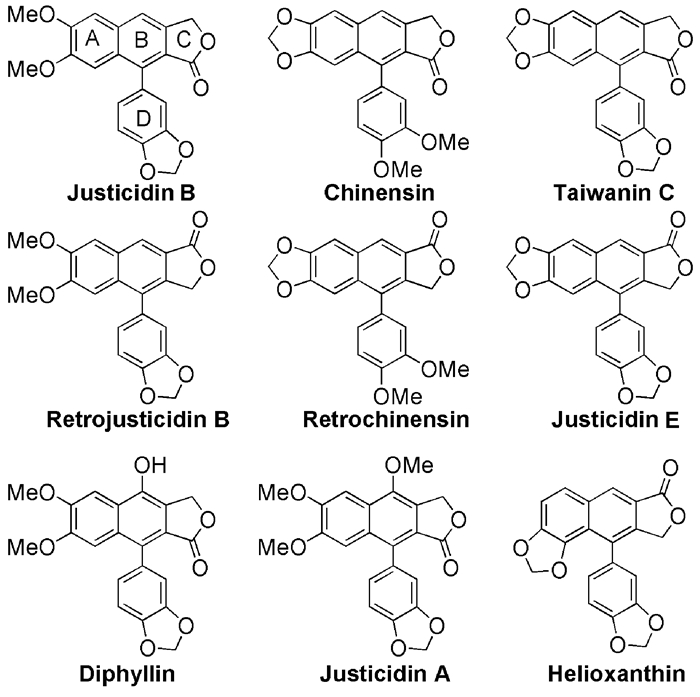
图式 1.
芳基萘类木脂素内酯代表化合物
Scheme 1.
Representative members of α-arylnaphthalene lignan lactones

芳基萘类木脂素内酯化合物是一类含有芳基萘骨架的天然产物,拥有广泛的生物活性[1],如磷酸二酯酶抑制活性[2]、抗菌[3]、抗病毒[4]、抗HIV病毒活性[5]、细胞毒[6]和抗肿瘤活性[7]等,从而引起了人们的高度重视,成为近年来较为活跃的研究领域之一。其中,代表性化合物Justicidin B[8]被报道可以抑制卵巢切除的小鼠体内的骨吸收,同时具有显著的抗病毒活性;而Retrojusticidin B及其衍生物具有强烈的人免疫缺陷病毒-1逆转录酶(HIV-1 RT)抑制活性[9]。该类化合物根据结构中的内酯环构型可以分为antero(type-A)型和retro(type-B)型。例如,化合物Justicidin B、Chinensin和Taiwanin C的内酯环是antero型,而化合物Retrojusticidin B、Retrochinensin和Justicidin E的内酯环是retro型。retro型的内酯化合物在自然界较为少见,其活性研究也相对较少。关于α-苯基萘类木脂素内酯化合物的提取分离、活性研究、化学合成和结构改造均有大量的报道[10]。图式 1示出了代表性的芳基萘类木脂素内酯化合物的结构式。但关于该类化合物合成方法的综述相对较少。本文对自1980年以来的α-苯基萘类木脂素内酯化合物的合成方法进行分类综述,旨在为该类化合物的合成和开发利用提供参考。
Stevenson等[11]报道了通过加热取代的芳香炔酸炔醇酯,直接发生分子内环化反应形成苯基萘木脂素内酯骨架,同时得到antero-型和retro-型两种产物(式(1))。该方法过程简单但收率不高,三步反应总收率约为34%~53%,且由于苯环上取代基的不同使得产物立体结构相对复杂,需要柱色谱进行分离,不便于大量制备。此外,他们[12]将该方法用于Justicidin E和Taiwanin C等天然产物的合成。随后,Stevenson等[13]将芳香炔醇替换为芳香烯醇,再加热芳香炔酸烯醇酯发生分子内环化得到α-苯基二氢萘木脂素内酯骨架(式(2))。环合过程中会产生多种环和产物,因此需要柱色谱分离,两步反应总收率约为35%~76%。类似地,唐建生等[14]报道了碳酸钾催化相同前体的Diels-Alder(DA)反应形成α-苯基二氢萘木脂素内酯骨架,关键的环合反应收率为65%~88%。二氢萘骨架通过氧化可以得到苯基萘木脂素骨架。以上三种方法过程简单但收率不高,起始原料需要实验室合成,产物立体结构相对复杂,不便于大量衍生物的制备。
|
|
(1) |
|
|
(2) |
Kudoh等[15]利用具有1, 7-二芳基-1, 6-二炔的缩醛结构发生分子内阴离子DA反应构建了α-苯基萘骨架。首先,由取代的苯丙炔醇和取代的苯丙炔醛形成缩醛中间体12。该中间体再发生分子内DA反应形成α-苯基萘骨架,环化反应收率63%。缩醛结构再经选择性还原、氧化的条件下分别形成antero-型和retro-型两种产物(式(3)),三步总收率约为22%~35%。该方法过程简、单条件温和且收率相对较高,能同时得到antero-型和retro-型两种产物,适用于该类化合物的大量制备。
|
|
(3) |
Basak等[16]以取代的二苯丙炔基醚15为关键中间体,利用Garratt-Braverman环化法直接构建α-苯基萘骨架,再经过KMnO4/CuSO4苄位氧化得到了包括Justicidin E和Tiwanin C在内的一系列α-苯基萘木脂素内酯化合物(式(4))。该方法收率较高,环化过程会产生多个产物,产物根据取代基的不同而产率不同,各个类型的产物产率约为15%~46%。环化产物的苄基位经KMnO4/CuSO4氧化能得到两个不同构型的产物。该方法与Kudoh等报道的方法类似,过程简单且能同时得到antero-型和retro-型两种产物,适用于该类化合物的大量制备。
Brummond等[17]报道的分子内脱氢DA反应可以进行antero-构型选择性的定向合成,芳环上的取代基导致有两种可能产物。关键中间体6在微波促进下以硝基苯为溶剂时可以得到一对脱氢产物(α-苯基萘木脂素内酯,二者比例为1.8:1),收率为90%(式(5));以DMF为溶剂时得到二氢产物(α-苯基二氢萘木脂素内酯,二者比例为1.5:1),收率为81%。该方法简单、高效,可用于α-苯基萘木脂素内酯衍生物的规模化合成。
|
|
(4) |
|
|
(5) |
Shin等[18]报道了通过DA反应区域选择性合成苯基萘木脂素内酯环的方法。该方法通过一步反应直接构建苯基萘内酯骨架,且能控制内酯环的构型。若以烯酸17和炔醇3成酯的中间体18在醋酸酐中发生分子内DA反应,可得到retro-型产物,环合反应收率为80%;若以炔酸1和烯醇20成酯的中间体21在相同的条件下反应则得到antero-型产物,环合反应收率为85%。DA反应的产物为α-苯基二氢萘内酯,经二氯二氰基苯醌(DDQ)氧化得到α-苯基萘脂素内酯,氧化反应收率约为70%。Shin等用该方法合成了Taiwanin C等天然木脂素类化合物(式(6))。该方法可以有效地控制产物的立体构型,但在反应过程中使用DDQ作为氧化剂使得产物纯化过程相对复杂。
Rodrigo等[19]通过异苯并呋喃前体与丁炔二酸二甲酯发生分子间DA反应构建了4位含有羟基取代的α-苯基萘骨架。其中,关键中间体24分子中含有缩醛结构和苄位醇羟基,该中间体在加热的情况下可以形成异苯并呋喃中间体。异苯并呋喃中间体不经分离,直接与丁炔二酸二甲酯发生分子间DA反应形成环氧化合物25。环氧化合物25在酸性条件下芳构化为4-羟基-α-苯基萘。他们运用该方法合成了Diphyllin等一系列α-苯基萘木脂素内酯类天然产物(式(7)),总收率约为35%。该方法可以得到立体选择性产物,但该方法使用正丁基锂,条件苛刻,且收率不高。随后,研究者利用该方法合成了一类新的α-苯基萘木脂素内酯化合物,并筛选了合成类似物的抗肿瘤活性[20, 21]。
|
|
(6) |
Szcze śniak等[22]以取代的苯甲酰胺28为原料,经过锂试剂和锌的处理在羧基的邻位引入一个碳原子,形成苯并呋喃内酯结构30。中间体30再在邻甲氧基苯基锂的作用下形成异苯并呋喃结构31,该中间体与丁炔二酸二甲酯发生分子间DA反应形成环氧化合物32。环氧化合物32在酸性条件下芳构化为4-羟基-α-苯基萘(式(8)),总反应收率约为10%。该路线较多地使用了有机锂试剂,不利于大规模制备的应用。
|
|
(7) |
|
|
(8) |
Padwa等[23]报道了利用串联Pummerer-Diels-Alder反应构建α-苯基萘骨架。以胡椒醇为原料经过溴代后分子内引入乙硫基,乙硫基再经氧化形成亚砜基。中间体36和乙酸酐发生Pummerer重排反应,生成不稳定的苯并异呋喃结构中间体;中间体不经分离直接与马来酸二甲酯发生DA反应形成环氧化合物37,该步反应收率85%。化合物37再经芳构化、还原得到4位无取代的α-苯基萘骨架39。若不经过还原过程,直接进行氧化、芳构化可得到4-羟基-α-苯基萘产物。内酯环的形成采用选择性还原的方法进行,可以方便地得到立体选择性的产物。Padwa利用该方法合成了Justicidin E和Taiwanin C等天然苯基萘类木脂素(式(9)),收率为56%(以化合物36计算)。该方法过程较长,以起始产物计算收率不高,多处需要柱色谱纯化,不适于大量制备。
|
|
(9) |
Avila等[24]报道芳基亲核取代反应构建α-苯基萘骨架,该方法由1, 4-二甲氧基-2-萘甲酸(41)为起始原料,经异噁唑保护羧基、苯基格氏试剂亲核进攻萘环的α-甲氧基从而引入α-苯基。苯基萘骨架43再经有机锂试剂的作用下引入一个碳,与原有的β位羧基成内酯环(式(10))。该方法使用格氏反应和有机锂试剂,条件苛刻,收率不高,且多需要柱色谱纯化,多步反应总收率不足20%。
Miyano等[25]报道了类似的格氏试剂芳基亲核取代反应。该方法先利用DA反应构建α位甲氧基取代的萘环骨架,得到关键中间体48;再用格氏试剂亲核取代中间体48的α-甲氧基,从而引入α苯基。α位亲核取代反应的顺利进行需要β-位存在酯基诱导效应,否则该反应无法进行。他们等利用该方法合成了Taiwanin C等天然产物(式(11)),由于该条路线较长,因此总收率不足20%。
|
|
(10) |
|
|
(11) |
范学森等[26]报道了一个高效合成α-氨基萘和α-苯基萘的方法。该方法通过2-炔基苯腈52与Reformatsky试剂53的串联反应一步构建α-氨基萘内酯骨架54。α-位的氨基再经重氮化、碘代反应转化为化合物55,中间体55再与苯硼酸试剂在钯催化下进行Suzuki-Miyaura偶联最终得到α-苯基萘木脂素内酯化合物。该方法简便、高效,适用于该类化合物的快速合成。他们将该方法用于Taiwanin C等天然产物的合成(式(12)),三步反应总收率高达60%。
|
|
(12) |
Shin等[27]通过分子内环化和Suzuki偶联反应构建了α-苯基萘内酯。该方法以3, 4-二甲氧基苯甲醛为起始原料,经过碳链延伸及成酯得到关键中间体58。中间体58再经安息香缩合反应、分子内环化反应得到α-羟基萘内酯化合物60。α-羟基经三氟甲磺酸化得到关键中间体三氟甲磺酸酯61,以起始原料计算中间体61的总收率为20%。中间体61再与不同取代的苯硼酸偶联得到α-苯基萘内酯类化合物,偶联反应的收率约为80%。该方法可以选择性地合成antero-型产物,且由参与反应的取代苯硼酸不同可以快速得到结构多样的产物(式(13))。
类似地,Mal等[28]以苯甲酸酯62作为受体,与2, 3-丁二烯酸乙酯在LDA的存在下一锅法合成萘并[c]呋喃酮60,该中间体收率为60%。得到化合物60后,再利用Shin等的方法合成得到Justicidin B(式(14))。该方法更为简便,收率更高,可以选择性合成antero-型产物,适合用于快速得到结构多样的类似物。
|
|
(13) |
|
|
(14) |
郭留城等[29]报道了一条以分子间DA反应和Suzuki偶联反应为关键步骤的α-苯基萘木脂素内酯化合物的合成路线。该路线以香草酸为起始原料,经选择性还原得到半缩醛中间体,该中间体不稳定,不经分离直接与丁炔二酸二甲酯进行DA反应得到环氧化物63。环氧化物63经芳构化、三氟甲磺酸酯化以47%的总收率得到关键中间体65。中间体65再与不同的苯硼酸进行Suzuki-Miyaura偶联反应构筑α-苯基萘骨架(偶联反应率45%~89%),最后经还原和Fetizon氧化反应实现了Justicidin B等6个天然产物的合成(式(15)),氧化反应收率为81%~97%。该方法可以同时得到antero-型和retro-型两种产物,并且适用于该类衍生物快速、多样化合成。中间体67在钌催化剂[30]的作用下将邻二羟甲基氧化可以选择性的得到antero-型产物。
Ham等[31]以保护的溴代苯甲醛68为起始原料经过取代、分子内环化,得到氰基异苯并呋喃酮关键中间体70。该中间体通过关键的Hauser-Kraus环化反应形成α-羟基萘内酯中间体71,α-羟基经三氟甲磺酸化得到关键中间体72,总收率65%。三氟甲磺酸酯72再与取代的苯硼酸偶联以高收率(约90%)得到α-苯基萘内酯类化合物(式(16))。该方法具有立体选择性,通过选择不同环化反应的亲核试剂,可以控制α-羟基取代的萘内酯中间体的构型,从而得到立体选择性产物。不足之处在于该方法中需要使用有机锂试剂和剧毒的氰化钾。
|
|
(15) |
|
|
(16) |
Iwasaki等[32]报道了通过共轭加成醛醇缩合反应合成了α-苯基萘木脂素内酯。该方法以保护的氰基苄醇73为原料在碱的作用下与丙烯酸甲酯和取代的苯甲醛进行分子间串联亲电反应得到关键中间体74。中间体74经过分子内环化、脱保护、再在碱性条件下引入一个碳,得到中间体77。中间体77极易在酸性条件下内酯化得到4-羟基-α-苯基萘木脂素内酯(式(17))。该方法由于起始分子内亲电反应产物复杂,必须经柱色谱分离,从而影响产率(不足20%),因此不适用于大量制备该类化合物。Iwasaki等应用该方法合成了Diphyllin等天然产物。
随后,他们[33]改进了上述路线,将丙烯酸甲酯替换为5H-呋喃-2-酮和取代的苯甲醛进行分子间串联亲电反应,得到关键中间体78。该中间体再在酸性条件下一锅反应得到4位无取代的α-苯基萘木脂素内酯类化合物(式(18)),收率有所提高。该方法步骤较少、操作简单,适用于该类化合物的快速合成。
|
|
(17) |
|
|
(18) |
Nishii等[34]报道了一条立体选择性芳构化合成α-苯基萘木脂素内酯的路线。该路线以二氯二甲基环丙烷甲酰氯81为起始产物与格氏试剂进行偶联,得到的酮类化合物82再与取代苯基锂试剂反应得到关键二氯环丙烷中间体83。该中间体在四氯化锡的作用下发生分子内芳构化形成α-苯基萘骨架中间体84,再经一系列的取代基变换得到α-苯基萘木脂素内酯(式(19))。该方法具有一定的立体选择性,但反应过程复杂且中间体的收率较低(总收率不到10%),加之反应过程与苯环上的取代基相关,因此不适用该类衍生物的快速合成。
|
|
(19) |
Balamurugan等[35]报道了一类金盐催化的分子内羰基与炔基之间的连续亲电反应和芳环化反应构建α-苯基萘骨架。该方法以取代的苯丙烯醇20为起始原料,先经过一系列的反应构建了苄基β酮酸苯丙炔酯93,收率38%。该关键中间体93在金盐的作用下发生分子内亲电反应和芳香化反应形成具有二苄基醚结构的α-苯基萘骨架16,环化反应收率为88%。最后,中间体16再经选择性氧化得到Justicidin E和Taiwanin C (式(20))。该方法由于步骤较长,因此收率不高(总收率约为24%),同时使用了钯、金等昂贵的金属催化剂,使得成本较高。
|
|
(20) |
Anastas等[36]在银催化下,通过取代的苯乙炔95、二氧化碳、和3-溴-1-苯基-1-丙炔之间一锅多组分分子间偶联反应直接构建苯基萘内酯骨架,同时得到antero-型(收率41%)和retro-型(收率25%)产物(式(21))。而后他们在该方法的基础进行了条件优化和底物广泛性扩展[37],将3-溴-1-苯基-1-丙炔替换成3-氯-1-苯基-1-丙炔和3-苯基-2-丙炔醇对甲基苯磺酸酯,仍然可以得到产物,而当底物为3-苯基-2-丙炔醇乙酸酯时无产物生成。该方法得到的antero-型产物相对较多,retro-型产物相对较少。他们运用该方法合成了一系列的天然产物[38]。该方法为一步反应,因此过程简单,但需要高温高压反应条件,产物需要柱色谱分离纯化,一定程度上限制了应用。
Narender等[39]通过银催化取代β-芳香酮酸酯与炔烃的碳氢键活化及官能团化反应构建了α-苯基萘骨架。该方法以二羰基化合物96和苯炔酯化合物97在银离子的催化下进行分子内碳氢键活化偶联(C(sp3)-H/C(sp)-H,C(sp2)-H/C(sp)-H),得到关键中间体98。反应以水为溶剂,具有条件温和、底物适应性广、区域选择性等特点,构建骨架单步反应收率在75%以上。再经选择性还原引入内酯环,得到α-苯基萘木脂素内酯。该方法过程相对简单,适用于合成4位羟基取代的苯基萘类木脂素内酯化合物的合成。他们将该方法应用于苯基萘类木脂素内酯化合物的合成,得到了天然产物Diphyllin和Justicidin A(式(22))。
|
|
(21) |
|
|
(22) |
Mori等[40]利用零价钯催化的芳炔前体和二炔化合物进行[2+2+2]环化反应,从而构建联芳基化合物。其中,芳炔前体为三氟甲磺酸苯酯99,二炔化合物为炔醇的芳炔酸酯或者炔基胺与芳炔酸形成的酰胺。该方法可用于一步构建4位取代的α-苯基萘类木脂素内酯骨架,单步环合反应收率最高可达78%。芳炔酸与炔醇的相对位置使其产物的内酯环构型具有选择性(retro-型)。环合反应得到的产物为4位氨甲酰基取代的α-苯基萘类木脂素内酯,需经过一系列的官能团转化才能得到目标产物,因此后续的步骤影响了该方法的总收率(式(23))。
Argade等[41]通过钯催化的芳炔和苯基取代的非对称共轭二烯[2+2+2]分子间环化反应构筑了具有二苄基醚结构的α-苯基萘骨架。其中,芳炔为取代的三氟甲磺酸苯酯106,其与苯基取代的非对称共轭二烯107反应引入α-苯基。该方法一步构建α-苯基萘骨架,环合反应收率为58%~66%,内酯环则由选择性还原而得到。该方法收率高、条件温和、底物适用性广,可用于α-苯基萘木脂素内酯Justicidin B和Retrojusticidin B的合成(式(24))。
|
|
(23) |
|
|
(24) |
Harrowven等[42]报道了一条分子间连续Horner-Emmons-Claisen反应构建α-苯基萘骨架的合成途径。该方法以藜芦醛为起始原料,经溴代、还原、偶联及氧化反应形成二酮结构关键中间体112。中间体112再与磷酰丁二酸二乙酯在碱性条件下发生连续的Witting-Horner反应形成α-苯基萘骨架,单步环合反应收率为73%。最终,由选择性还原二酯结构而形成内酯环,同时得到antero-型和retro-型两种产物。随后,他们[43]将此方法关键反应中的亲核试剂替换为丁二酸二乙酯,该反应依然能顺利进行,但收率有所降低(环合反应收率58%)。利用此方法可合成Justicidin B和Retrojusticidin B等一系列天然产物(式(25))。该方法条件温和、底物适用性广,但由于路线较长、收率不高,Horner-Emmons-Claisen反应产物需要柱色谱分离纯化,限制了其广泛应用。
|
|
(25) |
Takano等[44]报道了分子内连续环加成反应构建α-苯基萘骨架的方法。关键中间体116与顺丁烯二酸酐发生成酯、质子化、消除等反应,后续在加热条件下进行分子内连续的环加成反应直接形成α-苯基萘骨架,单步环合反应收率约为38%。酸酐117再经选择性还原和成环过程得到Justicidin E和Taiwanin C等α-苯基萘木脂素内酯类化合物(式(26))。该方法需要用到丙二硫醇和正丁基锂,条件苛刻;并且由116到117的反应过程复杂,因此收率较低且需要柱色谱分离纯化产物,因此应用范围有限。
|
|
(26) |
Shia等[45]由锰介导的串联自由基环化过程合成了α-苯基萘类木脂素。该方法的关键步骤是醋酸锰催化的分子内串联自由基环化过程,一步同时构建α-二氢苯基萘和内酯环,环化反应收率71%~89%。生成的二氢苯基萘中间体再在碘化钐的催化下脱去一分子的氢氰酸最终得到Justicidin E和Helioxanthin(式(27)),总收率为33%和44%。该方法收率高、具有区域选择性,可以选择性得到retro-型产物。
|
|
(27) |
现有的α-苯基萘类木脂素内酯化学合成策略主要解决苯基萘骨架的构筑和内酯环的形成。其中,DA反应已经是构建苯基萘骨架的较为有效的方法,内酯环构型选择性的合成依然是需要解决的问题。偶联法适用于快速合成取代基多样性的α-苯基萘类木脂素内酯化合物。分子间串联反应需要解决条件苛刻、提高收率与选择性的问题。总之,至今具有较好应用价值的该类化合物合成方法还很少。因此,在发展和完善现有合成策略的同时,还需开展α-苯基萘类木脂素内酯类化合物新的合成方法的研究,特别是发展简便的、选择性高的构建α-苯基萘内酯骨架结构的新方法研究,为α-苯基萘类木脂素内酯类化合物的生物活性研究及开发利用提供保障。
R B Teponno, S Kusari, M Spiteller. Nat. Prod. Res., 2016, 33(9):1044~1092. doi: 10.1039/C6NP00021E
T Ukita, Y Nakamura, A Kubo et al. J. Med. Chem., 1999, 42(7):1293~1305. doi: 10.1021/jm9807048
K Kawazoe, A Yutani, K Tamemoto et al. J. Nat. Prod., 2001, 64(5):588~591. doi: 10.1021/np000307b
D Janmanchi, C H Lin, J Y Hsieh et al. Bioorg. Med. Chem., 2013, 21(7):2163~2176. doi: 10.1016/j.bmc.2012.11.037
H J Zhang, E Rumschlag-Booms, Y F Guan et al. Phytochemistry, 2017, 136(Supplement C):94~100. http://www.ncbi.nlm.nih.gov/pubmed/28110956
(a) K Kang, S H Oh, J H Yun et al. Neoplasia, 2011, 13(11):1043~1057; (b) Y Ren, D D Lantvit, Y Deng et al. J. Nat. Prod., 2014, 77(6):1494~1504. doi: 10.1593/neo.11972
J Luo, Y Hu, W Kong et al. PloS One, 2014, 9(3):e93516. doi: 10.1371/journal.pone.0093516
S Hemmati, H Seradj. Molecules, 2016, 21(7):820~840. doi: 10.3390/molecules21070820
K S Sagar, C C Chang, W K Wang et al. Bioorg. Med. Chem., 2004, 12(15):4045~4054. doi: 10.1016/j.bmc.2004.05.036
J Y Pan, S L Chen, M H Yang et al. Nat. Prod. Res., 2009, 26(10):1251~1292. doi: 10.1039/b910940d
R Stevenson, J V Weber. J. Nat. Prod., 1989, 52(2):367~375. doi: 10.1021/np50062a024
R Stevenson, J V Weber. J. Nat. Prod., 1991, 54(1):310~314. doi: 10.1021/np50073a042
P T Anastas, R Stevenson. J. Nat. Prod., 1991, 54(6):1687~1691. doi: 10.1021/np50078a035
唐建生, 郑敏.化学试剂, 2011, 33(10):955~957. doi: 10.3969/j.issn.0258-3283.2011.10.027
T Kudoh, A Shishido, K Ikeda et al. Synlett, 2013, 24(12):1509~1512. doi: 10.1055/s-00000083
S Mondal, M Maji, A Basak. Tetrahed. Lett., 2011, 52(11):1183~1186. doi: 10.1016/j.tetlet.2011.01.011
L S Kocsis, K M Brummond. Org. Lett., 2014, 16(16):4158~4161. doi: 10.1021/ol501853y
J E Park, J Lee, S Y Seo et al. Tetrahed. Lett., 2014, 55(4):818~820. doi: 10.1016/j.tetlet.2013.12.014
(a) S O D Silva, C S Denis, R Rodrigo. Chem. Commun., 1980, 21(21):995~997; (b) H P Plaumann, J G Smith, R Rodrigo. Chem. Commun., 1980, 8(8):354~355.
X Lu, M G Bi, S Wu et al. J. Asian Nat. Prod. Res., 2012, 14(4):322~326. doi: 10.1080/10286020.2011.653561
J Hui, Z Yu, L Zhu. Med. Chem. Res., 2012, 21(12):3994~4001. doi: 10.1007/s00044-011-9937-1
J Epsztajn, A Jóźwiak, A K Szcześniak. Tetrahedron, 1993, 49(4):929~938. doi: 10.1016/S0040-4020(01)80334-3
(a) J E Cochran, A Padwa. J. Org. Chem., 1995, 60(13):3938~3939; (b) A Padwa, J E Cochran, C O Kappe. J. Org. Chem., 1996, 61(11):3706~3714.
A I Meyers, W B Avila. J. Org. Chem., 1981, 46(19):3881~3886. doi: 10.1021/jo00332a024
(a) T Hattori, H Tanaka, Y Okaishi et al. Chem. Commun., 1995, 26(3):235~241; (b) T Hattori, M Suzuki, Y Komuro et al. J. Chem. Soc., Perkin Transac., 1995, 12(12):1473~1474.
Y He, X Y Zhang, X S Fan. Chem. Commun., 2014, 50(42):5641~5643. doi: 10.1039/C4CC01738B
F Hayat, L Kang, C Y Lee et al. Tetrahedron, 2015, 71(19):2945~2950. doi: 10.1016/j.tet.2015.03.023
D Mal, S Jana. J. Org. Chem., 2016, 81(23):11857~11865. doi: 10.1021/acs.joc.6b02313
孟瑾, 杜利月, 郭留城.有机化学, 2016, 36(11):2723~2728. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201611019&dbname=CJFD&dbcode=CJFQ
Y Ishii, T Ikariya, M Saburi et al. Tetrahed. Lett., 1986, 27(3):365~368. doi: 10.1016/S0040-4039(00)84020-4
T Kim, K H Jeong, K S Kang et al. Eur. J. Org. Chem., 2017, 2017(13):1704~1712. doi: 10.1002/ejoc.v2017.13
(a) T Ogiku, M Seki, M Takahashi et al. Tetrahed. Lett., 1990, 31(31):5487~5490; (b) T Ogiku, S I Yoshida, T Kuroda et al. Synlett, 1992, 1992(8):651~652.
T Ogiku, S Yoshida, H Ohmizu et al. J. Org. Chem., 1995, 60(14):4585~4590. doi: 10.1021/jo00119a041
E Yoshida, D Yamashita, R Sakai et al. Synlett, 2010, (15):2275~2278. http://ci.nii.ac.jp/naid/120005055197/ja/
V Gudla, R Balamurugan. J. Org. Chem., 2011, 76(24):9919~9933. doi: 10.1021/jo201918d
N Eghbali, J Eddy, P T Anastas. J. Org. Chem., 2008, 73(17):6932~6935. doi: 10.1021/jo801213m
P Foley, N Eghbali, P T Anastas. Green. Chem., 2010, 12(5):888~892. doi: 10.1039/b913685a
P Foley, N Eghbali, P T Anastas. J. Nat. Prod., 2010, 73(5):811~813. doi: 10.1021/np900667h
G. Naresh, R Kant, T Narender. Org. Lett., 2015, 17(14):3446~3449. doi: 10.1002/chin.201550096/pdf
Y Sato, T Tamura, A Kinbara et al. Adv. Synth. Catal., 2007, 349(4/5):647~661.
R M Patel, N P Argade. Org. Lett., 2013, 15(1):14~17. doi: 10.1021/ol3028658
D C Harrowven, M Bradley, J L Castro et al. Tetrahed. Lett., 2001, 42(39):6973~6975. doi: 10.1016/S0040-4039(01)01436-8
S R Flanagan, D C Harrowven, M Bradley. Tetrahedron, 2002, 58(30):5989~6001. doi: 10.1016/S0040-4020(02)00616-6
S Takano, S Otaki, K Ogasawara. Tetrahed. Lett., 1985, 26(13):1659~1660. doi: 10.1016/S0040-4039(00)98577-0
T T Kao, C C Lin, K S Shia. J. Org. Chem., 2015, 80(13):6708~6714. doi: 10.1021/acs.joc.5b00866

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




























 下载:
下载:
 下载:
下载: