表 1
几种烯烃的氢化热
Table 1.
Hydrogenation heat of several olefins

共轭效应是有机化学中电子效应的一种,是阐释有机化合物某些性质、解决有机化学问题的基础理论之一。在共轭体系(π-π、p-π、p-p共轭)中,相邻的π键或p轨道之间相互影响,从而使体系中电子云密度重新分布,这种影响称为共轭效应(conjugative effect),标记为C。+C表示推电子共轭效应;-C表示拉电子共轭效应。共轭效应多见于多烯烃、α, β-不饱和羰基化合物、α, β-不饱和羧酸及其衍生物等有机化合物之中,其作用明显,应用广泛。
有机化学教科书中,通常是用氢化热的大小来说明共轭烯烃的稳定性[1]。如果一个分子含有多个双键,可以预计它的氢化热应是各个双键氢化热的总和。烯烃分子的氢化热越大,说明分子的内能越大而稳定性越差。例如表 1。
 下载:
导出CSV
下载:
导出CSV
| 烯烃名称 | 结构简式 | 氢化热/(kJ·mol-1) |
| 1-丁烯 | CH3CH2CH=CH2 | 126.8 |
| 1, 3-丁二烯 | CH2=CH-CH=CH2 | 238.9 |
| 1, 3-戊二烯 | CH2=CHCH=CHCH3 | 226 |
| 1, 4-戊二烯 | CH2=CHCH2CH=CH2 | 254 |
由表 1可知,共轭的1, 3-丁二烯分子中含有两个双键,理论上它的氢化热应该是1-丁烯的两倍,即126.8×2=253.6 kJ/mol,实测值是238.9kJ/mol,二者相差15.7kJ/mol;这意味着共轭双烯具有较低的内能,比一般烯烃稳定。1, 4-戊二烯和1, 3-戊二烯经氢化后都生成具有相同能量的戊烷,但它们的氢化热却相差28kJ/mol,说明共轭的1, 3-戊二烯比孤立的1, 4-戊二烯稳定,其内能差为28kJ/mol。
为何共轭烯烃较稳定呢?这与它的特殊结构有关。以1, 3-丁二烯为例,分子中的4个碳原子都是sp2杂化,处于同一平面,它们的p轨道相互平行,虽然在两个双键之间隔有一个单键,但由于位置较近,两个π键电子云可以在一定程度上相互作用而发生侧面重叠(图 1)。
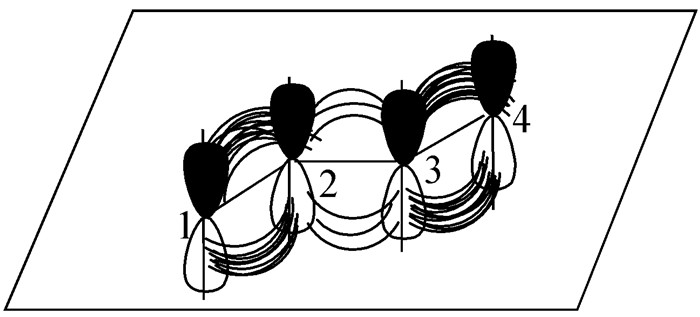
从图 1可看出,C1、C2和C3、C4原子未参与杂化p轨道的对称轴相互平行,侧面重叠分别形成π键;C2、C3的p轨道对称轴也相互平行,两个π键之间发生相互作用。这时π电子不是定域在C1=C2、C3=C4双键两个碳原子之间,而是发生离域,分布在四个碳原子之间,每一个电子不只受到两个碳核的束缚,而是受到4个碳核的束缚,因此增强了分子的稳定性。这种涉及π键之间的共轭即是π-π共轭,由于共轭作用降低的能量即是共轭能或离域能,相应的这个特殊结构体系就是共轭体系。因共轭体系的存在,导致分子内能降低,稳定性增大,并且随着共轭体系增大,也即共轭链增长,共轭能随之相应增大,分子的稳定性也就增大。
在共轭双烯或共轭多烯分子中,由于存在共轭效应,π电子离域在多个碳核控制的范围,其势能必然降低。对于1, 3-丁二烯分子,π电子离域在4个碳核控制的范围;因两个π键之间的相互作用使C2-C3之间的距离收缩,同时C1=C2与C3=C4之间的距离稍有拉伸,表现在键长的变化上。电子衍射法测定1, 3-丁二烯的结构发现,C2-C3单键的键长(0.148nm)[2]比普通的C-C单键(0.153nm)短,双键稍有拉长,但变化不大。这说明C2-C3单键已具有某些双键的性质,反映出共轭体系的特点,即共价键长的平均化作用。
共轭双烯或共轭多烯分子中共价键长平均化作用由共轭效应引起;共轭效应的存在,因轨道之间的相互作用,致使π电子的离域。离域电子活动范围的变大,导致整个共轭体系内电子云的分布变得均衡,实质上就是电子云的平均化变化;这种均衡变化使分子内出现了“和谐”状态,进而使分子的稳定性增大。共轭体系内电子云的平均化,体现出共轭效应的本质,表现为共价键长的平均化。
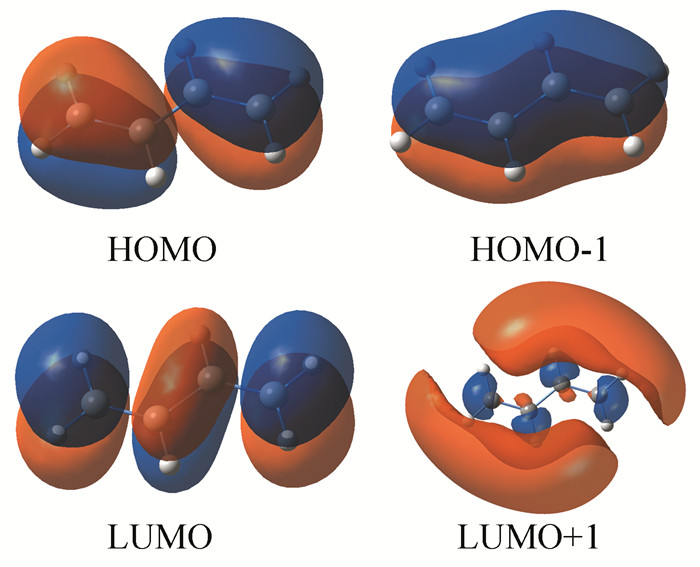
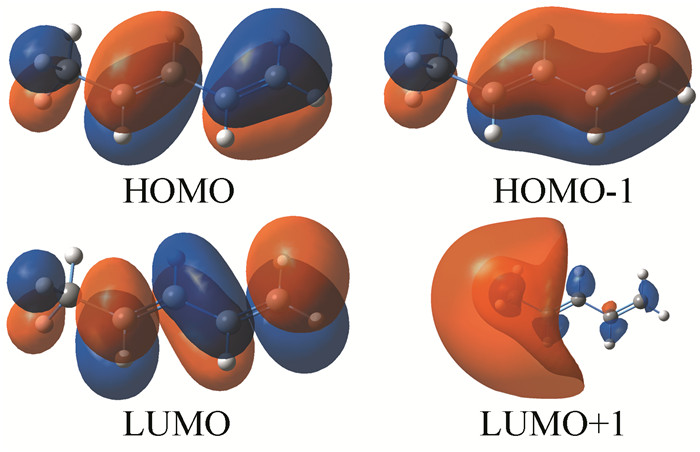
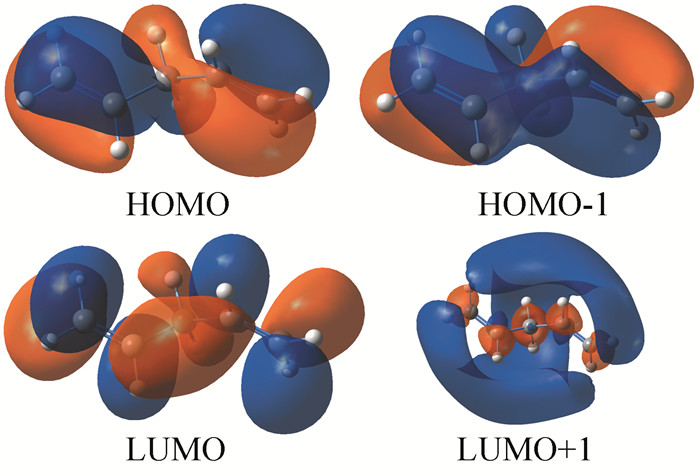
采用手工搭建方法构建了4个烯烃分子,在B3LYP/6-311+G(d, p)[3-4]水平上对该4个烯烃分子进行优化计算,给出了它们的最高占据分子轨道(HOMO)和最低空分子轨道(LUMO) (图 2~4),所有计算均采用Gaussian 09程序[5]。
分析图 2可看出,各烯烃分子轨道中的电子云分布变化较大,观察HOMO、LUMO、HOMO-1、LUMO+1等Kohn-Sham轨道发现,在1, 3-丁二烯分子中,由于双键之间形成共轭体系,π电子云之间相互重叠,形成一体化的电子云体系即电子云的平均化变化,π键电子不再定域而发生离域,存在分子内电荷转移现象,从而降低分子内能,并且使C2-C3之间的单键也缩短,C1-C2和C3-C4之间的双键拉长,趋于键长的平均化变化;这便是共轭效应的本质所在。对比分析图 3和图 4,1, 3-戊二烯分子内因有共轭大π键存在,HOMO、HOMO+1的电子云出现重叠现象,π键电子发生离域而显示共轭效应,从而降低了分子内能,稳定性较大;而在1, 4-戊二烯分子中,两个双键被两个C-C单键相隔,不能形成共轭体系,也无共轭效应,分子内能较高,稳定性较小。
有机化合物分子中存在共轭体系时,化学反应表现出较大变化。典型的反应体现在共轭烯烃的亲电加成和α, β-不饱和醛、酮等的加成反应中,如1, 3-丁二烯与Br2的亲电加成反应:
|
|
前一个反应中两个溴原子分别加到C1、C2(相邻的C)上,称为1, 2-加成;后一个反应中两个溴原子分别加到C1、C4上(共轭体系的首尾两端),同时双键转移至中间,称为1, 4-加成。
1, 3-丁二烯与其他亲电试剂的加成,与加Br2类似,也有1, 2-加成和1, 4-加成两种反应方式,相应生成两种产物。如氯化氢与1, 3-丁二烯的亲电加成反应:
|
|
以E+为亲电试剂,—Nu为亲核试剂,1, 3-丁二烯的亲电加成反应机理为:
|
|
从反应机理分析,第一步加上E+后生成碳正离子,由于烯丙基碳正离子的p-π共轭效应而存在共振离子,负性基团—Nu分别与之作用形成1, 2-加成和1, 4-加成产物。此外,两个共振离子也可以电子离域形式离子表示:
|
|
烯丙基碳正离子由于存在共轭效应而有较大的稳定性,正电荷主要分布在C2和C4上,C2上正电荷更稳定,负离子进攻C2和C4,分别得到1, 2-加成和1, 4-加成产物。对于氯化氢与1, 3-丁二烯的亲电加成反应,得如下所示产物(见下页)。
|
|
从能量角度分析,1, 2-加成属于动力学控制反应即速度控制,1, 4-加成属于热力学控制反应即平衡控制;通过对溴化氢与1, 3-丁二烯的加成反应研究[6]发现,1, 4-加成产物为主要产物。此种类型题常见于研究生招生考卷之中,列举三例如下:
例1:完成反应
|
|
例2:完成反应
|
|
例3:完成反应
|
|
例1和例2都是要求写出1, 2-加成与1, 4-加成产物,底物和试剂均是不对称分子,依据共轭效应比较易于完成;例3涉及两步反应,关键是第一步反应要判断正确,按1, 4-加成方式完成反应,之后变成单烯烃与HBr的加成反应,比较好判断。
对于α, β-不饱和醛、酮,因羰基与碳碳双键形成共轭体系而存在共轭效应,碳碳双键的存在影响着羰基的亲核加成反应活性与加成方式,同时还能发生亲电加成反应。如下式:
|
|
由于氧元素的电负性比碳元素的大,分子中出现偶极极化,1、3位带负电,2、4位带正电,以1, 2-位和1, 4-位两种方式进行亲核加成,生成1, 2-加成产物和1, 4-加成产物。需要注意的是,当带有氢原子的试剂(H+Nu-)与α, β-不饱和醛、酮进行1, 4-加成时,所生成的产物是烯醇结构,异构化得饱和的羰基化合物,相当于3, 4-加成,即对碳碳双键的加成,但本质上还属于1, 4-加成。
不同结构的醛、酮与不同的亲核试剂进行亲核加成反应时,因底物的结构差异和试剂的活性大小不同,1, 2-加成和1, 4-加成的倾向各不相同。底物的结构差异本质上也是活性大小不同。一般来说,底物中羰基附近空间位阻大,有利于1, 4-加成;空间位阻小,则有利于1, 2-加成。因此,α, β-不饱和醛倾向于1, 2-加成,而α, β-不饱和酮倾向于1, 4-加成。就亲核试剂而言,活性较高倾向于1, 2-加成,活性较低倾向于1, 4-加成。例如,α, β-不饱和酮与HCN反应,主要生成1, 4-加成产物:
|
|
而α, β-不饱和醛与HCN反应,则主要生产1, 2-加成产物:
|
|
格氏试剂活性较高,进行1, 2-加成的倾向较大,但底物结构也影响着加成方式。例如:
|
|
下面几例[7]反映的是试剂活性大小不同表现出不同的亲核加成倾向,从中可以找到一些规律:
|
|
受软硬酸碱规律支配,活性较高的格氏试剂、锂试剂等较硬,容易发生1, 2-位加成,而HCN和铜锂试剂是较软的Lewis碱,容易发生1, 4-加成。
此外,由于共轭效应的存在和影响,有机化合物的性质往往发生某些变化,如含有杂原子的五元杂环化合物噻吩、呋喃和吡咯:
|
|
它们的共同特征是杂原子上都带有处于p轨道上的孤对电子,与烯键形成共轭体系后共同构成5中心6电子环状大π键(以吡咯为例),并符合Hückel规则,因而具有芳香性;噻吩、吡咯、呋喃的芳香稳定化能分别为125.5、90.4、71.1 kJ/mol[8]。对于吡咯,本身是有弱碱性的,但由于N原子上的孤对电子参与形成环状大π键,致使N-H键极性增强而表现出弱酸性[9]:
|
|
在有机化学中,共轭效应的特殊性表现比较常见,在此不能一一列举,可在学习实践中不断总结,不断掌握。
王积涛, 张宝申, 王永梅等.有机化学(第二版).天津: 南开大学出版社, 2003, 90.
王积涛, 张宝申, 王永梅等.有机化学(第二版).天津: 南开大学出版社, 2003, 91.
A D Beck. J. Chem. Phys., 1993, 98(7): 5648~5659.
C Lee, W Yang, R G Parr. Phys. Rev. B, 1988, 37(2): 785~791.
M J Frisch, G W Trucks, H B Schlegel et al. Gaussian 09, Revision B. 01, Gaussian, Inc. Wallingford CT, 2010.
曾昭琼主编, 李景宁副主编.有机化学(上册, 第四版).北京: 高等教育出版社, 2004, 94.
王永梅, 王桂林编.有机化学提要·例题和习题.天津: 天津大学出版社, 1999, 181-182.
曾昭琼主编, 李景宁副主编.有机化学(下册, 第四版), 北京: 高等教育出版社, 2004, 205.
曾昭琼主编, 李景宁副主编.有机化学(下册, 第四版).北京: 高等教育出版社, 2004, 209.

图 1 1, 3-丁二烯中p电子和大π键示意图
Figure 1 Schematic diagram of p electrons and π bonds in 1, 3-butadiene
表 1 几种烯烃的氢化热
Table 1. Hydrogenation heat of several olefins
| 烯烃名称 | 结构简式 | 氢化热/(kJ·mol-1) |
| 1-丁烯 | CH3CH2CH=CH2 | 126.8 |
| 1, 3-丁二烯 | CH2=CH-CH=CH2 | 238.9 |
| 1, 3-戊二烯 | CH2=CHCH=CHCH3 | 226 |
| 1, 4-戊二烯 | CH2=CHCH2CH=CH2 | 254 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


















 下载:
下载: