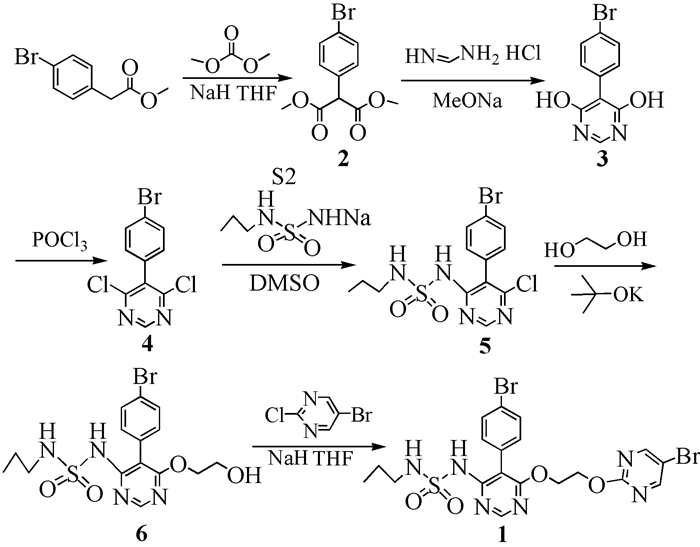
图式 1.
马西替坦的合成路线
Scheme 1.
Synthetic route of Macitentan

肺动脉高血压(PAH)是连接心脏与肺的动脉中产生的高血压,是一种慢性、进展性并使人衰弱的疾病,该疾病导致右侧心脏工作比正常情况困难,从而限制患者的运动能力并造成呼吸短促,严重可导致患者死亡或需肺移植[1]。内皮素作为当今所知的最强内源性缩血管物质,对维持基础血管张力与心血管系统稳态起着重要作用,具有良好的前景[2]。马西替坦{化学名为:N[-5-(4-溴苯基)-6-[2-[(5-溴嘧啶-2-基)氧]乙氧基]-嘧啶-4-基]-N′-丙基磺酰胺;分子式:C19H20Br2N6O4S;相对分子质量:588.273}是一种新型内皮素受体拮抗剂(ERA),其对内皮素A(ETA)受体和内皮素B(ETB)受体具有双重抑制作用,与内皮素受体结合后,可抑制由内皮素引起的血管收缩,降低肺动脉高压。马西替坦由爱可泰隆(Actelion)制药公司研制,商品名为Opsumit,该药先于2013年10月18日通过美国食品药品监督管理局(FDA)批准上市,后又于2013年12月20日通过欧洲药品管理局(EMA)批准上市用于治疗肺动脉高血压[3]。
目前,文献报道[4~15]的马西替坦合成路线主要有三条。原研公司埃科特莱茵研发了两条合成路线,第一条路线[4~7]为4-溴苯乙酸甲酯与碳酸二甲酯反应生成2-(4-溴苯基)丙二酸二甲酯(2),2与甲脒盐酸盐在甲醇钠的存在下关环生成5-(4-溴苯基)嘧啶-4, 6-二醇(3),3在N, N-二甲基苯胺的催化下,被三氯氧磷氯代为5-(4-溴苯基)-4, 6-二氯嘧啶(4),4与正丙胺磺酰胺钾盐发生亲核取代反应生成N-[5-(4-溴苯基)-6-氯嘧啶-4-基]-N′-丙胺基磺酰胺(5),5与乙二醇在强碱叔丁醇钾存在下反应生成N-[5-(4-溴苯基)-6-(2-羟乙基)嘧啶-4-基]-N′-丙胺基磺酰胺(6),6与2-氯-5-溴嘧啶在氢化钠存在下发生亲核取代反应生成最终产品马西替坦。第二条路线[8]为化合物5先与2-叔丁氧基乙醇反应,生成的产物再用四氯化钛脱去叔丁基生成化合物6,此条路线避免了相关杂质的生成,但是使用了在空气中极易发烟的四氯化钛以及价格较贵的2-叔丁氧基乙醇,使生产成本提高。第三条路线是李伟等的一项专利[9], 它由化合物4先和2-[(5-溴嘧啶-2-基)氧基]乙醇反应,再与正丙胺磺酰胺钾盐反应生成马西替坦,此条路线中2-[(5-溴嘧啶-2-基)氧基]乙醇的合成成本较高,导致总生产成本提高。本文对第一条路线进行了优化,通过实验确定了较佳的反应条件,尤其对中间体的纯化方法进行了研究,使中间体的纯度得到了明显提高,操作更加简单,适合于工业化生产。合成路线如图式 1所示。
14-溴苯乙酸甲酯、甲脒盐酸盐(纯度>98%,济南泰尔普化学有限公司);N-丙胺磺酰胺钠盐(纯度>99%,山东博洛德生物科技有限公司);5-溴-2-氯嘧啶(纯度>98 %,北京偶合);其他试剂均为市售分析纯级。
AVANCE-400核磁共振谱仪(TMS内标,德国Bruker公司);Trap VL型质谱仪(美国Agilent公司)。
向2L三口烧瓶中加入600mL四氢呋喃,分批加入34.9g(0.873mol)氢化钠,室温搅拌10min后,滴加100g(0.436mol)4-溴苯乙酸甲酯的四氢呋喃溶液300mL,滴加完毕,升温至45℃反应20min,然后降至室温,滴加78.7g(0.873mol)碳酸二甲酯,滴加完毕后室温搅拌12h。TLC检测反应完毕,降温至0℃,用25%的盐酸调节pH至7,浓缩除去四氢呋喃,用乙酸乙酯萃取(3×300mL),合并有机相,分别用300mL 1mol/L的盐酸和300mL饱和食盐水洗涤,经无水硫酸镁干燥后过滤,滤液减压蒸除溶剂得粘稠状固体,将其用160mL无水乙醇溶解,然后滴加80mL纯净水,搅拌30min,抽滤,烘干得78.8g类白色固体,收率62.9%。1H NMR(400MHz,DMSO-d6)δ7.59 (d,J=8.3 Hz,2H),7.34 (d,J=8.2 Hz,2H),5.11 (s,1H),3.68 (s,6H);ESI-MS m/z:287.0 [M+H]+。
将180mL无水甲醇加入2L三口烧瓶中,保持体系温度为10℃,慢慢加入14.4g(0.627mol)钠,加毕升至室温搅拌30min,然后滴加60g(0.209mol)化合物2的无水甲醇溶液(600mL),加毕,搅拌2h后加入20.2g(0.251mol)甲脒盐酸盐,搅拌24h。TLC检测反应完毕,将反应液浓缩,加入10%的柠檬酸,搅拌30min,抽滤,所得固体分别用100mL 10%的柠檬酸和100mL纯净水洗涤,然后再用无水乙醇50℃下搅拌4h,抽滤,40℃烘干,得37.5g类白色固体,收率67.2%。1H NMR (400MHz,DMSO-d6) δ8.10 (s,1H),7.61 (d,J=8.5 Hz,2H),7.45(d,J=8.6Hz,2H);ESI-MS m/z267.0 [M+H]+。
向500mL三口瓶中加入30g(0.112mol)化合物3、150mL三氯氧磷,然后慢慢滴加15mL N, N-二甲基苯胺,滴加完毕,升温至130℃反应2h。反应完毕,将反应液冷却至0℃左右,缓慢滴加300mL水,析出固体,抽滤,固体用5%的碳酸钾溶液(50mL×2)洗涤,然后将固体加入200mL丙酮中搅拌溶清,再慢慢滴加200mL纯净水,滴加完毕,搅拌1h,抽滤,烘干得22.5g类白色固体,收率65.8%。1H NMR(400MHz,DMSO-d6) δ8.97 (s,1H),7.76 (d,J=8.4Hz,2H),7.44 (d,J=8.4Hz,2H); ESI-MS m/z304.7 [M+H]+。
向1L三口瓶中加入20g(0.066mol)化合物4、200mL二甲基亚砜,搅拌溶清,再加入21.1g(0.132mol)正丙胺磺酰胺钠盐,25℃搅拌24h。TLC检测反应完毕,向反应体系中缓慢滴加600mL纯净水,滴加完毕,用醋酸调节溶液的pH至7,搅拌30min,抽滤,50℃下烘干得23.8g类白色固体,收率89.2%。1H NMR (400MHz,DMSO-d6) δ10.25 (s,1H),8.64 (s,1H),7.69 (d,J=8.4 Hz,2H),7.58 (s,1H),7.26 (d,J=8.4 Hz,2H),2.80 (t,J=7.1Hz,2H),1.41 (q,J=7.2Hz,2H),0.79 (t,J=7.4Hz,3H);ESI-MS m/z:406.9 [M+H]+。
向500mL三口瓶中加入20g(0.049mol)化合物5、16.5g(0.147mol)叔丁醇钾、100mL乙二醇,然后升温至100℃反应24h。TLC检测反应完毕,将反应液冷却至0℃,缓慢滴加10%柠檬酸溶液至pH=7,滴加过程中慢慢析出固体,滴加完毕搅拌30min,抽滤,固体用无水乙醇淋洗得18.2g类白色固体,收率86.2%。1HNMR(400MHz,DMSO-d6) δ9.78 (s,1H),8.48 (s,1H),7.62 (d,J=8.4Hz,2H),7.26 (d,J=8.4Hz,2H),7.23~7.16 (m,1H),4.73 (t,J=5.4Hz,1H),4.32 (t,J=5.2 Hz,2H),3.58 (q,J=5.2Hz,2H),2.79 (d,J=6.8Hz,2H),1.42 (m,J=7.3Hz,2H),0.80 (t,J=7.4Hz,3H);ESI-MS m/z432.9 [M+H]+。
向1L三口瓶中加入400mL四氢呋喃,分批加入4.2g(0.105mol)氢化钠,然后加入15g(0.035mol)化合物6,加毕,搅拌30min后加入8.1g(0.042mol)2-氯-5-溴嘧啶,然后升温至60℃反应2h。TLC检测反应完毕,将反应液冷却至0℃,用水淬灭氢化钠,将反应液浓缩得油状物,加入100mL水,用二氯甲烷(80mL×3)萃取,合并有机相,经无水硫酸镁干燥后过滤,滤液减压蒸除溶剂得粘稠状固体,将所得固体用乙酸乙酯/无水甲醇(体积比为1:2.5)混合溶剂重结晶得16.7g白色固体,收率81.2%。1H NMR (400MHz,DMSO-d6) δ:9.82 (s,1H),8.71 (s,2H),8.49 (s,1H),7.54 (d,J=8.1Hz,2H),7.24 (t,J=6.0 Hz,1H),7.15 (d,J=8.1Hz,2H),4.68~4.62 (m,2H),4.60~4.55 (m,2H),2.79 (q,J=6.7 Hz,2H),1.42 (q,J=7.3Hz,2H),0.80 (t,J=7.4 Hz,3H);ESI-MS m/z589.0 [M+H]+。
制备化合物2时,原研文献中氢化钠与4-溴苯乙酸甲酯的摩尔比为4:1,通过对反应机理的分析以及反应条件的尝试,最终采用氢化钠与4-溴苯乙酸甲酯的摩尔比为2:1,此比例即可保证反应完全,使成本降低且减小了实验的危险性。其次,按照原研文献得到的化合物2时副产物较多,对其纯化条件进行了研究,最终采用无水乙醇和水(体积比为2:1)的混合溶剂对粗品进行纯化,此方法能够去除杂质,得到纯度较高的产品。
制备化合物3时,按照原研文献得到的产品纯度不高,本实验对其纯化条件进行了研究,最终采用无水乙醇50℃下搅拌4h,这样得到的产品状态和纯度较好。
制备化合物4时,文献的后处理方法为浓缩除去三氯氧磷,加入纯净水,再用二氯甲烷萃取,然后浓缩有机相得到产品。本实验对反应后处理进行了改进,反应完毕后只需将反应液冷却,然后慢慢向反应液中滴加纯净水,便析出产品,操作更加简便,易于工业化。而且还对产品的纯化方法进行了研究,最终采用丙酮和水体积比为1:1的混合溶剂进行纯化。
制备化合物5时,文献的后处理方法为加入饱和NaCl溶液,用乙酸乙酯萃取,浓缩有机相得到产品,本实验对其进行了优化,直接滴加纯净水,再用醋酸调节溶液的pH至7,即可析出产品。和原研路线相比,操作简单,成本降低,而且产品的纯度明显提高。
制备化合物6时,对反应条件进行了改进,直接用乙二醇作为溶剂,和原研文献相比,避免了用乙酸乙酯萃取的过程,使操作更加简便,而且得到的产品纯度更高。
最后,对马西替坦的纯化条件进行了研究,结果表明,采用乙酸乙酯/无水甲醇(体积比为1:2.5)混合溶剂进行重结晶,可以大幅提高产物马西替坦的纯度。
本文通过六步反应合成了抗高压药物马西替坦,并对马西替坦的合成工艺进行了优化,使中间体的纯度大大提高,后处理操作更加简便,成本降低,适合较大规模生产。
M Hoeper, V Mclaughlin, A Dalaan et al. Lancet Respir. Med., 2016, 4(4):323~326. doi: 10.1016/S2213-2600(15)00542-1
G Feger, L Schilingl, H Ehrenreich et al. Res. Exp. Med., 1997, 196(6):327~337. doi: 10.1007/s004330050042
金盛飞, 程卯生.中国药物化学杂志, 2014, 24(2):166. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zgyh201402016&dbname=CJFD&dbcode=CJFQ
M Bolli, C Boss, C Binke et al. J. Med. Chem., 2012, 55(17):7849~7861. doi: 10.1021/jm3009103
M Bolli, C Boss, C Binke et al. US:20080004298, 2007.
C Boss, W Fischili, T Weller et al. WO:2006051502, 2006.
M Bolli, C Boss, A Trexander et al. US:20120142716, 2012.
A Stefan, F Jacques, S Ivan et al. WO:2014155304, 2014.
李伟, 高河勇, 陈琳. CN:105272923A, 2016.
王雪根, 何凌云, 郭莉芹等. CN:104447572A, 2015.
A Stefan, F Jacques, S Ivan et al. WO:2015121397, 2015.
A Stefan, F Jacques, S Ivan et al. WO:2015004265, 2015.
叶丁, 龚义, 丁诚等. CN:104311492A, 2014.
吴标, 凌林, 唐胜国等. CN:105461639A, 2016.
吴标, 凌林, 籍业等. CN:105461638A, 2016.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
 下载:
下载: