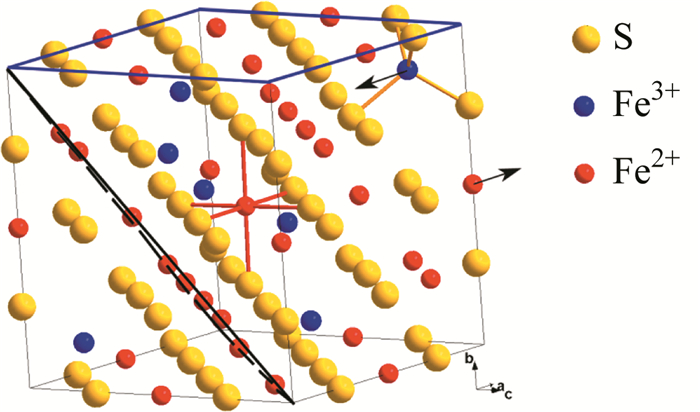
图 1.
Fe3S4晶体结构图:(001)和(111)位面分以蓝色和黑色标出;箭头方向为A、B的磁矩方向[85]

纳米材料和纳米结构通常是指在三维空间中至少有一维在1~100 nm的尺度范围内或者由其作为基本单元构成的材料。在众多纳米材料中,硫化铁纳米材料具有丰度高、价格低、毒性低的优点,在磁性、光学、电学的应用中极具有竞争力[1~3]。其中Fe3S4纳米粒子由于它优异的磁性和电学性质,在气感、可充电电池、热磁疗、吸附剂、催化剂等领域都有研究。硫化物Fe3S4和被广泛研究的磁铁矿(Fe3O4)有许多相似的地方,最早被Skinner等作为一种矿物报道出来[4, 5]。Fe3S4是和Fe3O4的晶格结构相似的铁磁性物质,同时含有Fe2+和Fe3+,是一种反尖晶石结构,包括四面体和八面体单元(图 1),其中硫形成立方密堆积阵列;一半的Fe3+占据了1/8的四面体体位,另一半的Fe3+和Fe2+共同占据了八面体的体位[6, 7]。四面体中的铁亚晶格磁畴与八面体中的铁亚晶格磁畴以反向平行方式排列,共同构成了Fe3S4的亚铁磁性[8]。Fe3S4的反尖晶石结构导致了八面体晶格中产生高自旋三价铁和亚铁的电子跃迁,产生半金属行为[9]。另外随着粒子尺寸的减小,磁性材料从多畴结构变为单畴结构,从而导致新的磁特性[10]。
Fe3S4与Fe3O4具有相似的化学成分和相同的反尖晶石结构,但它们的磁性在许多方面都不相同。在室温下Fe3S4的饱和磁化强度为59Am2 ·kg-1,略小于Fe3O4(90~92Am2 ·kg-1)。它们在室温下的易磁化晶轴也不相同,对于Fe3O4,[111]晶轴为它的易磁化轴,而Fe3S4的易磁轴为[100]晶轴。同时Fe3S4的居里温度(677K)也低于Fe3O4的(858K)。由于硫的离子半径要比氧的大,所以Fe3S4(9.876Å)的晶格常数也大于Fe3O4(8.397Å)。Fe3S4在A位点与B位点之间的交换耦合常数大约比Fe3O4(2.88meV)低1.03meV。这些都反映出了Fe3S4与Fe3O4在电子结构上面的差异,在研究其性质的时候要额外注意[11~13]。Wang等[14]研究表明,Fe3S4更适合自旋电子应用,与类似Fe3O4半导体相比,Fe3S4展现出了更好的导电性能。Roldan等[15]的研究表明,Fe3S4的硬度要比Fe3O4更高,但容易断裂,Fe-S相互作用具有比氧化物更多的共价特征。然而,高分辨率中子粉末衍射和极化中子衍射显示,Fe3S4的低温磁性和结构转变都不明显。两个研究小组根据第一性原理计算Fe3S4中是否存在Verwey跃迁得出了不同的结论,Fe3S4的电子结构和性能可能比我们预想的要复杂得多。
目前,Fe3S4纳米粒子还无法以商业规模量产[16],因此需要找到高效绿色可控的Fe3S4纳米粒子合成方法。本文就近十年Fe3S4纳米粒子的制备及相关应用研究进展进行综述。
目前,国内外制备Fe3S4纳米粒子的方法主要有沉淀法、热分解法、水热法等。这些制备方法的操作流程、设备各有不同,所得Fe3S4纳米粒子的粒径、形貌、性能也都大不相同。
沉淀法是一种操作简单的制备金属纳米颗粒的方法。根据溶液中含有的化合物离子的价态,沉淀法可以分为共沉淀法、氧化还原沉淀法。由于Fe3S4纳米粒子的亚稳态性质[17],利用沉淀法制备Fe3S4纳米粒子比较困难,因此报道较少,只有在较低的pH下或者厌氧环境中才可以通过沉淀法成功制备Fe3S4纳米粒子。已有的两例沉淀法制备Fe3S4纳米粒子的报道都只使用了Fe2+作为反应物,并没有加入其他的氧化剂来得到Fe3+,因此推测在反应阶段O2作为氧化剂将溶液中部分Fe2+氧化为Fe3+,从而进一步转换为Fe3S4纳米粒子,整个过程属于氧化还原沉淀法。
一般的制备过程是:在N2或者其他惰性气体的保护下,把Fe2+的盐溶液和S2-的硫化物盐溶液按比例充分混合均匀后,加入盐酸或乙酸溶液调节pH至3.0左右,保护产物不被分解,然后充分搅拌反应液,控制反应时间在10min左右,将分离出来的产物马上干燥,最终制得Fe3S4纳米粒子。
沉淀法制备的Fe3S4纳米粒子其影响微观形貌和尺寸的因素有很多,包括溶液的pH、反应温度、反应时间等,其中最关键的是pH和反应时间。Chang等[18]用共沉淀法在N2保护下将40mL 0.1mol/L的FeSO4 ·7H2O和40mL 0.1mol/L Na2S ·9H2O溶液混合,然后逐滴加入冰醋酸调节pH至3.0,搅拌反应5min后离心5min、冷干,制得粒径在50~100 nm的高度结晶的磁性Fe3S4纳米粒子。他们发现,在pH 4.0时制备出的样品的XRD谱中Fe3S4纳米粒子峰值明显低于在pH 3.0时制备的样品,在pH 5.0时制备的产物更是直接变为FeS;在反应时间为10min时会有高结晶度、高纯度的产物Fe3S4纳米粒子,随着反应时间的延长,产物中FeS2的产量也会增多。Moore等[16]尝试使用Chang等[18]的方法制备Fe3S4纳米粒子,却没能成功制备出结晶度高的Fe3S4产物。这也反映出了由于Fe3S4纳米粒子的亚稳态性,沉淀法制备具有局限性,不能高效率地合成出高结晶度、高纯度的单分散Fe3S4纳米粒子。
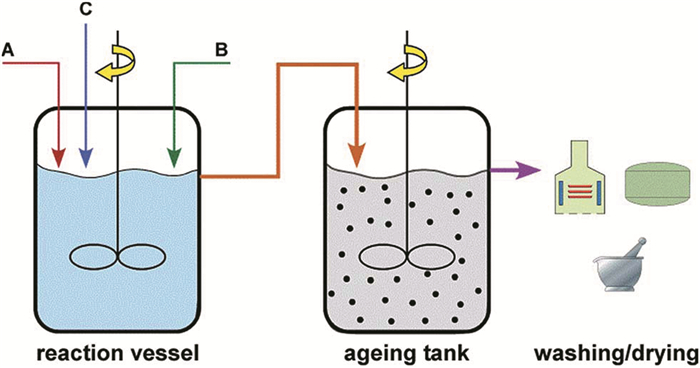
Simeonidis等[19]使用图 2所示的两步连续搅拌装置通过沉淀法探索Fe3S4纳米粒子的大规模生产途径,并在反应过程中加入了不同的功能化合物(十六烷基三甲基溴化铵(CTAB)、柠檬酸盐、葡聚糖)来修饰Fe3S4纳米粒子,制备出了粒径、饱和磁化强度不同的Fe3S4纳米粒子,具体数据参见表 1。制备过程为:将样品原料液1mol/L的A(FeSO4)和1mol/L的B(Na2S)以及10%的功能化合物,在一定的流速下(0.5~3 L ·h-1)同时连续注入第一个反应装置中,然后随着原料液的不断注入,让反应物流入第二个反应装置,在第二个反应装置中充分反应后流出并收集产物后,立即在100℃下干燥制得产品。反应溶液通过HCl溶液来调节溶液pH至3.5,反应时间控制在10min,防止产品进一步分解。该反应装置可以有效制备出粒径大致统一的Fe3S4纳米粒子,并且生产规模较大。
 下载:
导出CSV
下载:
导出CSV
| 产物 | 溶液A | 溶液B | pH | 官能团 | tr/min | Tr/Td/℃ | 粒径/nm | Ms/(Am2/kg) | IEP |
| Fe3S4 | FeSO4 | Na2S | 3.5 | - | 10 | 20/100 | 35 | 14 | 7.0 |
| Fe3S4 | FeSO4 | Na2S | 3.5 | citrate | 10 | 20/100 | 32 | 25 | 6.0 |
| Fe3S4 | FeSO4 | Na2S | 3.5 | CTAB | 10 | 20/100 | 38 | 1.5 | 7.2 |
| Fe3S4 | FeSO4 | Na2S | 3.5 | dextran | 10 | 20/100 | 31 | 26 | 6.7 |
| (tr:反应时间, Tr/Td:反应温度和干燥温度, Ms:饱和磁化强度, IEP:等电点) | |||||||||
反应原理为:
|
$ \text{FeS}{{\text{O}}_{4}}+\text{N}{{\text{a}}_{2}}\text{S}\xrightarrow{\text{pH}=3.5}\text{FeS}+\text{N}{{\text{a}}_{2}}\text{S}{{\text{O}}_{4}} $ |
|
$ {3{\rm{FeS}} + {{\rm{S}}^{2 - }} + {{\rm{O}}_2} + 4{{\rm{H}}^ + } \to {\rm{F}}{{\rm{e}}_3}{{\rm{S}}_4} \downarrow + 2{{\rm{H}}_2}{\rm{O}}} $ |
沉淀法的特点是反应时间短、设备工艺简单、原料成本低、成核易控制等,这种方法制得的Fe3S4纳米颗粒粒径一般较小(100nm以下),表面便于进行功能化修饰。但此方法受到的反应条件限制较多,由于反应速度快,产物容易团聚、不纯、易被氧化、磁性降低都是该方法容易出现的问题。一般情况下要在N2保护或无氧环境中反应,其中pH和反应时间是沉淀法合成Fe3S4的关键。沉淀法在形貌尺寸的调节、晶体形成高度控制等方面还有待提高。
水热法也称为热液法,广义上面可分为水溶剂热法(以水为溶剂)和溶剂热法(有机溶剂或水/有机溶剂的混合溶剂)。水热法是在高温、高压下进行的湿化学合成方法,近些年广泛应用于制备各种无机纳米材料,也是Fe3S4纳米粒子最常见的稳定的合成方法。水热法通常是于反应釜中加热溶于水或有机溶剂的反应物前体,在高温高压下导致反应物前体展现出与常温下不同的性质,并加速纳米颗粒的成核,导致生成更小尺寸的纳米粒子。
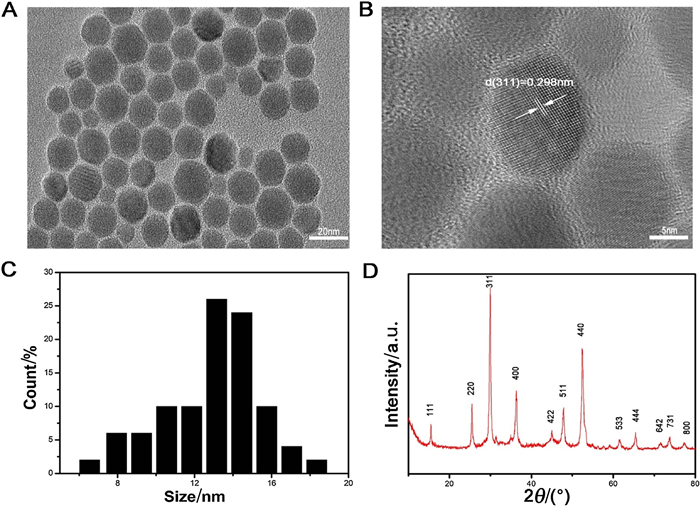
2019年,Liu等[20]以FeSO4 ·7H2O和L-半胱氨酸作为原料、聚乙烯吡咯烷酮(PVP)为助剂、乙二胺为尺寸控制剂,通过水热法制备了粒径13nm的单分散亲水Fe3S4纳米颗粒(见图 3)。其中乙二胺的存在是减小产物尺寸的主要原因。该方法制备的Fe3S4纳米颗粒具有良好的磁热性能和生物相容性,并在动脉炎症的治疗方面展现出优异的性质。Liu等[21]还以L-半胱氨酸、FeSO4溶液为原料,水为溶剂,通过简单的水热法成功制备了单一尺寸(50nm)的磁性Fe3S4纳米粒子,其在300K下的饱和磁化强度和矫顽力分别为35.5emu ·g-1和910Oe。
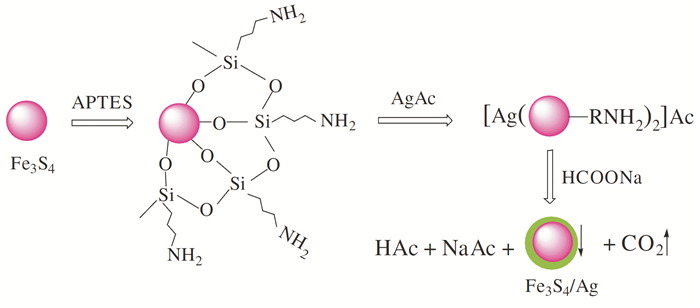
He等[22]以FeCl3 ·6H2O为铁源、Na2S ·9H2O为硫源、乙二醇为溶剂和还原剂,通过水热法成功制备出了直径175nm左右的磁性Fe3S4纳米颗粒,并将产物用氨丙基三乙氧基硅烷修饰后,合成出直径226nm左右的Fe3S4/Ag磁性纳米粒子,其可应用在抗菌方面。反应流程如图 4所示。反应原理:
|
$ \begin{array}{l} \begin{array}{*{20}{c}} {{{\rm{S}}^{2 - }} + 2{{\rm{H}}_2}{\rm{O}} \to {{\rm{H}}_2}{\rm{S}} + 2{\rm{O}}{{\rm{H}}^ - }}\\ 2\text{F}{{\text{e}}^{3+}}+{{\text{H}}_{2}}\text{S}\xrightarrow{\text{H}0\text{C}{{\text{H}}_{2}}\text{C}{{\text{H}}_{2}}\text{OH}}\text{2F}{{\text{e}}^{2+}}+\text{S}\downarrow +2{{\text{H}}^{+}}\\ \begin{array}{l} {\rm{2F}}{{\rm{e}}^{3 + }} + 3{{\rm{S}}^{2 - }} \to {\rm{F}}{{\rm{e}}_2}{{\rm{S}}_3} \downarrow \\ \;\;\;{\rm{F}}{{\rm{e}}^{2 + }} + {{\rm{S}}^{2 - }} \to {\rm{FeS}} \downarrow \end{array}\\ {{\rm{F}}{{\rm{e}}^{3 + }} + 3{\rm{O}}{{\rm{H}}^ - } \to {\rm{Fe}}{{({\rm{OH}})}_3}}\\ {{\rm{F}}{{\rm{e}}^{2 + }} + 2{\rm{O}}{{\rm{H}}^ - } \to {\rm{Fe}}{{({\rm{OH}})}_2}} \end{array}\\ \;\;\;\;\;\;\;\begin{array}{*{20}{c}} {2{\rm{Fe}}{{({\rm{OH}})}_3} + 3{{\rm{S}}^{2 - }} \to {\rm{F}}{{\rm{e}}_2}{{\rm{S}}_3} + 6{\rm{O}}{{\rm{H}}^ - }}\\ {{\rm{Fe}}{{({\rm{OH}})}_2} + {{\rm{S}}^{2 - }} \to {\rm{FeS}} + 2{\rm{O}}{{\rm{H}}^ - }}\\ \text{F}{{\text{e}}_{2}}{{\text{S}}_{3}}+\text{FeS}\xrightarrow{{{190}^{{}^\circ }}\text{C}}\text{F}{{\text{e}}_{3}}{{\text{S}}_{4}}\\ {2{\rm{Fe}}{{({\rm{OH}})}_3} + {\rm{Fe}}{{({\rm{OH}})}_2} \to {\rm{F}}{{\rm{e}}_3}{{\rm{O}}_4}} \end{array} \end{array} $ |
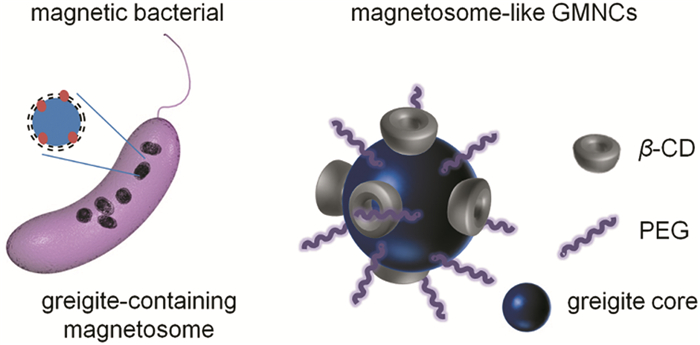
Feng等[23]用Fe(acac)3为铁源、硫代乙酰胺(CH3CSNH2)、作为硫源、乙二醇为溶剂,加入聚乙二醇(PEG)和β-环糊精(β-CD)作为表面活性剂和反应稳定剂,通过水热法制备了具有良好生物相容性、核磁共振成像能力和药物传递性的90nm左右的Fe3S4磁性纳米颗粒(如图 5)。其中PEG和β-CD起到了控制尺寸、稳定产物状态的关键性作用。
Hao等[24]将Fe(NO3)3 ·9H2O和3Ni(NO3)2 ·2H2O加入到苯甲醇中,强烈搅拌30min,然后加入硫代乙酰胺,继续搅拌30min;再将混合溶液移入高压釜中,220℃反应12h。产物经离心、洗涤、干燥后得到粒径在300nm左右的空心球形Fe3S4@NiS纳米颗粒,其具有优异的催化性能,可作为催化剂应用在析氧反应中。
Zhang等[25]通过调控反应溶剂乙二醇与水的比例,水热合成两种不同形态(片状和不规则颗粒状)的Fe3S4纳米产物,并发现尺寸较小的片状产物的饱和磁化强度要远低于颗粒状产物。在水热法合成Fe3S4纳米粒子的过程中,温度和反应溶剂都起到了关键的作用。当温度较低而其他条件不变时,产物会产生FeS2杂质;实验中当温度达到180℃时,产物中FeS2完全消失。溶剂改变时,会因介电常数、离子间的引力、溶质溶剂相互作用的改变而影响产物Fe3S4纳米粒子的晶格生长和成相。
还有一些三维立体形态(银耳状[26]、花簇状[27, 28]、桑葚状[29])的Fe3S4产物或Fe3S4复合产物可通过控制反应温度或者加入不同的表面活性剂经水热法合成出来,不同的形态、尺寸导致产物的性质发生了根本性的变化。
通过水热法制备出的产物纯度高、不易团聚,但是其操作过程比较复杂,对设备的要求较高,成本也会显著提高。
热分解法是制备Fe3S4纳米粒子常用的方法之一。通常是在有较高沸点的有机溶剂(油酸、脂肪酸、十六胺等)中加入含有Fe离子或S离子的前体,或者同时含有Fe、S离子的前体混合物,在较高温度下精确控制反应条件,制备出产物。
Beal等[30]将Fe(acac)2和十六胺混合,吹入N2,300℃下加热2h,所得溶液作为Fe源前体;S单质溶解在油胺(OA)中(每3.6mL OA中有1.8mmol硫单质),所得溶液作为S源前体;然后快速加入到Fe源前体溶液中,最后快速冷却到室温,用热乙醇稀释后离心、洗涤,得到尺寸在6.5±0.5 nm的单分散磁性Fe3S4纳米粒子,其在10和300 K下的饱和磁化强度分别为12和9 emu ·g-1。
Vanitha等[31]将单源前驱体[NnBu4]2[Fe4S4(SPh)4][32]加入已加热至实验温度的烷基胺(油胺、十二胺或十六胺)中搅拌12~16 h,精确控制反应条件,分别在不同温度(200、215和230 ℃)下制备出了Fe3S4纳米粒子。随着温度的升高,纳米粒子尺寸从4.5nm降至2.5nm。值得注意的是,在其他条件相同,温度变为180℃,溶剂改为辛胺时,产物则变为Fe7S8。小粒径产物可归因于高温促进了前体注入后的快速成核爆发,更高效消耗了单体。
Beal等[33]将单源前驱体[Fe(N-MeIm)6]S8(其中N-MeIm为N-甲基咪唑[34, 35])溶解在油胺中,随后快速注入到预热至300℃的油胺中,然后将反应溶液快速降至室温后离心,去除上清液后加入甲苯再次离心,去除上清液后再加入丙酮离心,干燥后得到颗粒尺寸在16±1 nm不规则形状的Fe3S4纳米粒子。随后,他们[36]使用Fe(acac)2为铁源、S单质为硫源、油胺为溶剂,在200℃下反应4h,制备了粒径12nm的磁性Fe3S4纳米粒子。
Vasilenko等[37]将[FeⅡFe2Ⅲ(CH3CO2)6] ·2H2O[38]和硫脲溶入四甘醇中,分别在250℃反应8h以及200℃下反应16和40 h,制备了粒径在10、20、30 nm左右的磁性Fe3S4纳米粒子。
Zhang等[39]合成Fe(Ddtc)3(Ddtc=二乙基二硫代氨基甲酸酯)[40]作为单源前驱体,分别以油胺/十八烯(1 :1)和油酸/油胺/十八烯(1 :1 :2)作为反应溶剂,经热分解法制备了粒径40~100 nm和30~100 nm的磁性Fe3S4纳米粒子,两种产品的饱和磁化率分别为31.3和36.9 emu ·g-1。Han等[41]先合成单源前驱体Fe(Ddtc)3,以油胺为反应溶剂,在280℃下制备了粒径100~500 nm、厚度为50nm的磁性Fe3S4纳米片。
Mlowe等[42]以哌啶二硫代氨基甲酸铁(Ⅲ)作为单源前驱体、油胺作为反应溶剂,在230℃下成功制备了50~200 nm的六边形磁性Fe3S4纳米片,并应用在气体感应研究方面。
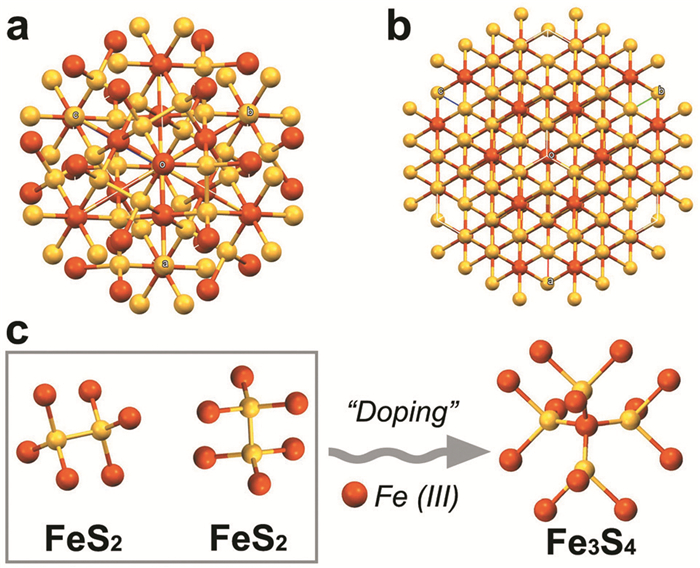
2015年,Li等[43]将Fe(acac)3、二苯醚和十八胺在真空条件下加热制得Fe源前体,再将硫单质和二苯醚在70℃下加热1h作为S源前体,最后将Fe源前体加热至220℃快速注入S源前体并继续反应3h,洗涤,干燥后,得到正多边形的厚度6.5nm、横向尺寸30~50 nm的磁性Fe3S4纳米片,产物的饱和磁化强度为25.6emu ·g-1。通过改变Fe/S的进料摩尔比、二苯醚/十八胺的体积比、温度和反应时间可以控制纳米片的成核数量和形态。在合成Fe3S4纳米片的过程中,首先形成了FeS2晶核,随后FeS2晶核与Fe(Ⅲ)进一步联合生长为Fe3S4纳米片,该反应的晶格结构变化过程如图 6所示。
Pattrick等[44]将单源前驱体二乙基二硫代氨基甲酸铁(Ⅲ)加入到预热至90℃并通氮气30min的油胺中,再将温度缓慢升至230℃并保持30min,冷却,洗涤,干燥后,得到50~100 nm的六边形磁性Fe3S4纳米粒子。通过添加不同比例的二乙基二硫代氨基甲酸盐,可将Ni和Zn元素掺杂进入产物中。
Yang等[45]使用三乙酰丙酮铁(Ⅱ)、乙二醇和硫代乙酰胺通过热解法制备出Fe3S4纳米粒子,将其保存在乙二醇中,进一步加入适量Cd(NO3)2 ·4H2O和Zn(NO3)2 ·6H2O,将Na2S ·9H2O的乙二醇溶液滴入上述体系,最终制得粒径在6~10 nm的球形磁性Cd0.2Zn0.8S@Fe3S4纳米粒子,复合产物具有核-壳结构,饱和磁化强度为35.02emu ·g-1,并具有良好的光催化剂性能。
通过高温热解法制备出来的Fe3S4纳米粒子通常具有较小的粒径,在室温下表现为超顺磁性。在其制备过程中温度、反应时间、溶剂和表面活性剂的选择为成功制备Fe3S4纳米粒子的关键因素。
高温热分解法制备出的Fe3S4磁性纳米粒子分散性好、粒径分布窄、尺寸和形貌容易控制,但其产物通常只溶于有机溶剂,生物相容性差;另外,有机金属前驱体价格较高,生产成本昂贵,不适合规模化生产,实际应用能力不佳。
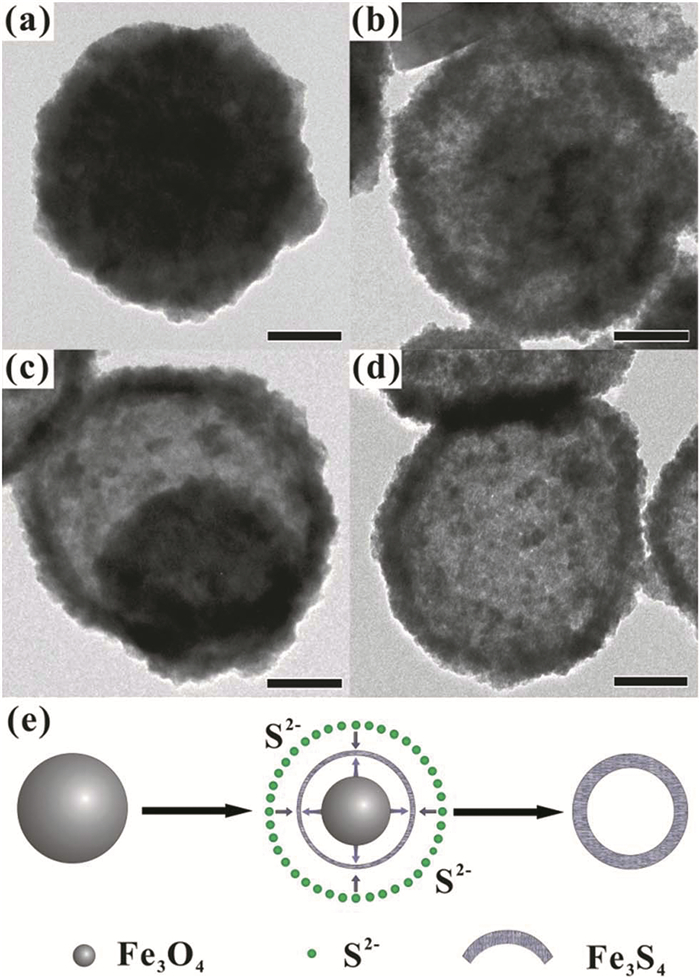
上述三种方法为合成磁性Fe3S4纳米粒子的常见方法,其优缺点对比见表 2。另外也有其他一些比较少用或者新颖的合成方法。2014年,Zheng等[46]以球形Fe3O4为模板,在乙醇中加入硫代乙酰胺和Fe3O4,于120℃下反应制备了直径400nm左右的空心介孔Fe3S4纳米球(反应过程中产物的变化过程见图 7)。由于其优异的纳米结构,在锂电池和水污染处理方面展现出较好的性能。
下载:
导出CSV
| 制备方法 | 反应时间 | 温度/℃ | 优点 | 缺点 |
| 沉淀法 | 10min | 20~100 | 制备过程简单,消耗低,颗粒小,可用于工业生产 | 容易团聚,掺杂,退化;形状和粒径不易控制 |
| 水热法 | 1~24 h | 160~220 | 纯度高,分散好,洁净度高,粒径统一 | 操作复杂,成本高 |
| 热解法 | 1~40 h | 90~300 | 分散好,粒径大小统一,粒径、形貌容易控制 | 环境友好性差,生产成本高 |
Paolella等[47]将FeCl2、Na2S2O3、硫单质、3-甲基邻苯二酚和十八胺溶解在十八烯中后置于50mL三颈瓶中,在N2保护下,80℃反应10min,再加热至200℃反应1h,制备了粒径10~20 nm、厚度4.5nm左右的多边形磁性Fe3S4纳米片。Lyubutin等[48]利用多元醇法通过调节Fe/S比制备了粒径9~20 nm的磁性Fe3S4纳米颗粒。Zhu等[49]先制备出粒径50~80 nm的Fe2O3模板,再通过水热法进一步将Fe2O3硫化得到了粒径500~700 nm的Fe3S4微粒。Liao等[50]用微量的六亚甲基四胺作为氧化剂和螯合剂,在125℃条件下加热FeSO4和硫脲混合溶液合成了磁性Fe3S4微片,并在静态磁场下组装成了3D花簇形态。
随着科技的不断进步,Fe3S4纳米粒子的制备方法也在不断完善,各种方法不断地交叉渗透,有利于开发新的更加经济简便的制备方法。
废水中重金属的高毒性和不可降解性对人类健康和环境造成严重威胁,磁性纳米吸附剂由于其表面积大、易分离已经引起了广泛的关注。金属硫化物含有丰富的硫,对重金属有很高的亲和力和选择性。其中Fe3S4由于其独特的磁性,是理想的重金属吸附剂[51~56]。影响Fe3S4纳米粒子吸附重金属能力的因素有很多,如反应温度、溶液pH、重金属离子浓度等[57~66]。
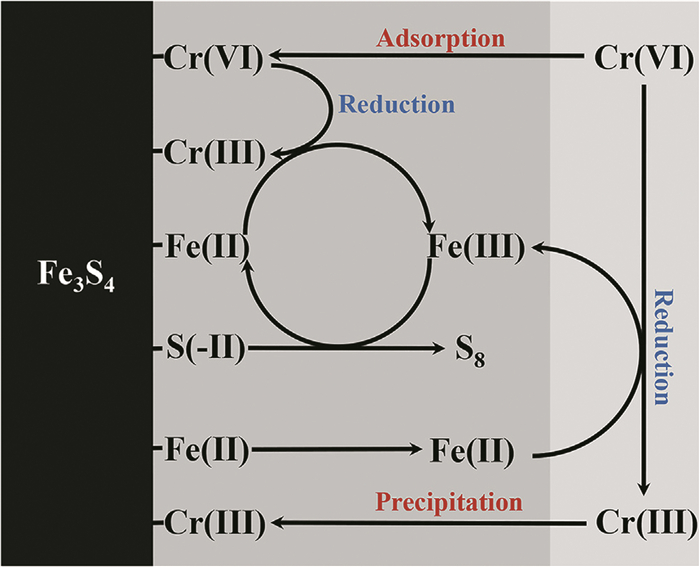
Liu等[67]利用水热法合成磁性Fe3S4纳米粒子,并将其用来去除水溶液中Cr6+,去除机理如图 8所示。他们系统研究了pH、Fe3S4 :Cr(Ⅵ)摩尔比、有无氧气和天然有机物存在对Cr(Ⅵ)去除效率的影响。结果表明,Cr(Ⅵ)的去除过程符合准一级反应动力学模型,并涉及表面吸附、表面还原和溶液还原。在pH 7.0时表面反应效率最高,可达到90%以上,并在重复使用三次后去除效率降低到50%;在pH 5.7条件下,Fe3S4 :Cr(Ⅵ)为1 :0.011到1 :0.171时去除效率达到100%,当Fe3S4 :Cr(Ⅵ)达到1 :0.228时,去除效率降低到72%;有无氧气和天然有机物对Cr(Ⅵ)去除效率没有影响。Yang等[68]通过水热法合成了花状磁性Fe3S4微粒,并研究其去除U(Ⅵ)和Cr(Ⅵ)的效率。在pH 5.0、298K下,Fe3S4微粒对U(Ⅵ)和Cr(Ⅵ)的单独最大去除量分别为423.0和231.3 mg ·g-1;而当U(Ⅵ)和Cr(Ⅵ)同时存在时,U(Ⅵ)和Cr(Ⅵ)的去除量分别提升为505.4和274.1 mg ·g-1。Fe3S4微粒通过不同的官能团吸附U(Ⅵ)和Cr(Ⅵ),U(Ⅵ)主要与硫官能团络合,Cr(Ⅵ)主要与氧官能团络合。U(Ⅵ)和Cr(Ⅵ)离子能被样品中的Fe(Ⅱ)和S2-部分还原为毒性小的U(Ⅳ)和Cr(Ⅲ)离子。两个反应过程均符合准二级反应动力学,化学吸附占主导。
Wang等[69]通过水热法合成了磁性Fe3S4/生物炭混合材料,并研究其对污水中Cr(Ⅵ)的去除效率及原理。当Cr(Ⅵ)初始浓度为20mg/L时,Fe3S4/生物炭质量比为30的该材料对Cr(Ⅵ)的最大去除效率为93%,最大去除能力为23.25mg ·g-1,反应过程遵循准一级反应动力学模型,反应过程包括了耦合吸附和氧化还原反应。Fe3S4/生物炭复合材料上的持久性自由基介导了Fe(Ⅲ)/Fe(Ⅱ)循环,从生物碳向Fe(Ⅲ)转移电子,从而实现了高效的Fe(Ⅲ)/Fe(Ⅱ)循环。Zhou等[70]通过改进的水热法合成了磁性Fe3S4-CTAB复合材料,并进行了去除水中重金属Cr(Ⅵ)的研究。当复合材料中CTAB的用量为0.75g时,所得产物比表面积最大,对Cr(Ⅵ)的最大吸附量为330.03mg ·g-1。吸附去除过程包括了吸附Cr(Ⅵ)到材料表面以及进一步的还原反应,吸附遵循准二级动力学模型。
2017年,Kong等[71]通过模板法合成磁性Fe3S4/还原氧化石墨烯(rGO)复合纳米材料,研究其对废水中重金属Pb(Ⅱ)的吸附过程。该复合材料对Pb(Ⅱ)的最大吸附量为285.71mg ·g-1。反应过程遵循准二级动力学模型,本质上是化学吸附,主要受吸附过程控制而不是扩散过程。在阳离子和阴离子共存的情况下,对Pb(Ⅱ)具有显著的选择性,腐殖酸的加入会促进复合材料对Pb(Ⅱ)的去除。Fe3S4/rGO对Pb(Ⅱ)去除效率的增强主要是Pb(Ⅱ)与表面吸附的Fe3S4纳米粒子之间的强选择性及Pb-S相互协同作用的结果。该复合材料还可有效去除废水中的As离子,去除效率可达98.18%;在循环使用5次后,去除效率仍可达到80%以上。
2015年,Kong等[72]通过热解法合成了磁性β环糊精(β-CD)-Fe3S4纳米复合材料,并用于对污水中Pb(Ⅱ)的去除实验。复合材料对Pb(Ⅱ)的最大吸附量达到256mg ·g-1,该过程遵循准二级动力学模型,吸附曲线遵循Langmuir曲线模型。并且其对Zn(Ⅱ)、Cd(Ⅱ)、Cu(Ⅱ)都有不同程度的吸附作用。Pb(Ⅱ)的去除主要是通过化学反应和表面吸附,反应机理为:
|
$ {\rm{F}}{{\rm{e}}_3}{{\rm{S}}_4} \leftrightarrow {\rm{F}}{{\rm{e}}^{2 + }} + 4{{\rm{S}}^{2 - }} + 2{\rm{F}}{{\rm{e}}^{3 + }} $ |
|
$ {\rm{P}}{{\rm{b}}^{2 + }} + {{\rm{S}}^{2 - }} \leftrightarrow {\rm{PbS}} $ |
Islam等[73]通过水热法合成出Fe3S4/聚吡咯复合材料,并应用于吸附水中的As(Ⅲ)和As(Ⅴ)。在pH 7.0时,其对As(Ⅲ)和As(Ⅴ)的吸附量分别达到15.21和15.18 mg ·g-1,反应过程遵循准一级动力学模型;As(Ⅲ)和As(Ⅴ)通过与复合物中Fe3S4的表面化学反应和与聚吡咯的离子交换而被去除。
Wang等[26]通过热分解法合成了银耳状磁性Fe3S4-C复合材料,并将其用于去除废水中的甲基蓝,最大吸附量为95mg ·g-1,反应过程符合准二级动力学模型,该复合材料经两次循环使用后,对甲基蓝的去除效率仍在90%以上。Zheng等[46]通过模板法合成了磁性介孔空心球型Fe3S4纳米材料,将其作为吸附剂进行去除对废水中甲基蓝和Pb(Ⅱ)的研究。当甲基蓝初始浓度为100mg/L时,Fe3S4纳米材料对甲基蓝的吸附量为29.3mg ·g-1,对Pb(Ⅱ)的最大吸附量为92.1mg ·g-1,两个过程都符合Freundlich吸附等温线模型。
Liu等[74]通过溶剂热法合成出磁性Fe3S4微粒,并将其应用于对水中洛克沙砷(ROX)的去除。结果显示:(1)Fe3S4微粒可在pH 3.6~6.6对ROX吸附和还原,ROX的去除过程遵循受pH严重影响的准二级动力学模型;(2)ROX的硝基被Fe3S4还原为含氨基的产物4-羟基-3-对氨基苯胂酸;(3)在中性和碱性pH下,ROX和4-羟基-3-对氨基苯胂酸优先被吸附在Fe3S4微粒表面而不是被还原;(4)ROX与Fe3S4之间的相互作用几乎不受共存阳离子、阴离子和天然有机质的作用影响。
Fe3S4纳米材料由于其独特的磁性、还原性、吸附性作为吸附剂有很好的潜力,但其在空气中不稳定容易转变为其他相、容易团聚等缺点也限制了其发展。合成Fe3S4纳米复合材料,用合适的官能团修饰来解决这些问题为该材料作为良好的吸附剂和还原剂提供了很好的前景[75, 76]。
锂离子电池由于具有高能量密度和快速充电能力,已经成功应用于我们日常生活的许多方面。Fe3O4因其应用在锂离子电池的高理论容量(924mAh ·g-1)而已经被广泛研究,虽然Fe3S4的理论容量为785mAh ·g-1,但Fe3S4与Fe3O4相比具有更高的电子导电性和更小的充电/放电循环过程中的体积膨胀,也常被作为电池负极材料进行研究。此外Fe和S之间的化学键比Fe和O之间的化学键弱,更有利于转化反应的进行[77~87]。Fe3S4或Fe3S4复合纳米材料的电化学性质对比见表 3。
下载:
导出CSV
| 材料 | 应用 | 电流密度/(A·g-1) | 循环次数 | 比容量/(mAh·g-1) | 参考文献 |
| Fe3S4八面体微晶 | 锂离子电池阳极材料 | 0.1 | 1/100 | 1161/563 | [85] |
| Fe3S4空心球型 | 锂离子电池阳极材料 | 0.2 | 100 | 750 | [41] |
| Fe3S4纳米片 | 锂离子电池阳极材料 | 0.1 | 1/100 | 1207/495 | [38] |
| Fe3S4八面体纳米粒子 | 钠离子电池阳极材料 | 20 | 3500 | 275 | [92] |
| Fe3S4纳米片 | 锂离子电池阳极材料 | 0.2 | 120 | 548 | [94] |
| Fe3S4 NPs@rGO | 锂离子电池阳极材料 | 0.1/1 | 100/800 | 950/720 | [90] |
| Iron sufides@rGO | 锂离子电池阳极材料 | 0.1/1 | 100/1000 | 1189.6/800 | [91] |
| Fe3S4微粒 | 可充电碱性水电池阳极材料 | 2/2 | 1/3000 | 110.6/83.8 | [95] |
| Fe3S4/Co9S8 | 锂离子电池阳极材料 | 0.1/2 | 100/1500 | 945/519 | [24] |
| Fe3S4@Li7P4S11 | 全固态锂电池阴极材料 | 0.1 | 200 | 1001 | [96] |
Li等[88]通过水热法合成了高纯度的八面体磁性Fe3S4微粒,并将其作为锂离子电池的负极材料,通过循环伏安法和恒流放电充电循环法测试了其电化学性能。其在第一个循环的放电容量高达1161mAh ·g-1,经过100次循环后的放电容量为563mAh ·g-1。容量的降低归因于该负极材料嵌入锂离子后体积发生较大变化,导致晶体解体,电极材料与集电体之间的连接丢失。第二次放电循环的反应式可以表示为:
|
$ \begin{array}{l} 1.87{\rm{V}}(1)2{\rm{FeS}} + 2{\rm{L}}{{\rm{i}}^ + } + 2{{\rm{e}}^ - } \leftrightarrow {\rm{L}}{{\rm{i}}_2}{\rm{Fe}}{{\rm{S}}_2} + {\rm{Fe }}\\ {\rm{1}}{\rm{. }}40{\rm{V}}(2){\rm{L}}{{\rm{i}}_2}{\rm{Fe}}{{\rm{S}}_2} + 2{\rm{Li}} + 2{{\rm{e}}^ - } \leftrightarrow {\rm{Fe}} + 2{\rm{L}}{{\rm{i}}_2}{\rm{S}} \end{array} $ |
单纯以Fe3S4作为锂离子电池负极材料时,材料的结构是不稳定的,在锂离子嵌入和脱嵌的过程中该负极材料会发生体积膨胀,晶体解体,粉化脱落,导致电池的循环性能变差,使Fe3S4作为锂离子负极材料的应用受到限制。通过合成不同形态和结构的Fe3S4纳米微粒或者与其他电化学活跃、表面积大的材料复合可以有效解决该问题[89~92]。Zheng等[46]通过模板法合成了介孔空心球形Fe3S4纳米粒子,其用作锂离子电池负极材料时显示出高的可逆容量,在0.2A ·g-1下经100个放电/充电循环后容量为750mAh ·g-1。Guo等[93]通过水热法合成了包裹在rGO中的Fe3S4@rGO复合材料。将其作为锂离子电池负极材料,在0.1A ·g-1下100次循环后的可逆容量达950mAh ·g-1,1A ·g-1下800次循环后可逆容量仍达720mAh ·g-1。Fe3S4@rGO优异的电化学性能归因于Fe3S4纳米粒子与rGO基体的协同作用:(1)Fe3S4可以扩大rGO的层间距,增加Li存储量,增加石墨烯与电解质之间的双层电容;(2)rGO的加入使Fe3S4纳米粒子的尺寸变小,为Li的存储提供更多的电活性位点,并在循环过程中有效限制Fe3S4纳米粒子的体积变化;(3)电极材料表面形成的聚合物/凝胶状薄膜在第一次放电后会产生赝电容效应,这对增强可逆性能有一定的贡献。
此外,Xu等[94]使用水热法合成了FeS2球形微粒、Fe3S4立方体微粒和rGO的复合材料iron sulfides@rGO(图 12)。将该复合材料作为锂离子电池负极材料,其在100mA ·g-1下的初始充放电容量达1476.2mAh ·g-1,经过100次循环的容量为1189.6mAh ·g-1;在1A ·g-1下经1000次循环后,其仍保有800mA ·g-1的可逆容量。
Fe3S4也可作为其他离子电池材料进行相关研究。Li等[95]合成出磁性Fe3S4纳米片,并作为钠离子电池负极材料。其在0.2A ·g-1时展现出548mAh ·g-1的可逆容量,在20A ·g-1电流密度下经过3500次循环后,其比容量仍达275mAh ·g-1。Cao等[28]合成了3D花状Fe3S4微粒并将其作为镍氢电池负极材料的一部分,验证了其电化学储氢能力,其在室温下的放电容量为214mAh ·g-1。
Zhang等[96]使用水热法合成MgH2-Fe3S4复合材料,并研究了其储氢性能。复合材料对氢的吸附重量比纯MgH2高三倍,氢的解吸率比纯MgH2快了2倍。此外脱氢温度也比没有加入Fe3S4的MgH2低了90K,展现出了其良好的储氢能力。
纳米技术在克服传统方法的治疗、诊断和检测癌症时所面临的许多问题方面拥有巨大的潜力。目前,磁性纳米粒子在癌症方面的应用可以分为诊断和治疗两部分。在诊断方面,Fe3S4磁性纳米粒子能够用于核磁共振成像(MRI),可以提供肿瘤的各种信息,并在药物载体传递中判断药物是否递送到目标细胞。在治疗方面,Fe3S4纳米粒子可应用于磁热疗、光热疗和磁性靶向给药。磁热治疗主要是通过注射将磁性纳米粒子送往肿瘤部位,在外加交流磁场的作用下,纳米粒子的磁畴方向发生变化,并通过磁滞损耗、尼尔弛豫和布朗弛豫等机制产热,进而实现治疗肿瘤的效果。光热治疗是指通过注射将磁性纳米粒子送往特定部位,利用不同磁性纳米粒子吸收特定波长的光引起的表面等离子体共振(SPR)产生热能,进而治疗肿瘤。磁性靶向给药是指将药物负载到经过修饰的磁性纳米粒子表面,然后通过静脉或动脉注入人体内,在外加磁场的引导作用下,将药物定向运输到指定部位[99~112]。
Feng等[23]利用β-CD和PEG合成具有类似磁小体结构和磁性具有仿生设计的Fe3S4纳米粒子(GMNCs)。该纳米粒子具有良好的生物相容性,MRI能力和药物传递能力。GMNCs的横向弛豫强度达到94.8mM-1 ·S-1,磁性靶向给药对化疗药物阿霉素的包封率达到58.7%。此外,还可通过静脉注射负载阿霉素的GMNCs对小鼠肿瘤进行强化治疗。
随着生物药学的应用发展,作为一种理想的纳米诊疗材料不仅需要同时实现诊断和肿瘤治疗(磁热疗、光热疗、磁性靶向药物传递),还应当对肿瘤的微环境有积极的反应。2019年,Liu等[20]通过水热法合成了具有良好光热性、生物相容性的磁性Fe3S4纳米粒子,并通过以其为基础的磁热疗和光热疗协同作用治疗动脉粥样硬化和血管狭窄。在小鼠模型的体内实验表明,磁热疗与光热疗协同作用能有效清除浸润的炎症巨噬细胞和进一步阻止动脉狭窄的形成。该Fe3S4纳米粒子除了具备将光和磁的刺激转化为热的双重能力外,还具有良好的T2加权核磁共振成像性能(52.8mM-1 ·S-1)。
2018年,Guan等[113]通过热分解法合成了PVP包裹的磁性Fe3S4纳米片(120±18nm),发现其在磁靶向作用下可在肿瘤内有效积累,并在三周内逐渐转化为5nm左右的小颗粒,在正常生理条件下发挥其治疗作用后有效从体内排除。分散在水中的该复合纳米片具有较强的近红外吸收特性、良好的光热转换效率(64.3%)和良好的T2加权核磁共振成像性能(71.3mM-1 ·S-1)。此外,该材料可因为光热疗产生的局部高温促进其与肿瘤微环境中过量的H2O2实现芬顿过程产生·OH,并抑制肿瘤的生长和光热疗后的复发,实现了光热疗和化学动态治疗协同治疗肿瘤。
除了上面的应用外,Fe3S4纳米材料还应用在气体感应[42]、催化剂[45, 114~117]、电磁波吸收材料[118]等方面。由于其特殊的纳米尺寸效应、磁性和还原性使其广泛应用于环境、医药、能源储存、催化等方面,另外Fe3S4还对古地磁学的研究有重要的作用。
Fe3S4作为Fe3O4的硫化物对照物,Fe3O4已发表的研究成果会为Fe3S4提供极具价值的研究方向,对Fe3S4基本性质的研究也会起到参考作用。同时,对Fe3S4的深入研究也可以有助于理解Fe3O4的研究缺失部分,如探索Fe3S4在高压情况下的形状、磁性、电子的改变,有助于更好地理解Fe3O4在极端条件下的磁性和电子行为[119]。在实际应用中Fe3O4已经被应用于涂料、着色剂、塑料、皮革、汽车面漆、高磁记录材料、吸附剂、催化剂等多个方面,而Fe3S4在许多方面的研究深度和广度还远不及Fe3O4,这一定程度是由于难以合成高纯度Fe3S4,导致其在应用方面的研究较少。因此,开创新的Fe3S4合成方法,合成出高纯度、高结晶度、形貌可控的Fe3S4晶体是对Fe3S4进行深入研究的关键。
本文总结了近十年来Fe3S4纳米粒子的制备方法以及每种合成方法的优劣势,希望随着科技的不断进步,各种方法不断地进行交叉渗透,创新出制备过程更加简单、性能结构更加优异的纳米粒子。目前在制备Fe3S4纳米粒子的过程中仍旧存在颗粒易团聚、颗粒尺寸不均匀、分散不均匀、产量低等一些问题,导致了Fe3S4纳米材料的广泛应用受到阻碍。今后的研究应当从Fe3S4纳米粒子的微观结构、形貌出发,满足精确控制产品形貌、大小等要求。将Fe3S4纳米材料与其他先进材料或者有特定官能团的材料复合,合成功能更加多样化的复合材料,这对拓展其在更多潜在领域的应用具有现实意义。
Hutchison J E. ACS Nano, 2008, 2(3): 395-402. doi: 10.1021/nn800131j
Sharma V K, Yngard R A, Lin Y. J. Colloid Interf. Sci., 2009, 145: 83-96. doi: 10.1016/j.cis.2008.09.002
Xi C, Zeng Y, Rui X, et al. ACS Nano, 2012, 6: 4713-4721. doi: 10.1021/nn2045714
Devey A J, Grau-Crespo R, de Leeuw N H. Phys. Rev. B, 2009, 79: 195126. doi: 10.1103/PhysRevB.79.195126
Skinner B J, Erd R C, Grimaldi F S. Am. Mineral., 1964, 46: 543-555.
Spender M R, Coey J M D, Morrish A H. Can. J. Phy., 1972, 50(19): 2313-2326. doi: 10.1139/p72-306
Gibbs G V, Cox D F, Rosso K M, et al. J. Phys. Chem. B, 2007, 111(8): 1923-1931. doi: 10.1021/jp065086i
Chang L, Rainford B D, Stewart J R, et al. J. Geophys. Res. Atmos., 2009, 114: B07101.
Roberts A P, Chang L, Rowan C J, et al. Rev. Geophys., 2011, 49: RG1002.
Muxworthy A R, Williams W, Roberts A P, et al. Geochem. Geophys. Geosy., 2013, 14: 5430-5441. doi: 10.1002/2013GC004973
Chang L, Roberts A P, Tang Y, et al. J. Geophys. Res. Earth, 2008, 113: B06104.
Li P, Xia C, Zhang Q, et al. J. Appl. Phys., 2015, 117: 223903. doi: 10.1063/1.4922578
Zhang B, de Wijs G A, de Groot R A. Phys. Rev. B, 2012, 86(2): 020406. doi: 10.1103/PhysRevB.86.020406
Wang J, Cao S H, Wu W, et al. Phys. Scripta, 2011, 83: 045702. doi: 10.1088/0031-8949/83/04/045702
Roldan A, Santos-Carballal D, de Leeuw N H. J. Chem. Phys., 2013, 138: 204712. doi: 10.1063/1.4807614
Moore J, Nienhuis E, Ahmadzadeh M, et al. AIP Adv., 2019, 9: 035012. doi: 10.1063/1.5079759
Goodenough J B. Mater. Res. Bull., 1978, 13: 1305-1314. doi: 10.1016/0025-5408(78)90121-6
Chang Y S, Savitha S, Sadhasivam S, et al. J. Colloid Interf. Sci., 2011, 363: 314-319. doi: 10.1016/j.jcis.2010.06.069
Simeonidis K, Liebana-Vinas S, Wiedwald U, et al. RSC Adv., 2016, 6: 53107-53117. doi: 10.1039/C6RA09362K
Liu J C, Guo X, Zhao Z, et al. Appl. Mater. Today, 2020, 18: 100457. doi: 10.1016/j.apmt.2019.100457
Liu X G, Feng C, Bi N N, et al. Ceram. Int., 2014, 40: 9917. doi: 10.1016/j.ceramint.2014.02.087
He Q G, Huang C Y, Liu J. Nanosci. Nanotechnol., 2014, 6(1): 10-17.
Feng M, Lu Y, Yang Y, et al. Sci. Rep., 2013, 3: 2994. doi: 10.1038/srep02994
Hao Z W, Wei P K, Kang H Z, et al. J. Electro. Chem., 2019, 850: 113436. doi: 10.1016/j.jelechem.2019.113436
Zhang Z J, Chen X Y. J. Alloys Compd., 2009, 488: 339-345. doi: 10.1016/j.jallcom.2009.08.127
Wang X B, Cai W P, Wang G Z, et al. CrysEngComm, 2013, 15: 2956. doi: 10.1039/c3ce26856j
Luo J L, Hu Y B, Xiao L, et al. Nanotechnology, 2020, 31: 085708. doi: 10.1088/1361-6528/ab53c4
Cao F, Hu W, Zhou L, et al. Dalton Transac., 2009, 42: 9246-9252.
Liu Q, Chen Z Z, Qin R, et al. Electrochim. Acta, 2019, 304: 405-414. doi: 10.1016/j.electacta.2019.03.034
Beal J H L, Prabakar S, Gaston N, et al. Chem. Mater., 2011, 23: 2514-2517. doi: 10.1021/cm2002868
Vanitha P V, O'Brien P. J. Am. Chem. Soc., 2008, 130: 17256. doi: 10.1021/ja8078187
Que L, Holm R H, Mortenson L E. J. Am. Chem. Soc., 1975, 97(2): 463-464. doi: 10.1021/ja00835a064
Beal J H L, Etchegoin P G, Tilley R D. J. Phys. Chem. C, 2010, 114: 3817-3821. doi: 10.1021/jp910354q
Ramli E Rauchfuss T B, Stern C L. J. Am. Chem. Soc., 1990, 112(10): 4043-4044. doi: 10.1021/ja00166a054
Rauchfuss T B. Inorg. Chem., 2004, 43: 14-26. doi: 10.1021/ic0343760
Beal J H L, Etchegoin P G, Tilley R D. J. Solid State Chem., 2012, 189: 57-62. doi: 10.1016/j.jssc.2012.01.015
Vasilenko I V, Cador O, Ouahab L, et al. Theor. Exp. Chem., 2010, 46: 322-327. doi: 10.1007/s11237-010-9160-z
Oh S M, Henderickson D N, Hassett K L, et al. J. Am. Chem. Soc., 1985, 107: 8009-8018. doi: 10.1021/ja00312a035
Zhang Y J, Du Y P, Xu H R, et al. CrystEngComm, 2010, 12: 3658-3663. doi: 10.1039/c002824j
Chen X, Wang Z, Wang X, et al. Inorg. Chem., 2005, 44(4): 951-954. doi: 10.1021/ic049049m
Han W, Gao M Y. Cryst. Growth Des., 2008, 8: 1023-1030. doi: 10.1021/cg701075u
Mlowe S, Osman N S E, Moyo T, et al. Mater. Chem. Phys., 2017, 198: 167-176. doi: 10.1016/j.matchemphys.2017.06.012
Li T T, Li H H, Wu Z N, et al. Nanoscale, 2015, 7: 4171-4178. doi: 10.1039/C4NR06927G
Pattrick R A D, Coker V S, Akhtar M, et al. Mineral Mag., 2017, 81: 857-872. doi: 10.1180/minmag.2016.080.114
Yang S, Zhang C H, Cai Y Q, et al. J. Alloys Compd., 2018, 735: 1955-1961. doi: 10.1016/j.jallcom.2017.11.301
Zheng J, Cao Y, Cheng C, et al. J. Mater. Chem. A, 2014, 2: 19882-19888. doi: 10.1039/C4TA05148C
Paolella A, George C, Povia M, et al. Chem. Mater., 2011, 23: 3762-3768. doi: 10.1021/cm201531h
Lyubutin I S, Starchikov S S, Lin C R, et al. J. Nanopart. Res., 2013, 15: 1397. doi: 10.1007/s11051-012-1397-0
Zhu Y Z, Yun X Y, Wu S L, et al. Ionics, 2020, 26: 105-113. doi: 10.1007/s11581-019-03237-5
Liao T Q, Wang W, Song Y L, et al. J. Mater. Sci. Technol., 2015, 31: 895-900.
Zhu M Y, Diao G W. Nanoscale, 2011, 3(7): 2748-2767. doi: 10.1039/c1nr10165j
Gautam R K, Tiwari I. Chemosphere, 2020, 245: 125553. doi: 10.1016/j.chemosphere.2019.125553
Wang X B, Liu J, Xu W Z. Colloid. Surf. A, 2012, 415: 288-294. doi: 10.1016/j.colsurfa.2012.09.035
Mendez A, Fernandez F, Gasco G. Desalination, 2007, 206: 147-153. doi: 10.1016/j.desal.2006.03.564
Edwin V A. Eur. J. Chem., 2008, 5: 844-852.
Ma L J, Wang Q, Islam S M, et al. J. Am. Chem. Soc., 2016, 138: 2858-2866. doi: 10.1021/jacs.6b00110
Peng Q M, Guo J X, Zhang Q R, et al. J. Am. Chem. Soc., 2014, 136: 4113-4116. doi: 10.1021/ja500506k
Chandra V, Park J, Chun Y, et al. ACS Nano, 2010, 4: 3979-3986. doi: 10.1021/nn1008897
Jiang W J, Cai Q, Wu W, et al. Environ. Sci. Technol., 2014, 48: 8078-8085. doi: 10.1021/es405804m
Fu Y, Wang J Y, Liu Q, et al. Carbon, 2014, 77: 710-721. doi: 10.1016/j.carbon.2014.05.076
Jiang W, Wang W F, Pan B C, et al. ACS Appl. Mater. Interf., 2014, 6: 3421-3426. doi: 10.1021/am405562c
Zhang C, Sui J H, Li J, et al. Chem. Eng. J., 2012, 210: 45-52. doi: 10.1016/j.cej.2012.08.062
Li G L, Zhao Z S, Liu J Y, et al. J. Hazard. Mater., 2011, 192: 277-283.
Li B Y, Zhang Y M, Ma D X, et al. Nat. Commun., 2014, 5: 5537. doi: 10.1038/ncomms6537
Shao D, Ren X, Wen J, et al. J. Hazard. Mater., 2016, 302: 1-9. doi: 10.1016/j.jhazmat.2015.09.043
Hoon Y J, Bjorn K, Joel D B, et al. Environ. Sci. Technol., 2007, 41: 7699-7705. doi: 10.1021/es070289l
Liu W, Jin L D, Xu J, et al. Chem. Eng. J., 2019, 359: 564-571. doi: 10.1016/j.cej.2018.11.192
Yang S Y, Li Q, Chen L, et al. Chem. Eng. J., 2020, 385: 123909. doi: 10.1016/j.cej.2019.123909
Wang X D, Xu J, Liu J, et al. Sci. Total Environ., 2020, 700: 134414. doi: 10.1016/j.scitotenv.2019.134414
Zhou Y X, Zhao Y T, Wu X G, et al. RSC Adv., 2018, 8: 31568. doi: 10.1039/C8RA06534A
Kong L, Li Z C, Huang X Q, et al. J. Mater. Chem. A, 2017, 5: 19333. doi: 10.1039/C7TA05389D
Kong L, Yan L L, Qu Z, et al. J. Mater. Chem. A, 20153: 15755-15763. doi: 10.1039/C5TA03442F
Islam M, Patel R. Separat. Sci. Technol., 2017, 52: 2835-2852.
Liu W, Ai Z H, Dahlgren R A, et al. Chem. Eng. J., 2017, 330: 1232-1239. doi: 10.1016/j.cej.2017.07.176
Na L, Fu F, Lu J, et al. Environ. Pollut., 2017, 220: 1376-1385. doi: 10.1016/j.envpol.2016.10.097
Yantasee W, Warner C L, Sangvanich T, et al. Environ. Sci. Technol., 200741: 5114-5119. doi: 10.1021/es0705238
Jia X L, Chen Z, Cui X, et al. ACS Nano, 2012, 6: 9911-9919. doi: 10.1021/nn303478e
Li X Y, Fu N Q, Zou J Z, et al. Electrochim. Acta, 2017, 225: 137-142. doi: 10.1016/j.electacta.2016.12.127
Park A R, Jeon K J, Park C M. Electrochim. Acta, 2018, 265: 107-114. doi: 10.1016/j.electacta.2018.01.158
Zhang L, Lu L, Zhang D C, et al. Electrochim. Acta, 2016, 209: 423-429. doi: 10.1016/j.electacta.2016.05.106
Bracamonte M V, Primo E N, Luque G L, et al. Electrochim. Acta, 2017, 258: 192-199. doi: 10.1016/j.electacta.2017.10.034
Lu Y, Yu L, Lou X W. Chem, 2018, 4: 972-996. doi: 10.1016/j.chempr.2018.01.003
Zhang R P, Wang Y, Jia M Q, et al. Appl. Surf. Sci., 2018, 437: 375-383. doi: 10.1016/j.apsusc.2017.12.110
Yin L X, Chai S M, Huang J F, et al. Electrochim. Acta, 2017, 238: 168-177. doi: 10.1016/j.electacta.2017.03.183
Dunn B, Kamath H, Tarascon J M. Science, 2011, 334: 928-935. doi: 10.1126/science.1212741
Rui X, Tan H, Yan Q. Nanoscale, 2014, 6: 9889-9924. doi: 10.1039/C4NR03057E
Xia X, Zhang Y, Chao D, et al. Nanoscale, 2014, 6: 5008-5048. doi: 10.1039/C4NR00024B
Li G W, Zhang B M, Yu F, et al. Chem. Mater., 2014, 26: 5821-5829. doi: 10.1021/cm501493m
Li H J, Su Q M, Kang J W, et al. Mater. Res. Bull., 2018, 108: 106-112. doi: 10.1016/j.materresbull.2018.08.042
Li J C, Ma Z, Chi Y, et al. J. Mater. Sci., 2017, 52: 1573-1580. doi: 10.1007/s10853-016-0451-1
Guo S P, Li J C, Ma Z, et al. J. Mater. Sci., 2017, 52: 2345-2355. doi: 10.1007/s10853-016-0527-y
Li J C, Xue H G, Guo S P. Funct. Mater. Lett., 2017, 10: 1750054. doi: 10.1142/S1793604717500540
Guo S P, Li J C, Xiao J R, et al. ACS Appl. Mater. Interf., 2017, 9: 37694-37701. doi: 10.1021/acsami.7b10406
Xu Q T, Li J C, Xue H G, et al. J. Power Sources, 2018, 396: 675-682. doi: 10.1016/j.jpowsour.2018.06.088
Li Q D, Wei Q L, Zuo W B, et al. Chem. Sci., 2017, 8: 160-164. doi: 10.1039/C6SC02716D
Zhang W, Cheng Y, Han D, et al. Energy, 2015, 93: 625-630. doi: 10.1016/j.energy.2015.09.080
Zheng J, Cao Y, Fu J R, et al. J. Alloy Compd., 2016, 668: 27-32. doi: 10.1016/j.jallcom.2016.01.189
Zhang Q, Mwizerwa J P, Wan H L, et al. J. Mater. Chem. A, 2017, 5: 23919. doi: 10.1039/C7TA07972A
Sharma S K, Shrivastava N, Rossi F, et al. Nano Today, 2019, 29: 100795. doi: 10.1016/j.nantod.2019.100795
Rivas J, Bañobre L M, Piñeiro R Y, et al. J. Mag. Mag. Mater., 2012, 324: 3499-3502. doi: 10.1016/j.jmmm.2012.02.075
Mehran M, Kordbacheh A A, Ghobadi A, et al. Comput. Bio. Med., 2020, 120: 103741. doi: 10.1016/j.compbiomed.2020.103741
Dutz S, Hergt R. Int. J. Hyperther., 2013, 29: 790-800. doi: 10.3109/02656736.2013.822993
Krishnan K M. IEEE Trans. Mag., 2010, 7: 2523-2558.
Hergt R, Andra W, d'Ambly C G, et al. IEEE Trans. Mag., 1998, 34: 3745. doi: 10.1109/20.718537
Oh J, Yoon H, Park J H. Bio. Eng. Lett., 2013, 3: 67-73. doi: 10.1007/s13534-013-0097-8
Jain P K, Huang X, El-Sayed I H, et al. Plasmonics, 2007, 3: 107-118.
Robinson R, Gerlach W, Ghandehari H. J. Control. Release, 2015, 220: 245-252. doi: 10.1016/j.jconrel.2015.10.036
Gong F, Hong Y Z, Papavassiliou D V, et al. Nanotechonlogy, 2014, 20: 205101.
Sadat M E, Baghbador M K, Dumn A W, et al. Appl. Phys. Lett., 2014, 9: 091903.
Moroz P, Jones S K, Gray B N. Int. J. Hyperthermia, 2002, 18: 267-284. doi: 10.1080/02656730110108785
Wang J Q, Liu G. Angew. Chem. Int. Ed., 2018, 57: 3008-3010. doi: 10.1002/anie.201711705
Choi H S, Liu W H, Misra P, et al. Nat. Biotechnol., 2007, 25: 1165-1170. doi: 10.1038/nbt1340
Guan G Q, Wang X, Li B, et al. Nanoscale, 2018, 10: 17902. doi: 10.1039/C8NR06507A
Choe Y J, Byun J Y, Kim S H, et al. Appl. Catal. B, 2018, 233: 272-280. doi: 10.1016/j.apcatb.2018.03.110
Ding C P, Yan Y H, Xiang D S, et al. Mikrochim. Acta, 2016, 183: 625-631. doi: 10.1007/s00604-015-1690-6
Lin X L, Shih K M, Chen J H, et al. Chem. Eng. J., 2019, 391: 1385-8947.
Sharifvaghefi S, Zheng Y. Can. J. Chem. Eng., 2018, 96: 231-240. doi: 10.1002/cjce.23027
Wu H J, Liu J L, Liang H S, et al. Chem. Eng. J., 2020, 393: 124743. doi: 10.1016/j.cej.2020.124743
Huang S X, Kang D, Wu X, et al. Sci. Rep., 2017, 7: 46334. doi: 10.1038/srep46334


图 2 用于Fe3S4金属纳米粒子大规模合成装置的缩略图。A和B对应金属和硫化物试剂对反应器的进料;C表示功能分子的进料[14]
Figure 2 Sketch of the large-scale production setup for synthesis of metal nanoparticles. Streams A and B corresponding to the feed of reactor by metal and sulfide reagents; Stream C indicates the addition of functional molecules or seed nanoparticles [14]



图 6 Fe和S原子在FeS2(a)和Fe3S4(b)的具体排列。(c)伴随着Fe配位环境的变化,Fe(Ⅲ)掺杂时从FeS2到Fe3S4的结构演化示意图[38]
Figure 6 The specific arrangement of Fe and S atoms in FeS2(a) and Fe3S4(b); (c)The schematic structural evolution from FeS2 to Fe3S4 as doping Fe(Ⅲ), which is accompanied with the variation of Fe coordination environment [38]

表 1 Fe3S4纳米粒子的合成参数和性质[14]
Table 1. Synthesis parameters and properties of Fe3S4 nanoparticles
| 产物 | 溶液A | 溶液B | pH | 官能团 | tr/min | Tr/Td/℃ | 粒径/nm | Ms/(Am2/kg) | IEP |
| Fe3S4 | FeSO4 | Na2S | 3.5 | - | 10 | 20/100 | 35 | 14 | 7.0 |
| Fe3S4 | FeSO4 | Na2S | 3.5 | citrate | 10 | 20/100 | 32 | 25 | 6.0 |
| Fe3S4 | FeSO4 | Na2S | 3.5 | CTAB | 10 | 20/100 | 38 | 1.5 | 7.2 |
| Fe3S4 | FeSO4 | Na2S | 3.5 | dextran | 10 | 20/100 | 31 | 26 | 6.7 |
| (tr:反应时间, Tr/Td:反应温度和干燥温度, Ms:饱和磁化强度, IEP:等电点) | |||||||||
 下载: 导出CSV
下载: 导出CSV
表 2 Fe3S4纳米粒子各种制备方法的优缺点
Table 2. Advantages and disadvantages of various preparation methods of Fe3S4 nanoparticles
| 制备方法 | 反应时间 | 温度/℃ | 优点 | 缺点 |
| 沉淀法 | 10min | 20~100 | 制备过程简单,消耗低,颗粒小,可用于工业生产 | 容易团聚,掺杂,退化;形状和粒径不易控制 |
| 水热法 | 1~24 h | 160~220 | 纯度高,分散好,洁净度高,粒径统一 | 操作复杂,成本高 |
| 热解法 | 1~40 h | 90~300 | 分散好,粒径大小统一,粒径、形貌容易控制 | 环境友好性差,生产成本高 |
下载: 导出CSV
表 3 Fe3S4纳米材料及复合材料之间的电化学性质对比
Table 3. Comparison of electrochemical properties between Fe3S4 nanomaterials and Fe3S4composite materials
| 材料 | 应用 | 电流密度/(A·g-1) | 循环次数 | 比容量/(mAh·g-1) | 参考文献 |
| Fe3S4八面体微晶 | 锂离子电池阳极材料 | 0.1 | 1/100 | 1161/563 | [85] |
| Fe3S4空心球型 | 锂离子电池阳极材料 | 0.2 | 100 | 750 | [41] |
| Fe3S4纳米片 | 锂离子电池阳极材料 | 0.1 | 1/100 | 1207/495 | [38] |
| Fe3S4八面体纳米粒子 | 钠离子电池阳极材料 | 20 | 3500 | 275 | [92] |
| Fe3S4纳米片 | 锂离子电池阳极材料 | 0.2 | 120 | 548 | [94] |
| Fe3S4 NPs@rGO | 锂离子电池阳极材料 | 0.1/1 | 100/800 | 950/720 | [90] |
| Iron sufides@rGO | 锂离子电池阳极材料 | 0.1/1 | 100/1000 | 1189.6/800 | [91] |
| Fe3S4微粒 | 可充电碱性水电池阳极材料 | 2/2 | 1/3000 | 110.6/83.8 | [95] |
| Fe3S4/Co9S8 | 锂离子电池阳极材料 | 0.1/2 | 100/1500 | 945/519 | [24] |
| Fe3S4@Li7P4S11 | 全固态锂电池阴极材料 | 0.1 | 200 | 1001 | [96] |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: